 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSelective P4 activation by an organometallic nickel(I) radical: formation of a dinuclear nickel(II) tetraphosphide and related di- and trichalcogenides†‡
Stefan
Pelties
a,
Dirk
Herrmann
a,
Bas
de Bruin
b,
František
Hartl
c and
Robert
Wolf
*a
aUniversity of Regensburg, Institute of Inorganic Chemistry, 93040 Regensburg, Germany. E-mail: robert.wolf@ur.de
bUniversity of Amsterdam, Van't Hoff Institute for Molecular Sciences, Science Park 904, 1098 XH Amsterdam, The Netherlands
cUniversity of Reading, Department of Chemistry, Whiteknights, Reading, RG6 6AD, UK
First published on 14th May 2014
Abstract
The reaction of the 17e nickel(I) radical [CpNi(IDipp)] (1, IDipp = 1,3-bis(2,6-diisopropylphenyl)imidazolin-2-ylidene) with P4 results in a nickel tetraphosphide [{CpNi(IDipp)}2(μ-η1:η1-P4)] with a butterfly-P42− ligand; related chalcogenides [{CpNi(IDipp)}2(μ-E2)] (E = S, Se, Te) and [{CpNi(IDipp)}2(μ-E3)] (E = S, Se) are formed with S8, Se∞ and Te∞.
The P4 molecule is the most reactive allotrope of phosphorus; its activation and transformation by transition metal compounds has attracted substantial interest over the years.1 While many low-valent metal complexes, e.g. transition metal carbonyls or anionic metalates, react with P4, it is still challenging to design highly selective transformations.2,3
White phosphorus is able to efficiently trap organic and main group element radicals.4 Therefore, one potential solution to the selectivity issue is to use a radical pathway in transition metal-mediated P4 transformations. While 2nd and 3rd row metalloradicals are well-established,5 nickel(I) radicals have attracted significant attention recently.6,7 Importantly, Driess et al. have shown that reactions of β-diketiminato nickel(I) complexes with P4 yield dinuclear complexes [(LRNi)2(μ-η3:η3-P4)] (LR = HC[CMeN(2,6-R2C6H3)]2 with R = Et, iPr).8 The P–P bond activation in the doubly η3-coordinated ligand is reversible and occurs without the reduction of P4 to formally P42−.
We have been interested in designing new reactive nickel(I) radicals for element–element bond activations. We now report the synthesis of complexes 1–3§ featuring an NHC and a cyclopentadienyl ligand, and an initial reactivity study of complex 1 with P4 and related small molecules.
Complexes 1–3 are accessible according to Scheme 1 by the reduction of the appropriate nickel(II) halides with KC8 in THF.¶1H NMR monitoring shows that 1–3 are formed very selectively; they can be isolated as yellow crystalline solids in modest to high yields. Single X-ray structure analyses (ESI‡) revealed that the nickel centre is surrounded by the carbene carbon and one η5-coordinated Cp or Cp* moiety. No further significant interactions between nickel and the diisopropylphenyl groups are apparent. Nonetheless, the cyclopentadienyl ligand is tilted with respect to the nickel carbene bond with an angle Ccarbene–Ni–(C5R5)centroid of 154.3(1)° for 1, 151.9(1)° for 2 and 164.6(1)° for 3.§
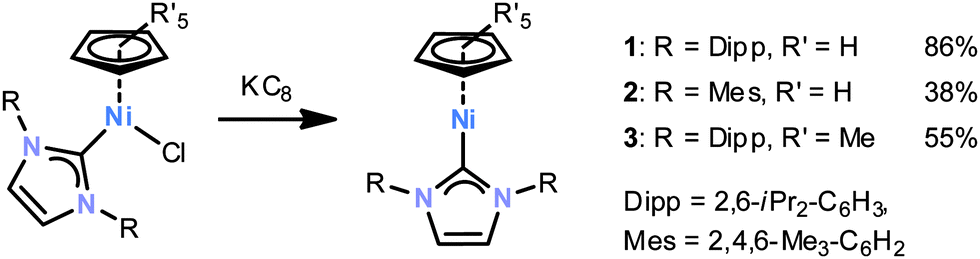 | ||
| Scheme 1 Synthesis of nickel(I) complexes 1−3. | ||
Cyclic voltammograms show one electrochemically quasi-reversible wave at E1/2 = −1.02 and −1.06 V vs. Fc/Fc+ for Cp-substituted 1 and 2, respectively, and a reversible wave at −1.18 V vs. Fc/Fc+ for the Cp* complex 3 (ESI‡). UV/vis-spectroelectrochemistry (see Fig. 1 for 1) confirms that these processes correspond to chemically reversible oxidations of neutral 1–3 to stable cationic nickel(II) complexes, which probably bind THF in the case of 1 and 2. Indeed, the preparative oxidation of 1 with [Cp2Fe]PF6 affords the THF adduct [(C5H5)Ni(IDipp)(THF)]PF6 (1-THF) (ESI‡).§
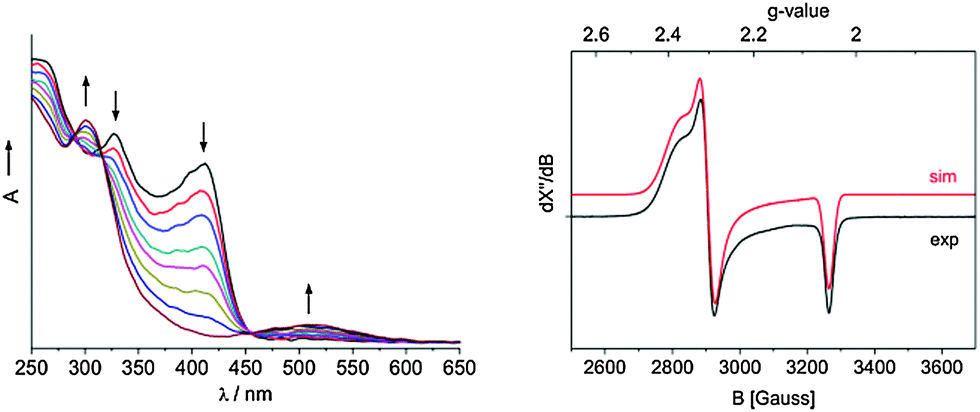 | ||
| Fig. 1 Left: UV/Vis monitoring of the oxidation of 1 performed at −0.83 V vs. Fc/Fc+ within an OTTLE cell equipped with a Pt minigrid working electrode, THF/TBAH under Ar, 293 K. Right: experimental and simulated X-band EPR spectrum of 1 in frozen THF. Freq. 9.3646 GHz, 0.063 mW, 20 K, mod. 4 Gauss; g-tensor parameters obtained from simulations and DFT calculations (b3-lyp, def2-TZVP) are: g11 = 2.377 (2.220), g22 = 2.306 (2.187), g33 = 2.050 (2.078) (DFT-calculated values in parentheses). | ||
Complexes 1–3 show identical magnetic moments of 2.3(1), 2.3(1), and 2.2(1) μB in [D8]THF, which indicate the presence of one unpaired electron per molecule. The EPR spectrum of 1 is characteristic for an S = 1/2 system and reveals a rhombic g-tensor with significant deviations from ge pointing to metalloradical character. DFT calculated g11 and g22 values are somewhat smaller than the experimental ones, but show a similar rhombicity (Fig. 1).
Initial reactivity studies of 1 established its behavior as a typical metal-centered radical. The reactions of phenyl disulfide and TEMPO with 1 in THF afforded the known thiolate [(C5H5)Ni(SPh)(IDipp)] (4)9 and the new TEMPO adduct 5 in quantitative yield (Fig. 2). The molecular structure of 5 shows a side-on η2-coordinated TEMPO ligand and an η1-coordinated Cp ligand at the distorted square planar nickel(II) atom. The structural parameters agree with presence of a formally anionic TEMPO− ligand.10 A sharp 1H NMR singlet at 5.93 ppm is observed for the Cp moiety even at −90 °C presumably due to rapid haptotropic migration.
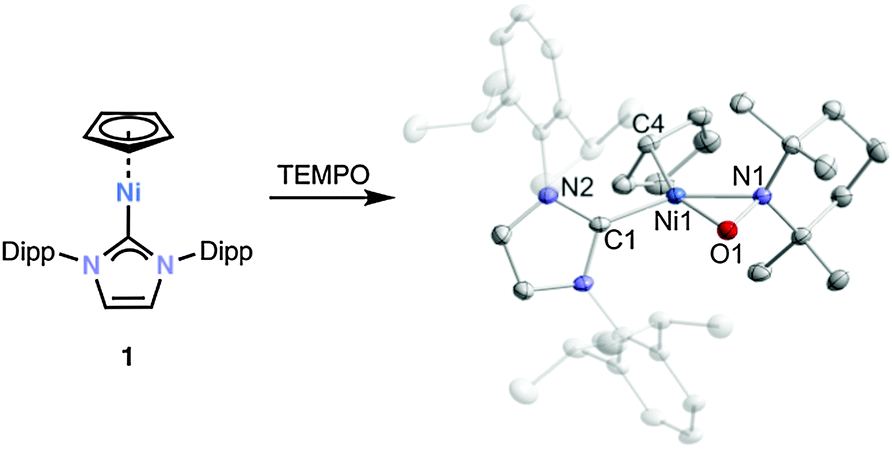 | ||
| Fig. 2 Reaction of 1 with TEMPO and solid-state molecular structure of [(C5H5)Ni(TEMPO)(IDipp)] (5). The hydrogen atoms are omitted for clarity. Thermal ellipsoids are drawn at 40% level. Selected bond lengths [Å] and angles [°]: Ni1–O1 1.8408(14), Ni1–N1 1.9581(16), N1–O1 1.3989(20), Ni1–C1 1.8824(19), Ni1–C4 2.034(2), C1–Ni1–O1 104.50(7), O1–Ni1–N1 43.07(6), C1–Ni1–C4 97.104(4), N1–Ni1–C4 115.325(2). | ||
We next investigated the reactivity of 1 with the heavier chalcogens. The reaction with S8 (1/8 equivalents) gave the blue disulfide 6-S and the purple trisulfide 7-S (Fig. 3) in a 7![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 ratio according to 1H NMR analysis. 6-S is soluble in n-hexane and diethyl ether and can thus be separated from 7-S by extraction and subsequent crystallisation (ESI‡). Disulfide-bridged dinuclear complexes with an M–S–S–M motif are well-known,11 while complexes with an unsupported μ-S32− bridge are still rather scarce.11a,b,12 The structure of 7-S shows a similar S1–S2–S3 angle and S–S bond lengths as the structure of [{(C5H5)Fe(CO)2}2(μ-S3)].11a Diselenide 6-Se (31% isolated) is the major reaction product of 1 with one equivalent of elemental selenium. A 1H NMR spectrum of the reaction mixture (THF, room temperature) shows that 6-Se is formed in more than 80% yield whereas the triselenide 7-Se is a minor by-product. Ditelluride 6-Te was the only product to be detected after stirring 1 with one equivalent of grey tellurium for seven days. It was isolated as a dark brown crystalline solid in 31% yield. The molecular structures of 6-Se, 6-Te and 7-Se are analogous to the corresponding sulfides 6-S and 7-S (ESI‡).
:3 ratio according to 1H NMR analysis. 6-S is soluble in n-hexane and diethyl ether and can thus be separated from 7-S by extraction and subsequent crystallisation (ESI‡). Disulfide-bridged dinuclear complexes with an M–S–S–M motif are well-known,11 while complexes with an unsupported μ-S32− bridge are still rather scarce.11a,b,12 The structure of 7-S shows a similar S1–S2–S3 angle and S–S bond lengths as the structure of [{(C5H5)Fe(CO)2}2(μ-S3)].11a Diselenide 6-Se (31% isolated) is the major reaction product of 1 with one equivalent of elemental selenium. A 1H NMR spectrum of the reaction mixture (THF, room temperature) shows that 6-Se is formed in more than 80% yield whereas the triselenide 7-Se is a minor by-product. Ditelluride 6-Te was the only product to be detected after stirring 1 with one equivalent of grey tellurium for seven days. It was isolated as a dark brown crystalline solid in 31% yield. The molecular structures of 6-Se, 6-Te and 7-Se are analogous to the corresponding sulfides 6-S and 7-S (ESI‡).
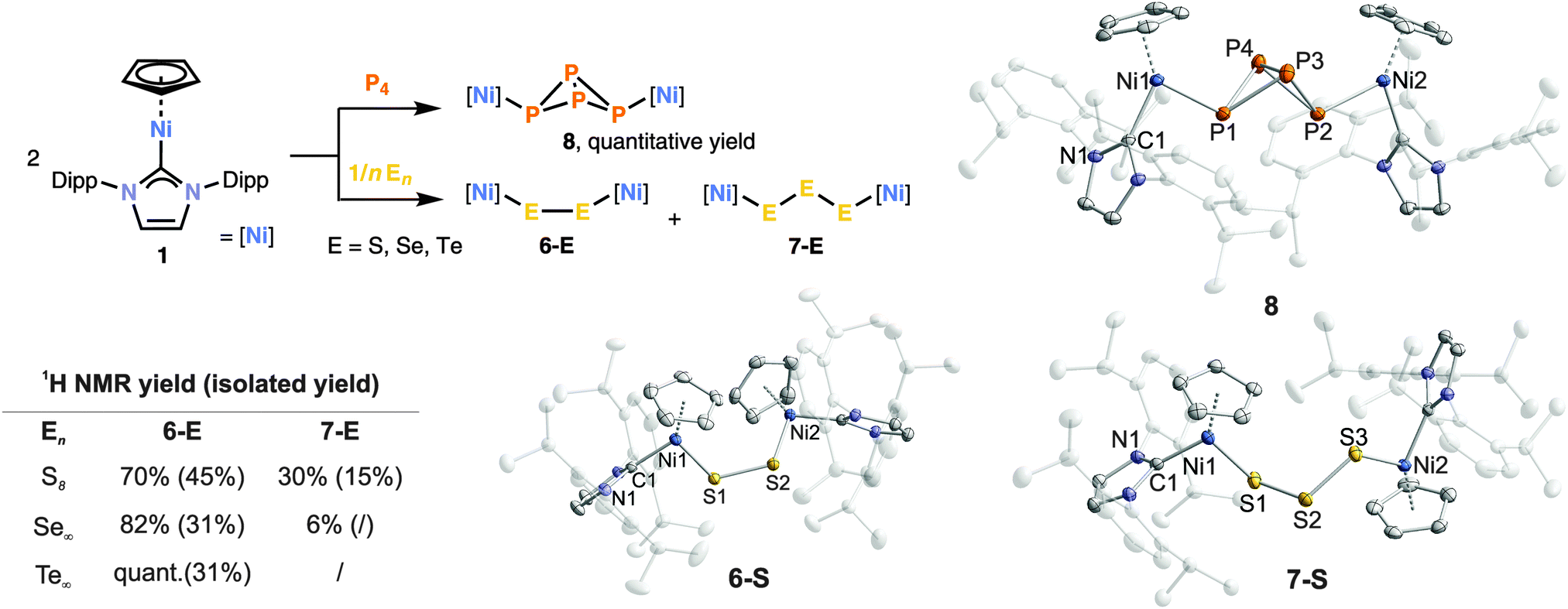 | ||
| Fig. 3 Left: reactions of 1 with P4, S8, Se∞ and Te∞. Right: solid-state molecular structures of the products 6-S, 7-S and 8. The hydrogen atoms are omitted for clarity. Thermal ellipsoids are drawn at 40% level. Selected bond lengths [Å] and angles [°]: 6-S: Ni1–S1/Ni2–S2 2.1800(1)/2.1797(1), S1–S2 2.0476(1), Ni1–S1–S2–Ni2 78.601(5), 7-S: Ni1–S1/Ni2–S3 2.1936(6)/2.1748(5), S1–S2/S2–S3 2.0561(7)/2.0522(7), S1–S2–S3 111.58(3), 8: Ni1–P1/Ni2–P2 2.2107(6)/2.2103(6), P1–P3/P4 2.2334(7)/2.2111(7), P3–P4 2.1649(7), P1–P2 2.8897(8). | ||
Considering that a mixture of at least two products is formed with sulfur and selenium, it was gratifying to discover that complex 1 reacts with P4 in a highly selective fashion in THF at room temperature, giving tetraphosphide 8 as the sole product. The reaction is instantaneous, and compound 8 can be isolated as an analytically pure, dark purple powder in quantitative yield simply by removing the solvent. Its molecular structure (Fig. 3) shows an exo/exo configuration for the two [(C5H5)Ni(IDipp)] units. The P–P bond lengths (2.2111(7)–2.2334(7) Å) are very similar to those in P4 (P–P 2.21 Å). The 31P{1H} NMR spectrum shows two triplets at δ = −307.4 and −45.8 ppm with 1JP–P = −190.5 Hz. These values are similar to those of [{CpRFe(CO)2}2(μ-η1:η1-P4)] (CpR = C5H3-1,3-tBu2, C5H2-1,2,4-tBu3, C5H-iPr4, C5Me5) and [{Cp*Cr(CO)3}2(μ-η1:η1-P4)], which also display a tetraphospha-[1.1.0]bicyclobutane framework.13
In conclusion, we have prepared rare mononuclear cyclopentadienyl nickel(I) complexes 1–3 with significant metalloradical character.6,7 This feature was successfully utilized for the high-yield synthesis of the novel tetraphosphido complex [{(C5H5)Ni(IDipp)}2(μ-η1:η1P4)] (8), which features an uncommon μ-η1:η1-bridging P42− ligand.14 Further reactivity studies of 1–3 and 8 are in progress; the results will be reported in due course.
We thank Christian Hoidn, Christian Preischl and Philipp Büschelberger for preparing 1–3 as part of their BSc projects. Financial support by the DFG and NWO (NWO-VICI 016.122.613) is gratefully acknowledged.
Notes and references
- (a) B. M. Cossairt, N. A. Piro and C. C. Cummins, Chem. Rev., 2010, 110, 4164 CrossRef CAS PubMed; (b) M. Caporali, L. Gonsalvi, A. Rossin and M. Peruzzini, Chem. Rev., 2010, 110, 4178 CrossRef CAS PubMed; (c) M. Scheer, G. Balázs and A. Seitz, Chem. Rev., 2010, 110, 4236 CrossRef CAS PubMed.
- (a) G. L. Simon and L. F. Dahl, J. Am. Chem. Soc., 1973, 95, 2175 CrossRef CAS; (b) O. J. Scherer, H. Sitzmann and G. Wolmershäuser, Angew. Chem., Int. Ed. Engl., 1985, 24, 351 CrossRef; (c) O. J. Scherer and T. Brück, Angew. Chem., Int. Ed. Engl., 1987, 26, 59 Search PubMed; (d) O. J. Scherer, M. Swarowsky, H. Swarowsky and G. Wolmershäuser, Angew. Chem., Int. Ed. Engl., 1988, 27, 694 CrossRef; (e) M. Scheer and U. Becker, Chem. Ber., 1996, 129, 1307 CrossRef CAS.
- (a) E. Urnežius, W. W. Brennessel, C. J. Cramer, J. E. Ellis and P. von R. Schleyer, Science, 2002, 295, 832 CrossRef PubMed; (b) E.-M. Schnöckelborg, J. J. Weigand and R. Wolf, Angew. Chem., Int. Ed., 2011, 50, 6657 CrossRef PubMed.
- (a) D. H. R. Barton and J. Zhu, J. Am. Chem. Soc., 1993, 115, 2071 CrossRef CAS; (b) D. H. Barton and R. A. Vonder Embse, Tetrahedron, 1998, 54, 12475 CrossRef CAS; (c) S. L. Hinchley, C. A. Morrison, D. W. H. Rankin, C. L. B. Macdonald, R. J. Wiacek, A. Voigt, A. H. Cowley, M. F. Lappert, G. Gundersen, J. A. C. Clyburne and P. P. Power, J. Am. Chem. Soc., 2001, 123, 9045 CrossRef CAS PubMed; (d) N. A. Giffin, A. D. Hendsbee, T. L. Roemmele, M. D. Lumsden, C. C. Pye and J. D. Masuda, Inorg. Chem., 2012, 51, 11837 CrossRef CAS PubMed.
- B. de Bruin, D. G. H. Hetterscheid, A. J. J. Koekkoek and H. Grützmacher, Prog. Inorg. Chem., 2007, 247 CrossRef CAS.
- (a) P. L. Holland, T. R. Cundari, L. L. Perez, N. A. Eckert and R. J. Lachicotte, J. Am. Chem. Soc., 2002, 124, 14416 CrossRef CAS PubMed; (b) N. A. Eckert, A. Dinescu, T. R. Cundari and P. L. Holland, Inorg. Chem., 2005, 44, 7702 CrossRef CAS PubMed; (c) B. R. Dible, M. S. Sigman and A. M. Arif, Inorg. Chem., 2005, 44, 3774 CrossRef CAS PubMed; (d) C. A. Laskowski and G. L. Hillhouse, J. Am. Chem. Soc., 2008, 130, 13846–13847 CrossRef CAS PubMed; (e) D. Bai, P. Wei and D. W. Stephan, Organometallics, 2005, 24, 5901 CrossRef; (f) C. J. E. Davies, M. J. Page, C. E. Ellul, M. F. Mahon and M. K. Whittlesey, Chem. Commun., 2010, 46, 5151 RSC; (g) M. Vogt, B. de Bruin, H. Berke, M. Trincado and H. Grützmacher, Chem. Sci., 2011, 2, 723 RSC; (h) K. Zhang, M. Conda-Sheridan, S. R. Cooke and J. Louie, Organometallics, 2011, 30, 2546 CrossRef CAS PubMed; (i) S. Nagao, T. Matsumoto, Y. Koga and K. Matsubara, Chem. Lett., 2011, 40, 1036 CrossRef CAS; (j) C. A. Laskowski, D. J. Bungum, S. M. Baldwin, S. A. Del Ciello, V. M. Iluc and G. L. Hillhouse, J. Am. Chem. Soc., 2013, 135, 18272 CrossRef CAS PubMed; (k) M. J. Page, W. Y. Lu, R. C. Poulten, E. Carter, A. G. Algarra, B. M. Kariuki, S. A. Macgregor, M. F. Mahon, K. J. Cavell, D. M. Murphy and M. K. Whittlesey, Chem. – Eur. J., 2013, 19, 2158 CrossRef CAS PubMed; (l) R. C. Poulten, M. J. Page, A. G. Algarra, J. J. Le Roy, I. López, E. Carter, A. Llobet, S. A. Macgregor, M. F. Mahon, D. M. Murphy, M. Murugesu and M. K. Whittlesey, J. Am. Chem. Soc., 2013, 135, 13640 CrossRef CAS PubMed.
- J. Wu, A. Nova, D. Balcells, G. W. Brudvig, W. Dai, M. L. M. Guard, N. Hazari, P.-H. Lin, R. Pokhrel and M. K. Takase, Chem. – Eur. J., 2014, 18, 5327 CrossRef PubMed.
- S. Yao, Y. Xiong, C. Milsmann, E. Bill, S. Pfirrmann, C. Limberg and M. Driess, Chem. – Eur. J., 2010, 16, 436 CrossRef CAS PubMed.
- D. A. Malyshev, N. M. Scott, N. Marion, E. D. Stevens, V. P. Ananikov, I. P. Beletskaya and S. P. Nolan, Organometallics, 2006, 25, 446 CrossRef.
- (a) M. H. Dickman and R. J. Doedens, Inorg. Chem., 1982, 21, 682 CrossRef CAS; (b) M. K. Mahanthappa, K.-W. Huang, A. P. Cole and R. M. Waymouth, Chem. Commun., 2002, 502 RSC; (c) D. Isrow and B. Captain, Inorg. Chem., 2011, 50, 5864 CrossRef CAS PubMed; (d) D. G. H. Hetterscheid, J. Kaiser, E. Reijerse, T. P. J. Peters, S. Thewissen, A. N. J. Blok, J. M. M. Smits, R. de Gelder and B. de Bruin, J. Am. Chem. Soc., 2005, 127, 1895 CrossRef CAS PubMed.
- Selected examples: (a) M. A. El-Hinnawi, A. A. Aruffo, B. D. Santarsiero, D. R. McAlister and V. Schomaker, Inorg. Chem., 1983, 22, 1585 CrossRef CAS; (b) N. Zhu, S. Du, X. Wu and J. Lu, Angew. Chem., Int. Ed. Engl., 1992, 31, 87 CrossRef; (c) M. Emirdag-Eanes and J. A. Ibers, Inorg. Chem., 2001, 40, 6910 CrossRef CAS PubMed; (d) J. T. York, E. C. Brown and W. B. Tolman, Angew. Chem., Int. Ed., 2005, 44, 7745 CrossRef CAS PubMed; (e) J. Hu, G. Liu, Q. Jiang, R. Zhang, W. Huang and H. Yan, Inorg. Chem., 2010, 49, 11199 CrossRef CAS PubMed; (f) L.-P. Wei, Z.-G. Ren, L.-W. Zhu, W.-Y. Yan, S. Sun, H.-F. Wang, J.-P. Lang and Z.-R. Sun, Inorg. Chem., 2011, 50, 4493 CrossRef CAS PubMed; (g) E. M. Matson, M. D. Goshert, J. J. Kiernicki, B. S. Newell, P. E. Fanwick, M. P. Shores, J. R. Walensky and S. C. Bart, Chem. – Eur. J., 2013, 19, 16167 CrossRef PubMed; (h) J. Wallick, C. G. Riordan and G. P. A. Yap, J. Am. Chem. Soc., 2013, 135, 14972 CrossRef CAS PubMed.
- (a) R. Steudel, M. Kustos and A. Prenzel, Z. Naturforsch., B: J. Chem. Sci., 1997, 52, 79 CAS; (b) E. Galardon, H. Daguet, P. Deschamps, P. Roussel, A. Tomas and I. Artaud, Dalton Trans., 2013, 42, 2817 RSC.
- (a) L. Weber and U. Sonnenberg, Chem. Ber., 1991, 124, 725 CrossRef CAS; (b) P. Jutzi and S. Opiela, J. Organomet. Chem., 1992, 431, C29 CrossRef CAS; (c) O. J. Scherer, G. Schwarz and G. Wolmershäuser, Z. Anorg. Allg. Chem., 1996, 622, 95 CrossRef; (d) O. J. Scherer, T. Hilt and G. Wolmershäuser, Organometallics, 1998, 17, 4110 CrossRef CAS; (e) C. Schwarzmaier, PhD thesis, University of Regensburg, 2012 Search PubMed.
- For related work on P4 activation by Ni0 complexes, see: B. Zarzycki, T. Zell, D. Schmidt and U. Radius, Eur. J. Inorg. Chem., 2013, 2051 CrossRef CAS , and literature cited therein.
Footnotes |
| † Dedicated to the memory of Prof. Michael F. Lappert. |
| ‡ Electronic supplementary information (ESI) available. Full experimental details, electrochemical, EPR and crystallographic data. CCDC 995931–995941 and 999501. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4cc02601b |
| § During the preparation of this manuscript, Hazari et al. reported the synthesis and characterization of 1, 1-THF and closely related mono- and dinuclear species by a different synthetic route.7 Based on DFT calculations, the bending of the Ccarbene–Ni–(C5H5)centroid angle in the structure of 1 was attributed to the asymmetric spin density distribution. |
| ¶ The hydride complex [(C5H5)NiH(IDipp)] (1-H) was identified as a minor by-product (<5%) of the synthesis of 1. Compound 1-H was prepared independently and features a distinct molecular structure from 1; see the ESI‡ for details. |
| This journal is © The Royal Society of Chemistry 2014 |