 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Guest-induced symmetry lowering of an ionic clathrate material for carbon capture†
S.
Muromachi
*a,
K. A.
Udachin
b,
K.
Shin
c,
S.
Alavi
bd,
I. L.
Moudrakovski
d,
R.
Ohmura
e and
J. A.
Ripmeester
b
aMethane Hydrate Research Centre, National Institute of Advanced Industrial Science and Technology (AIST), 16-1 Onogawa, Tsukuba, 305-8569 Japan. E-mail: s-muromachi@aist.go.jp
bNational Research Council of Canada, Canada
cKorea Advanced Institute of Science and Technology (KAIST), Korea
dUniversity of Ottawa, Canada
eKeio University, Japan
First published on 29th May 2014
Abstract
We report a new lattice structure of the ionic clathrate hydrate of tetra-n-butylammonium bromide induced by guest CO2 molecules, which is found to provide high CO2 storage capacity. The structure was characterized by a set of methods, including single crystal X-ray diffraction, NMR, and MD simulations.
Gas capture by guest–host compounds is a technology which has a possibility to mitigate a specific gas from a gas mixture with low energy consumption and not accompanied by an irreversible chemical reaction.1 Ionic clathrate hydrates are a class of guest–host materials in which large ionic guest species, primarily substituted ammonium or phosphonium salts, are incorporated into a solid ice-like hydrogen-bonded water framework.2 They are generally formed from aqueous solutions, and have higher melting temperatures than ice, which makes these materials suitable for applications such as cool-energy storage and gas separation/storage.3–6 The interest in ionic clathrate hydrates lies in the fact that the small dodecahedral (D) water cages formed in the lattice are available for occupancy by small gas-phase guest species, e.g., CO2, CH4 and H2, generally under milder conditions as compared to the pure small-guest canonical clathrate hydrates. The tetra-n-butylammonium bromide (TBAB) + CO2 hydrate has been a very popular system for investigation and there have been a number of reports on the modeling of the TBAB + CO2 hydrate.7 However, the work reported here reinforces the need to have good structural information as the currently available models for the ionic hydrates depend on a number of unfounded assumptions. For example, at a CO2 gas pressure of 4 MPa, the TBAB ionic hydrate, encapsulating CO2 guests, is formed at 291 K which is ∼10 K higher than the temperature needed to synthesize the pure CO2 clathrate hydrate at the same pressure.4 In the canonical clathrate hydrates, several guest-induced modifications of the host water framework have been reported.8 Such host-framework rearrangements in the presence of a gaseous guest allow the most efficient incorporation of the guest in the resulting structure. The occupancy of CO2 as a guest substance in hydrate cages is affected by guest–host and/or guest–guest interactions,9,10 however, a lattice structural transition induced by CO2 has not been reported.
Herein, we report a new CO2-driven ionic clathrate-hydrate structure transformation which leads to efficient CO2 encapsulation. To see how the guest gas molecules stabilize/destabilize the ionic clathrate hydrate, we performed single crystal X-ray diffraction (SCXRD) measurements at 100 K on the TBAB + CO2 ionic clathrate hydrate. The crystals were synthesized from an aqueous TBAB solution with mole fraction xTBAB = 0.0064 under CO2 pressure P = 1.08 MPa at temperature T = 282.65 K. The experimental details and a figure of the crystal structure are given in the ESI.† The crystallographic information is summarized in the footnote.‡ The TBA cation is encapsulated in super cages derived from four of the large canonical clathrate cages, and Br− replaces water molecules in the lattice.2 Unexpectedly, in the presence of CO2, only the orthorhombic Imma crystal structure was found, whereas the TBAB hydrate formed without a guest gas has the tetragonal (TBAB·26H2O) and orthorhombic (TBAB·38H2O) phases as the most stable phases, with yet other phases also observed. The determined space group Imma has the same point group but a different lattice from Pmma observed for TBAB·38H2O.11 The symmetry of the Imma structure is lower because of the doubling of the unit cell size in the direction of the b-axis compared to the pure Pmma hydrate. This lattice change evidently arises from the asymmetrical distributions of the incorporated CO2 molecules in the dodecahedral (D) cages of the Imma hydrate and is not related to structural changes in the TBAB and water framework upon CO2 encapsulation. The CO2 molecules occupy the two symmetry-distinct dodecahedral-cages as shown in Fig. 1 (and Fig. S1 of the ESI†), and include the highly distorted cages DA and more regular dodecahedral cages DB. The chemical formula of the empty host framework can be written as TBAB·38H2O·DA·2DB.
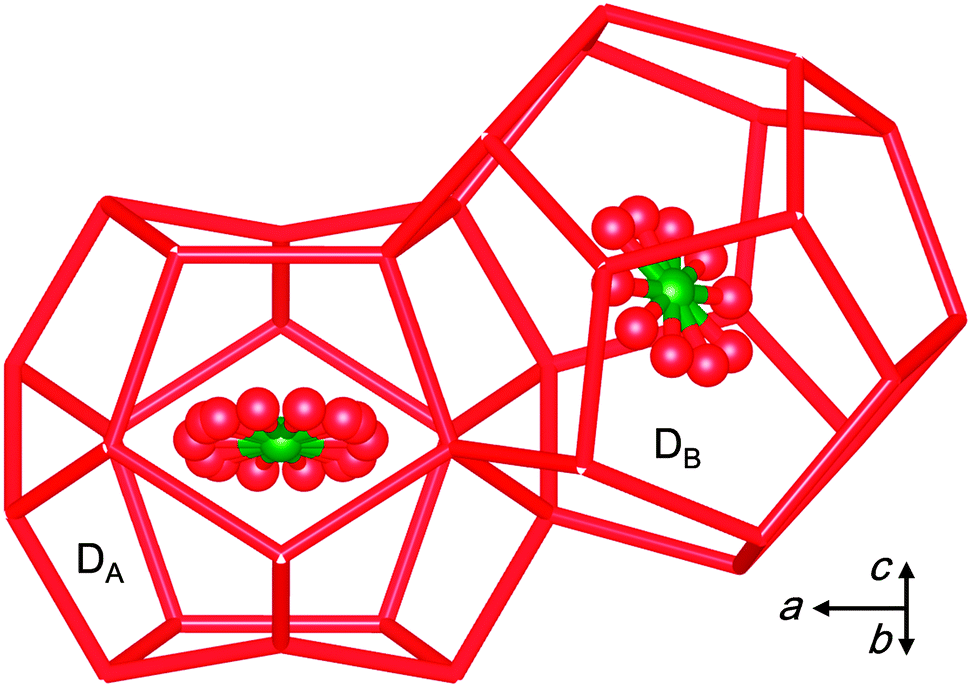 | ||
| Fig. 1 The highly distorted DA and regular shaped DB dodecahedral cages in the TBAB + CO2 structure. The arrow denotes the direction of the unit cell axes. | ||
In the crystal structure, the fraction of DA cages occupied by CO2 is almost twice that of the DB cages, i.e., 0.867 and 0.490, respectively. As shown in Fig. 1, the water molecules forming the DA cage are displaced by the n-butyl chains of the [TBA]+ ion and pushed inward by the Br− ions. As a result, the lattice is highly distorted along a direction perpendicular to the plane of the confined CO2 molecules. This clearly differs from the structure of D cages holding CO2 in canonical clathrate hydrate phases. The DA and DB cages have similar volumes, i.e., 153 and 160 Å3, respectively. As reported in Table S2 of the ESI,† three crystal samples of the TBAB + CO2 hydrate formed under the different P-T-x conditions had the same orthorhombic Imma structure. The preference of an orthorhombic hydrate structure over the tetragonal structure in the presence of CO2 (or small-guest) can be rationalized by considering the unit cell volume per 512 cages, i.e., 76H2O/6 D cages for the orthorhombic phase and 164H2O/10 D cages for the tetragonal TBAB phase.2,6 Thus application of Le Chatelier's principle suggests that the orthorhombic structure gives the largest pressure drop and the greatest stabilization effect as a result of CO2 incorporation into the D cages.
To study the CO2 guest environments and occupancies in the DA and DB cages, CP-MAS 13C NMR spectra of the TBAB + CO2 solid phase were obtained, as shown in Fig. 2. This sample was made by exposing the solid TBAB hydrate to CO2 gas under pressure; see the ESI† for details of the sample preparation. The spectra demonstrate a complex overlap of the isotropic signals and spinning sidebands near 125 ppm of CO2 guests in three distinct sites and TBAB in the lattice of the ionic clathrate. The ratio of the integrated intensities for the hydrate cage signals with isotropic chemical shifts at 124.87 and 125.13 ppm is less than unity, i.e., 0.54 (30/55), which is consistent with the relationship between the total number of CO2 guests in the DA and DB cages determined by the SCXRD, N(DA)/N(DB) = 0.867/(2 × 0.490) = 0.88. Both the signal intensities in the NMR spectrum and the CO2 occupancies in the SCXRD structure indicate a smaller CO2 population in the more abundant DB cages.
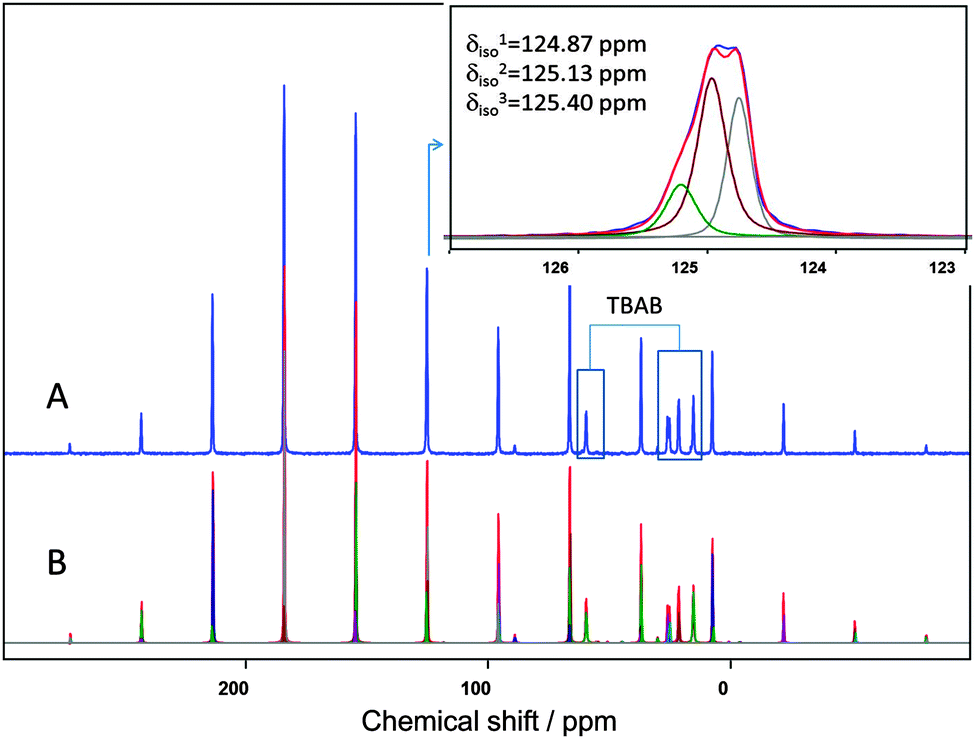 | ||
| Fig. 2 (A) Experimental 13C CP MAS spectrum of the 13CO2·TBAB·38H2O hydrate obtained under a field of 9.4 T (Larmor frequency of 100.67 MHz) at 173 K and a spinning speed of 2975 Hz. (B) Simulation of the experimental spectrum with a model accounting for the spectral intensity spread in the spinning sidebands. | ||
The isotropic chemical shifts for CO2 residing in D cages in this work are close to the ∼128 ppm chemical shifts of CO2 guests previously reported in other clathrate hydrate phases.7 The difference in chemical shifts from 128 ppm in D cages reported for other hydrates to ∼125 ppm in the presently found DA and DB cages can be related to the smaller sizes of the latter cages, or to the presence of the charged Br− and [TBA]+ groups in close proximity, and also to the fact that previous chemical shift data were reported for stationary samples thus offering lower accuracy because of the broad powder patterns. The assignment of the low intensity signal at 125.40 ppm is less certain, and could arise from a different ionic clathrate phase formed under the non-equilibrium sample preparation conditions of the powder NMR sample. This is verified by powder X-ray diffraction (PXRD) measurements of the TBAB + CO2 phases formed under conditions similar to the preparation of the NMR samples. The PXRD patterns given in the ESI† show that the sample contained both orthorhombic and tetragonal phases, and thus the two main signals for CO2 (natural abundance) appearing at 124.85 and 125.06 ppm of the 13C NMR spectrum shown in Fig. S5 (ESI†) should originate from the orthorhombic phase and the additional small peak at 125.30 ppm from a small fraction of the tetragonal phase, respectively.
Guest dynamics were also studied using the MD simulation methodology with details outlined in the ESI.† The CO2 guests were found to have restricted, but different spatial distributions/motions in DA and DB cages. The sampled orientations of the CO2 guest molecules in the DA and DB cages with respect to the lattice b-direction (corresponding to the flattened direction of the DA cage) are reported in the ESI.†
The dynamics of the CO2 guest motion were also studied using the orientation autocorrelation function (OACF) around the molecular axis. If ϕ(t) is the rotation angle of the unit vector along the direction of the CO2 molecular axis at time t, μ(t), with respect to the vector orientation at time 0, μ(0), the OACF defined as the ensemble average of the first moment of the Legendre polynomial for the rotation angle, 〈P1(t)〉, is presented as,
〈P1(t)〉 = 〈μ(t)·μ(0)〉 = 〈cos![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) θ(t)〉 θ(t)〉 | (1) |
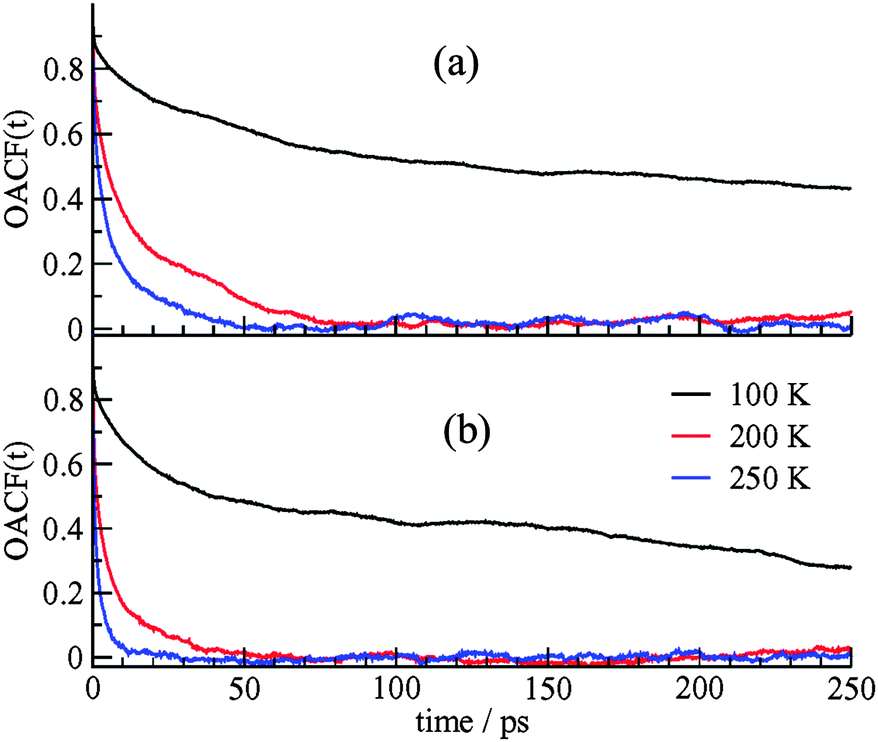 | ||
| Fig. 3 The OACF for CO2 molecules in (a) the DA cages, (b) the DB cages. The size of the DA cages is smaller and the orientation correlations decay more slowly for molecules in these cages. | ||
The simulations show that the lattice vibrations of tetra-n-butylammonium and bromide groups about their equilibrium positions are small and they are kept in place by electrostatic and van der Waals interactions with the neighboring water molecules. This is particularly interesting with regard to the Br− lattice vibrations as this ion may seem to be relatively free to move in the ionic hydrate lattice. The lattice site of injection of the Br− anion can be the site of Bjerrum L-defects12 in the neighboring water molecule lattice. In the simulation, water molecules show relatively slow rotational dynamics in the lattice. This is consistent with experiments since the 13C NMR powder patterns are broad and look distributed at around 200 K, much the same as for the large cage in the sI hydrate at this temperature. The water dynamics in the double hydrate of tetrahydrofuran + CO2 are much faster at temperatures around 200 K.10
The presence of CO2 guests lowered the symmetry of the original TBAB hydrate structure formed without small guest molecules. The CO2 gas storage capacities in the two types of D cages in the structure differ significantly because of the size and shape differences of the cages. This demonstrates that the shape of the guest molecules is a secondary factor for determining the gas storage capacity of the ionic clathrates in addition to the primary factor of molecular size. From a practical point of view, our experiments show that upon exposure to relatively mild CO2 pressures and controlled temperature/composition conditions, it is possible to selectively drive the formation of the ionic clathrate hydrate towards the orthorhombic TBAB·38H2O·nCO2 phase which is the most effective one for carbon capture. We note that the large difference in gas-holding capacity of the DA and DB small cage types in the hydrate is of key importance in developing satisfactory models that will account for a variety.
This study was subsidized in part by the Keio University Global Center of Excellence Program “Center for Education and Research of Symbiotic, Safe and Secure System Design”. SM acknowledges the financial support provided by the Japan Society for the Promotion of Science (JSPS) through the program “Grant-in-Aid for JSPS Fellows of the Ministry of Education, Culture, Sports, Science, and Technology (MEXT)” (Grant No. 23-56572).
Notes and references
- Comprehensive Supramolecular Chemistry, Supramolecular Technology, ed. D. N. Reinhoudt, Pergamon, Oxford, 1996, vol. 10 CrossRef CAS PubMed; D. M. D'Alessandro, B. Smit and J. R. Long, Angew. Chem., Int. Ed., 2010, 49, 6038 CrossRef CAS PubMed; S. D. Kenarsari, D. Yang, G. Jiang, S. Zhang, J. Wang, A. G. Russell, Q. Wei and M. Fan, RSC Adv., 2013, 3, 22739 RSC.
- M. Bonamico, G. A. Jeffrey and R. K. McMullan, J. Chem. Phys., 1962, 37, 2219 CrossRef CAS PubMed; R. K. McMullan, G. A. Jeffrey and M. Bonamico, J. Chem. Phys., 1963, 39, 3295 CrossRef PubMed; D. W. Davidson, in Water. A Comprehensive Treatise, ed. F. Franks, Plenum Press, New York, NY, 1973 Search PubMed; G. A. Jeffrey, in Inclusion Compd., ed. J. L. Atwood, J. E. D. Davies and D. D. MacNicol, Academic Press, London, 1984, ch. 5, vol. 1 Search PubMed.
- A. Chapoy, R. Anderson and B. Tohidi, J. Am. Chem. Soc., 2007, 129, 746 CrossRef CAS PubMed; N. H. Duc, F. Chauvy and J.-M. Herri, Energy Convers. Manage., 2007, 48, 1313 CrossRef PubMed; D. Zhong and P. Englezos, Energy Fuels, 2012, 26, 2098 CrossRef.
- N. Ye and P. Zhang, J. Chem. Eng. Data, 2012, 57, 1557 CrossRef CAS.
- H. Oyama, W. Shimada, T. Ebinuma, Y. Kamata, S. Takeya, T. Uchida, J. Nagao and H. Narita, Fluid Phase Equilib., 2005, 234, 131 CrossRef CAS PubMed; K. Sato, H. Tokutomi and R. Ohmura, Fluid Phase Equilib., 2013, 337, 115 CrossRef PubMed.
- S. Muromachi, S. Takeya, Y. Yamamoto and R. Ohmura, CrystEngComm, 2014, 16, 2056 RSC.
- X. Liao, X. Guo, Y. Zhao, Y. Wang, Q. Sun, A. Liu, C. Sun and G. Chen, Ind. Eng. Chem. Res., 2013, 52, 18440 CrossRef CAS; W. Lin, D. Dalmazzone, W. Fürst, A. Delahaye, L. Fournaison and P. Clain, J. Chem. Eng. Data, 2013, 58, 2233 CrossRef; A. Joshi, P. Mekala and J. S. Sangwai, J. Nat. Gas Chem., 2012, 21, 459 CrossRef; P. Paricaud, J. Phys. Chem. B, 2011, 115, 288 CrossRef PubMed; M. Kwaterski and J.-M. Herri, Fluid Phase Equilib., 2014, 371, 22 CrossRef PubMed; P. Babu, W. I. Chin, R. Kumar and P. Linga, Ind. Eng. Chem. Res., 2014, 53, 4878 CrossRef.
- K. Shin, S. Choi, J.-H. Cha and H. Lee, J. Am. Chem. Soc., 2008, 130, 7180 CrossRef CAS PubMed; K. A. Udachin, C. I. Ratcliffe, G. D. Enright and J. A. Ripmeester, Angew. Chem., Int. Ed., 2008, 47, 9704 CrossRef PubMed; K. Shin, Y. Kim, T. A. Strobel, P. S. R. Prasad, T. Sugahara, H. Lee, E. D. Sloan, A. K. Sum and C. A. Koh, J. Phys. Chem. A, 2009, 113, 6415 CrossRef PubMed; K. Shin, K. A. Udachin, I. L. Moudrakovski, D. M. Leek, S. Alavi, C. I. Ratcliffe and J. A. Ripmeester, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 8441 Search PubMed; K. Shin, R. Kumar, K. A. Udachin, S. Alavi and J. A. Ripmeester, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 14785 CrossRef PubMed; K. Shin, I. L. Moudrakovski, M. D. Davari, S. Alavi, C. I. Ratcliffe and J. A. Ripmeester, CrystEngComm, 2014 10.1039/c3ce41661e.
- J. A. Ripmeester and C. I. Ratcliffe, Energy Fuels, 1998, 12, 197 CrossRef CAS; K. A. Udachin, C. I. Ratcliffe and J. A. Ripmeester, J. Phys. Chem. B, 2001, 105, 4200 CrossRef; S. Alavi, P. Dornan and T. K. Woo, ChemPhysChem, 2008, 9, 911 CrossRef PubMed.
- S. Alavi and J. A. Ripmeester, J. Chem. Phys., 2012, 137, 054712 CrossRef CAS PubMed; K. Tezuka, R. Shen, T. Watanabe, S. Takeya, S. Alavi, J. A. Ripmeester and R. Ohmura, Chem. Commun., 2013, 49, 505 RSC.
- L. A. Gaponenko, S. F. Solodovnikov, Y. A. Dyadin, L. S. Aladko and T. M. Polyanskaya, Zh. Strukt. Khim., 1984, 25, 175 CAS; W. Shimada, M. Shiro, H. Kondo, S. Takeya, H. Oyama, T. Ebinuma and H. Narita, Acta Crystallogr., Sect. C, 2005, 61, o65 Search PubMed.
- N. Bjerrum, Science, 1952, 115, 385 CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. A, 1990, 46, 467 CrossRef; G. M. Sheldrick, Acta Crystallogr., Sect. A, 1993, 49(suppl), C53 Search PubMed.
- K. Momma and F. Izumi, J. Appl. Crystallogr., 2011, 44, 1272 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental and computational methods and supplementary results of single crystal and powder XRD, NMR and MD simulations. CCDC 963037. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4cc02111h |
| ‡ C17.85H111BrNO41.69, Mr = 1087.28, crystal dimensions: 0.6 × 0.4 × 0.2, orthorhombic Imma, a = 21.0197(7), b = 25.2728(8), c = 12.0096(4) Å, V = 6379.8(4) Å3, Z = 4, T = 100.0(1) K, ρcalcd = 1.132 g cm−3, μ = 0.730, radiation and wavelength: Mo Kα and 0.71070 Å, 2θmax = 72.76, no. of measured and independent reflections: 87521 and 8002, Rint = 0.0363, final R(I > 2σ(I)) indices: R1 = 0.0266, wR2 = 0.0589. The structure was solved by direct methods using the SHELXTL suite of programs13 and visualized by VESTA.14 CCDC 963037. |
| This journal is © The Royal Society of Chemistry 2014 |