 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A trinuclear Zn3(OAc)4-3,3′-bis(aminoimino)binaphthoxide complex for highly efficient catalytic asymmetric iodolactonization†
Takayoshi
Arai
*a,
Noriyuki
Sugiyama
a,
Hyuma
Masu
b,
Sayaka
Kado
b,
Shinnosuke
Yabe
c and
Masahiro
Yamanaka
c
aDepartment of Chemistry, Graduate School of Science, Chiba University, 1-33 Yayoi, Inage-ku, Chiba, Japan. E-mail: tarai@faculty.chiba-u.jp; Fax: +81 43 290 2889
bCenter for Analytical Instrumentation, Chiba University, 1-33 Yayoi, Inage-ku, Chiba, Japan
cDepartment of Chemistry, Rikkyo University, 3-34-1 Nishi-Ikebukuro, Toshima-ku, Tokyo, Japan
First published on 24th April 2014
Abstract
A 3,3′-bis(aminoimino)BINOL ligand was newly designed and synthesized for the formation of a trinuclear Zn complex upon reaction with Zn(OAc)2. Using the harmony of the tri-zinc atoms, 1 mol% Zn3(OAc)4-3,3′-bis(aminoimino)binaphthoxide catalyzed asymmetric iodolactonization in up to 99.9% ee.
In nature, many metalloenzymes containing multiple metal centers show remarkable catalytic activity not observed in non- or monometallic active sites. The multiple metal ions in the multinuclear metalloenzymes act cooperatively to produce maximum activity. For example, phosphatidylcholine-preferring phospholipase C from Bacillus cereus (PC-PLCBc) catalyzes the hydrolysis of phospholipids. The active site of PC-PLCBc contains three Zn2+ ions, and the multiple zinc atoms are bridged by Asp122 and a water (or hydroxide) molecule.1 Employing multi-nuclear cooperative effects also becomes an important concept for the design of artificial “catalysts”,2 after pioneering works on the development of lanthanide-containing heterobimetallic asymmetric catalysts.3 While the bimetallic system in asymmetric catalyses has typically been produced using the force of self-assembly, the rational design of tri- or higher multinuclear complexes is still difficult even in cutting-edge chemistry. Here, we report a designer multinuclear metal catalyst for catalyzed asymmetric iodolactonization.
Iodolactonization,4–10 a type of halolactonization,11–13 represents a powerful synthetic tool for generating iodine-functionalized cyclic compounds in a single reaction. The resulting iodolactones are both versatile and useful, and have applications as synthetic intermediates in the total synthesis of natural products, as well as in the production of biologically significant pharmaceutical compounds and agricultural chemicals. Jacobsen pioneered an organo-catalytic version of a useful asymmetric iodolactonization reaction using an urea catalyst.4 More recently, Johnston reported an asymmetric iodolactonization catalyzed by a bis(amidine) (BAM)-based Brønsted acid.5 Within the limited examples of metal-catalyzed asymmetric iodolactonization, Gao reported the use of a mononuclear Co-salen complex as a Lewis acid catalyst for asymmetric iodolactonization.8,9 We have also reported an asymmetric iodolactonization strategy using a newly developed PyBidine(L1)-Ni(OAc)2 catalyst.10 The designer chiral ligands are prepared to form a multinuclear metal catalyst for use in asymmetric iodolactonization, through the story described in Scheme 1.
 | ||
| Scheme 1 The development of the chiral ligand of the multinuclear metal catalyst for use in asymmetric iodolactonization, and preliminary results on asymmetric iodolactonization using L1 and L2. | ||
Based on the mononuclear L1–Ni(OAc)2 catalyst, the imidazolidine ligand L2, with a chiral BINOL-backbone, was designed for a dinuclear metal complex. 3,3′-bis(imidazolidine)BINOL L2, prepared in situ by the condensation of monobenzyl (R,R)-diphenylethylenediamine with (R)-3,3′-formylbinaphthol, was applied to complex formation using 2 equiv. of metal acetate. In the asymmetric iodolactonization of 5-phenylhex-5-enoic acid (1a), although the L2–Ni(OAc)2 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 complex gave iodolactone 2a with 41% ee, the L2–Zn(OAc)2 1:2 complex drastically improved the optical yield of 2a to 98% ee. The diastereomeric 3,3′-bis(imidazolidine)BINOL, derived from (S,S)-diphenylethylenediamine with (R)-3,3′-formylbinaphthol, gave a 92% yield of 2a with 85% ee. The successful development of an efficient asymmetric catalyst encouraged further study to determine the structure of the L2–Zn(OAc)2 complex. The 1H-NMR study revealed that both L2 and the L2–Zn(OAc)2 1:2 complex existed as complex mixtures. The isolation of pure L2 required great effort, but using the 1:2 Zn(OAc)2 catalyst with isolated L2 gave 2a with a significantly lower selectivity of 62% ee compared to that obtained using the catalyst generated in situ. The imidazolidine ring of L2 prepared in situ was hypothesized to be in equilibrium with the opened aminoimino-form as shown in Scheme 2a, meaning that the 3,3′-bis(aminoimino)BINOL is a promising candidate for providing an effective Zn(OAc)2 catalyst.
:2 complex gave iodolactone 2a with 41% ee, the L2–Zn(OAc)2 1:2 complex drastically improved the optical yield of 2a to 98% ee. The diastereomeric 3,3′-bis(imidazolidine)BINOL, derived from (S,S)-diphenylethylenediamine with (R)-3,3′-formylbinaphthol, gave a 92% yield of 2a with 85% ee. The successful development of an efficient asymmetric catalyst encouraged further study to determine the structure of the L2–Zn(OAc)2 complex. The 1H-NMR study revealed that both L2 and the L2–Zn(OAc)2 1:2 complex existed as complex mixtures. The isolation of pure L2 required great effort, but using the 1:2 Zn(OAc)2 catalyst with isolated L2 gave 2a with a significantly lower selectivity of 62% ee compared to that obtained using the catalyst generated in situ. The imidazolidine ring of L2 prepared in situ was hypothesized to be in equilibrium with the opened aminoimino-form as shown in Scheme 2a, meaning that the 3,3′-bis(aminoimino)BINOL is a promising candidate for providing an effective Zn(OAc)2 catalyst.
 | ||
| Scheme 2 (a) The equilibrium of L2 between the imidazolidine and aminoimino forms. (b) The design and synthesis of the 3,3′-bis(tert-aminoimino)BINOL ligand (L3). | ||
To eliminate the cyclized imidazolidine from the equilibrium, the 3,3′-bis(tert-aminoimino)BINOL ligand (L3) in Scheme 2b was redesigned. The newly designed L3 can capture two or three metals in the flexible bis(aminoimino)binaphthol pocket.14 Reaction of (R)-3,3′-formylbinaphthol and (1R,2R)-2-(isoindolin-2-yl)-1,2-diphenylethan-1-amine (A)15 in ethanol proceeded smoothly at 80 °C to form the imine, and a cold ethanol wash of the resulting precipitate gave L3 as the sole product.16 The effect of changing the ratio of Zn(OAc)2 to L3 on catalyst performance was examined in Table 1. With an L3:Zn(OAc)2 ratio of 1:1 to 1:3, the iodolactone 2a was produced with 99% ee (entries 1–3). However, stopping the reaction within 2 h revealed that a 1:1 L3:Zn(OAc)2 ratio resulted in a slower reaction than using 1:2 or 1:3 ratios. The catalytic activity of several analogs was also investigated in Table 1. The diastereomer of L3 (L4) only gave 68% ee of 2a (entry 4). When one aminoimino functional group was eliminated from L3, the L5–Zn(OAc)2 catalyst yielded a product with 89% ee, although catalytic activity was significantly reduced (entry 5). Removal of the axial chirality of the binaphthyl skeleton resulted in a trace amount of 2a (entry 6). These results suggest that at least one set of zinc atoms, existing at the appropriate position, harmonizes cooperatively to produce iodolactones in a highly enantioselective manner.
:Zn(OAc)2 ratio and the analogs on catalytic activity
|
|
|||||
|---|---|---|---|---|---|
| Entry | Ligand | X (mol%) | Time (h) | Yield (%) | ee (%) |
| 1 | L3 | 1 | 2 | 19 | 99 |
| 2 | L3 | 2 | 2 | 64 | 99.6 |
| 3 | L3 | 3 | 2 | 69 | 99.6 |
| 4 | L4 | 3 | 2 | 9 | 68 |
| 5 | L5 | 2 | 24 | 34 | 89 |
| 6 | L6 | 1 | 24 | Trace | –– |

|
|||||

Results of investigations into the scope and generalization of the catalytic asymmetric iodolactonization are shown in Table 2.
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | R1 | R2 | n | Time (h) | Yield (%) | ee (%) |
|
a 0.1 mol% catalyst was used.
b 5 mol% catalyst was used.
c The solvent was toluene:CH2Cl2 = 4:1.
|
||||||
| 1 | C6H5 | H | 1 | 20 | >99 | 99.5 |
| 2a | C6H5 | H | 1 | 20 | >99 | 98 |
| 3 | p-BrC6H4 | H | 1 | 6 | >99 | 99.8 |
| 4 | p-ClC6H4 | H | 1 | 16 | >99 | 99.8 |
| 5 | p-FC6H4 | H | 1 | 15 | >99 | 94 |
| 6 | p-CF3C6H4 | H | 1 | 12 | >99 | 99.9 |
| 7 | p-MeC6H4 | H | 1 | 17.5 | >99 | 93 |
| 8 | m-MeC6H4 | H | 1 | 8 | >99 | 99.7 |
| 9b | o-MeC6H4 | H | 1 | 18 | >99 | 99.4 |
| 10 | p-MeOC6H4 | H | 1 | 12 | >99 | 82 |
| 11c | C6H5 | H | 0 | 9 | 96 | 87 |
| 12 | c-C6H11 | H | 1 | 4 | >99 | 99.3 |
| 13 | Me | H | 1 | 20 | 92 | 94 |
| 14 | C6H5 | Me | 1 | 24 | 74 | 99 |

Using 1 mol% L3–Zn3(OAc)4 catalyst, a variety of 5-arylhex-5-enoic acid substrates were converted quantitatively to the corresponding chiral gluconolactones with excellent enantioselectivity. For example, the p-trifluoromethyl-substituted compound was obtained in 99.9% ee (entry 6). The relatively less reactive substrate, having an o-substituent on the benzene ring, was used with 5 mol% catalyst to give the product in >99% yield with 99.4% ee (entry 9). The reaction of 4-phenylpent-4-enoic acid gave the γ-butyrolactone with 87% ee (entry 11). 5-Cyclohexylhex-5-enoic acid was also transformed successfully with 99.3% ee (entry 12). It should be emphasized that only 0.1 mol% L3–Zn3(OAc)4 catalyst gave 98% ee of 2a with a quantitative yield (entry 2).
For accessing the catalytic structure of the L3–Zn(OAc)2 complex, ESI-MS analysis of the catalyst solution suggested the presence of a multi-nuclear zinc complex (Fig. 1). An ion peak at m/z = 1028.3031 attributed to [L32–Zn3]2+ was observed, even when mixing L3 and Zn(OAc)2 in a 1:1 ratio. When L3 and Zn(OAc)2 were mixed in a 1:3 ratio, ESI-MS analysis showed a new peak at m/z = 1301.2389, which suggested the formation of a trinuclear zinc complex with L3.
 | ||
| Fig. 1 ESI-MS spectra of (a) L3 + Zn(OAc)2 (1:1), (b) L3 + Zn(OAc)2 (1:3), and (c) L3–Zn3(OAc)4 + 1a (1:10). | ||
A single crystal was obtained from the reaction of the 1:3 mixture of L3 and Zn(OAc)2 in methanol, and X-ray crystallographic analysis revealed the structure of the complex to be trinuclear Zn3(OAc)4-3,3′-bis(aminoimino)binaphthoxide, as shown in Fig. 2.
 | ||
| Fig. 2 X-ray crystallographic analysis of the trinuclear Zn3(OAc)4-3,3′-bis(aminoimino)binaphthoxide complex (L3–Zn3(OAc)4): (a) the side view, and (b) the front view of the complex. Yellow and purple colored atoms are coordinated acetyl carbons. Hydrogen atoms and solvent molecules are omitted for clarity. | ||
In L3–Zn3(OAc)4, the two end zinc atoms make the complex hexa-coordinated, and the central zinc atom is part of a tetrahedral coordination sphere. For complex formation, one Zn(OAc)2 reacted with L3 to give the central zinc binaphthoxide, and the two remaining Zn(OAc)2 were coordinated, one at each end, by the aminoimino functionality of L3. Alternatively, both end zinc atoms formed a mixed acetoxy-binaphthoxide, and the central zinc remained as Zn(OAc)2. Because X-ray crystallography and DFT calculations of L3–Zn3(OAc)4 suggest σ-bond characteristics of the central zinc with phenolic oxygens, complex formation could be explained via zinc-binaphenoxide [Zn–O: 1.949(3) or 1.962(3) Å]. In the L3–Zn3(OAc)4 complex, each of the acetoxy anions bridges the central zinc and the end zinc atoms, which restricts the conformation of the L3–Zn3 complex. The isolated crystalline Zn3(OAc)4-3,3′-bis(aminoimino)binaphthoxide produced a clean 1H-NMR spectrum at −40 °C (Fig. 3) and 1 mol% catalyst promoted the asymmetric iodolactonization to give 2a with 99% ee.
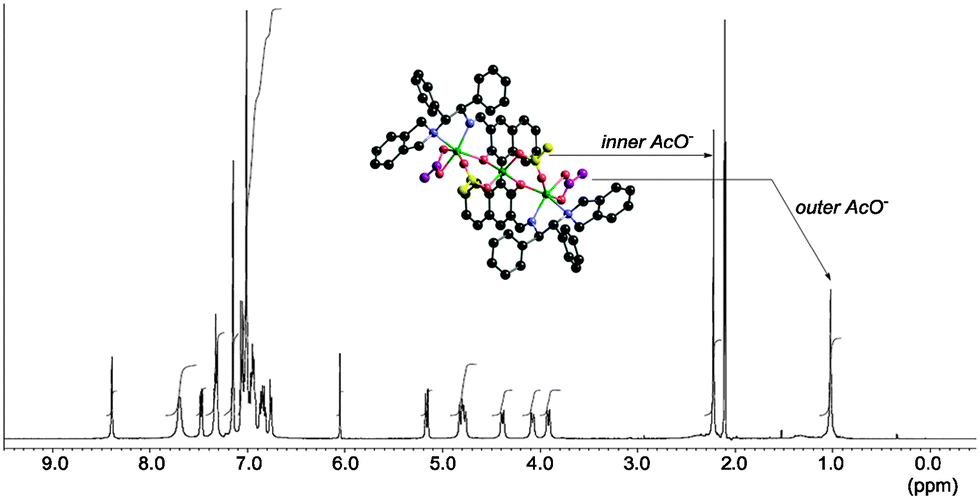 | ||
| Fig. 3 1H-NMR spectrum of the L3–Zn3(OAc)4 complex in toluene-d8 at −40 °C. | ||
Regarding the catalytic role of L3–Zn3(OAc)4, after mixing L3–Zn3(OAc)4 and 10 equiv. of 1a, all acetoxy anions of L3–Zn3(OAc)4 were replaced with 1a to give the ion peak at m/z = 1691.4724, corresponding to [L3–Zn3(CO2(CH2)3C(CH2)Ph)3]+ (Fig. 1c). This exchange suggests that the zinc-carboxylate of 1a is generated by the L3–Zn3(OAc)4 catalyst as a vital intermediate in the highly enantioselective iodolactonization reaction. However, two types of acetoxy anions were observed in the 1H-NMR spectrum of L3–Zn3(OAc)4, at 1.02 and 2.22 ppm. The up-field peak was assigned by the DFT-GIAO calculation to the outer acetoxy anions indicated in purple. Because the peak at 1.02 ppm becomes broader than the peak at 2.22 ppm, the outer acetoxy anions would smoothly accept the exchange with substrate 1a. Based on these experimental analyses on the interaction of L3–Zn3(OAc)4 with 1a, a plausible transition state for the L3–Zn3(OAc)4-catalyzed iodolactonization is proposed by DFT computed molecular modeling (Fig. 4).17
 | ||
| Fig. 4 A plausible transition state of L3–Zn3(OAc)4-catalyzed iodolactonization: TS1 for (R)-iodolactone 2a, TS2 for (S)-2a. | ||
The zinc-carboxylate of 1a is generated on the outer zinc atom. In the cyclic transition state of the iodolactonization, the benzene ring of 1a keeps away from the naphthyl ring of L3 to avoid the steric repulsion observed in TS2 (the red curves in Fig. 4). From the TS1 depicted in Fig. 4, the stereoselective formation of (R)-iodolactone 2a is explained well. Because multiple zinc atoms are important for getting high catalytic activity as shown in Table 1, the central zinc atom would also contribute to enhancing the zinc-carboxylate formation of 1a and/or to accelerating the nucleophilic cyclization of the zinc-carboxylate.
In conclusion, using a newly designed 3,3′-bis(aminoimino)BINOL ligand, the trinuclear Zn3(OAc)4-3,3′-bis(aminoimino)binaphthoxide complex (L3–Zn3(OAc)4) was prepared. The harmony of the tri-Zn centers in L3–Zn3(OAc)4 showed outstanding catalytic activity for the iodolactonization reaction to yield products in quantitative yields with excellent enantioselectivity.18
This work was supported by a Grant-in Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science and Technology (Japan), by the Chiba University Iodine Research Project and by the Workshop on Chirality in Chiba University (WCCU).
Notes and references
- E. Hough, L. K. Hansen, B. Birknes, K. Jynge, S. Hansen, A. Hordvik, C. Little, E. Dodson and Z. Derewenda, Nature, 1989, 338, 357 CrossRef CAS PubMed.
- Multimetallic Catalysts in Organic Synthesis, ed. M. Shibasaki and Y. Yamamoto, Wiley-VCH, Weinheim, 2004 Search PubMed.
- M. Shibasaki, H. Sasai and T. Arai, Angew. Chem., Int. Ed. Engl., 1997, 36, 1236 CrossRef.
- G. E. Veitch and E. N. Jacobsen, Angew. Chem., Int. Ed., 2010, 49, 7332 CrossRef CAS PubMed.
- M. C. Dobish and J. N. Johnston, J. Am. Chem. Soc., 2012, 134, 6068 CrossRef CAS PubMed.
- J. E. Tungen, J. M. J. Nolsøe and T. V. Hansen, Org. Lett., 2012, 14, 5884 CrossRef CAS PubMed.
- C. Fang, D. H. Paull, J. C. Hethcox, C. R. Shugrue and S. F. Martin, Org. Lett., 2012, 14, 6290 CrossRef CAS PubMed.
- Z. Ning, R. Jin, J. Ding and L. Gao, Synlett, 2009, 2291 CAS.
- L. Filippova, Y. Stenstrfm and T. V. Hansen, Tetrahedron Lett., 2014, 55, 419 CrossRef CAS PubMed.
- T. Arai, S. Kajikawa and E. Matsumura, Synlett, 2013, 2045 CrossRef CAS PubMed.
- Recent reviews on catalytic asymmetric halocyclization: (a) G. F. Chen and S. M. Ma, Angew. Chem., Int. Ed., 2010, 49, 8306 CrossRef CAS PubMed; (b) A. Castellanos and S. P. Fletcher, Chem. – Eur. J., 2011, 17, 5766 CrossRef CAS PubMed; (c) C. K. Tan, L. Zhou and Y.-Y. Yeung, Synlett, 2011, 1335 CAS; (d) U. Hennecke, Chem. – Asian J., 2012, 7, 456 CrossRef CAS PubMed; (e) S. E. Denmark, W. E. Kuester and M. T. Burk, Angew. Chem., Int. Ed., 2012, 51, 10938 CrossRef CAS PubMed.
- Catalytic asymmetric bromolactonization: (a) K. Murai, T. Matsushita, A. Nakamura, S. Fukushima, M. Shimura and H. Fujioka, Angew. Chem., Int. Ed., 2010, 49, 9174 CrossRef CAS PubMed; (b) K. Murai, A. Nakamura, T. Matsushita, M. Shimura and H. Fujioka, Chem. – Eur. J., 2012, 18, 8448 CrossRef CAS PubMed; (c) D. H. Paull, C. Fang, J. R. Donald, A. D. Pansick and S. F. Martin, J. Am. Chem. Soc., 2012, 134, 11128 CrossRef CAS PubMed; (d) W. Zhang, S. Zheng, N. Liu, J. B. Werness, I. A. Guzei and W. Tang, J. Am. Chem. Soc., 2010, 132, 3664 CrossRef CAS PubMed; (e) W. Zhang, N. Liu, C. M. Schienebeck, K. Decloux, S. Zheng, J. B. Werness and W. Tang, Chem. – Eur. J., 2012, 18, 7296 CrossRef CAS PubMed; (f) L. Zhou, C. K. Tan, X. Jiang, F. Chen and Y.-Y. Yeung, J. Am. Chem. Soc., 2010, 132, 15474 CrossRef CAS PubMed; (g) X. Jiang, C. K. Tan, L. Zhou and Y.-Y. Yeung, Angew. Chem., Int. Ed., 2012, 51, 7771 CrossRef CAS PubMed; (h) C. K. Tan, L. Zhou and Y.-Y. Yeung, Org. Lett., 2011, 13, 2738 CrossRef CAS PubMed; (i) H. J. Lee and D. Y. Kim, Tetrahedron Lett., 2012, 53, 6984 CrossRef CAS PubMed; (j) C. K. Tan, C. Le and Y.-Y. Yeung, Chem. Commun., 2012, 48, 5793 RSC.
- Catalytic asymmetric chlorolactonization: (a) D. C. Whitehead, R. Yousefi, A. Jaganathan and B. Borhan, J. Am. Chem. Soc., 2010, 132, 3298 CrossRef CAS PubMed; (b) R. Yousefi, D. C. Whitehead, J. M. Mueller, R. J. Staples and B. Borhan, Org. Lett., 2011, 13, 608 CrossRef CAS PubMed; (c) and also see, ref. 12e.
- Selected examples of multinuclear metal catalysts: (a) N. Kumagai, S. Matsunaga, T. Kinoshita, S. Harada, S. Okada, S. Sakamoto, K. Yamaguchi and M. Shibasaki, J. Am. Chem. Soc., 2003, 125, 2169 CrossRef CAS PubMed; (b) B. M. Trost, H. Ito and E. R. Silcoff, J. Am. Chem. Soc., 2001, 123, 3367 CrossRef CAS; (c) Y. Xiao, Z. Wang and K. Ding, Chem. – Eur. J., 2005, 11, 3668 CrossRef CAS PubMed; (d) B. Y. Lee, H. Y. Kwon, S. Y. Lee, S. J. Na, S. Han, H. Yun, H. Lee and Y.-W. Park, J. Am. Chem. Soc., 2005, 127, 3031 CrossRef CAS PubMed; (e) T. Ohshima, T. Iwasaki and K. Mashima, Chem. Commun., 2006, 2711 RSC; (f) R. M. Thomas, P. C. B. Widger, S. M. Ahmed, R. C. Jeske, W. Hirahata, E. B. Lobkovsky and G. W. Coates, J. Am. Chem. Soc., 2010, 132, 16520 CrossRef CAS PubMed; (g) S. Takizawa, T. Katayama and H. Sasai, Chem. Commun., 2008, 4113 RSC; (h) N. V. Kaminskaia, B. Spingler and S. J. Lippard, J. Am. Chem. Soc., 2000, 122, 6411 CrossRef CAS.
- T. Arai, M. Watanabe and A. Yanagisawa, Org. Lett., 2007, 9, 3595 CrossRef CAS PubMed.
- B. Saito and T. Katsuki, Angew. Chem., Int. Ed., 2005, 44, 4600 CrossRef CAS PubMed.
- The transition structure was partially optimized with the B3LYP method using 631LAN basis set consisting of LANL2DZ for Zn and 6-31G* for the rest. The L3–Zn3(OAc)3 moiety was only fully optimized while the iodolactone 2a was frozen at the transition structure model of the Zn cation catalyzed iodolactonization.
- H. Nakatsuji, Y. Sawamura, A. Sakakura and K. Ishihara, Angew. Chem., Int. Ed. DOI:10.1002/anie.201400946.
Footnote |
| † Electronic supplementary information (ESI) available: Procedural and spectral data. CCDC 956608. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4cc02415j |
| This journal is © The Royal Society of Chemistry 2014 |