 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIonic liquids for energy, materials, and medicine
M.
Smiglak
a,
J. M.
Pringle
b,
X.
Lu
c,
L.
Han
c,
S.
Zhang
*c,
H.
Gao
c,
D. R.
MacFarlane
*d and
R. D.
Rogers
*e
aPoznan Science and Technology Park, Adam Mickiewicz University Foundation, 61-612 Poznan, Poland
bARC Centre of Excellence for Electromaterials Science, Deakin University, Burwood, VIC 3125, Australia
cInstitute of Process Engineering, Chinese Academy of Sciences, Beijing 100190, China. E-mail: sjzhang@ipe.ac.cn
dSchool of Chemistry, Monash University, Clayton, Victoria 3800, Australia. E-mail: douglas.macfarlane@monash.edu
eCenter for Green Manufacturing and Department of Chemistry, The University of Alabama, Tuscaloosa, AL 35487, USA. E-mail: rdrogers@ua.edu
First published on 16th May 2014
Abstract
As highlighted by the recent ChemComm web themed issue on ionic liquids, this field continues to develop beyond the concept of interesting new solvents for application in the greening of the chemical industry. Here some current research trends in the field will be discussed which show that ionic liquids research is still aimed squarely at solving major societal issues by taking advantage of new fundamental understanding of the nature of these salts in their low temperature liquid state. This article discusses current research trends in applications of ionic liquids to energy, materials, and medicines to provide some insight into the directions, motivations, challenges, and successes being achieved with ionic liquids today.
Introduction
The recent ChemComm web themed issue on ionic liquids1 highlighted the continued evolution of research into the fundamental nature and new applications of ionic liquids (ILs). This evolution has meant that ILs are now not only considered as important alternative, and occasionally “Green”, solvents, but as materials with unique and tuneable properties which can be adjusted by selecting appropriate ions for a specific need. This unique ability to tune the properties, often with just selection of the ions rather than covalent modification, makes ILs attractive for a range of applications, from materials science to electrochemistry and from catalysis to medicinal chemistry. In this article, we discuss current research trends and potential future directions in applications of ILs in three major fields of societal importance: energy, materials, and medicines. Our intent is not to provide comprehensive reviews of these fields, rather to provide some insight into the directions, motivations, challenges, and successes being achieved today. In each case more detailed reviews are available to which the interested reader is referred for more in depth discussion.Ionic liquids – impact on old and new energy technologies
1. Cleaning of the old: CO2 and SO2 capture and separation by ionic liquids
In this context, ILs increasingly are a major focus of attention in developing second generation technologies for CO2 capture and separation. Existing CO2 capture technology is hampered by the energy cost of the absorption–desorption cycle and the chemical cost of the absorber lost to evaporation. IL-based absorbers have the potential to solve both of these issues by harnessing the intrinsic low volatility of the IL and careful tuning of the absorber functionality. However, the cost of these materials remains a major impediment and recent work has focused on how to achieve the high absorption capacity on a g(CO2)/g(IL) basis in an IL that is intrinsically of low cost to manufacture. At present, research on ILs for CO2 capture revolves mostly around four main groups of ILs; conventional (or commonly known and used) ILs, functionalized ILs (in the cation or in the anion), supported IL membranes, and IL-based mixtures.
As the CO2 solubility in conventional ILs is typically not sufficient for large-scale use, focus has shifted to functionalized ILs with high CO2 capacity. The first amine functionalized IL for CO2 capture was reported by Davis et al., (Fig. 1a); this IL could capture 0.5 mole CO2 per mole of IL in 3 h under ambient pressure.4 This limiting stoichiometry is expected on the basis of carbamate formation by reaction of the CO2 with the amine functional group. However, its viscosity was found to be too high to capture CO2 on an industrial scale.
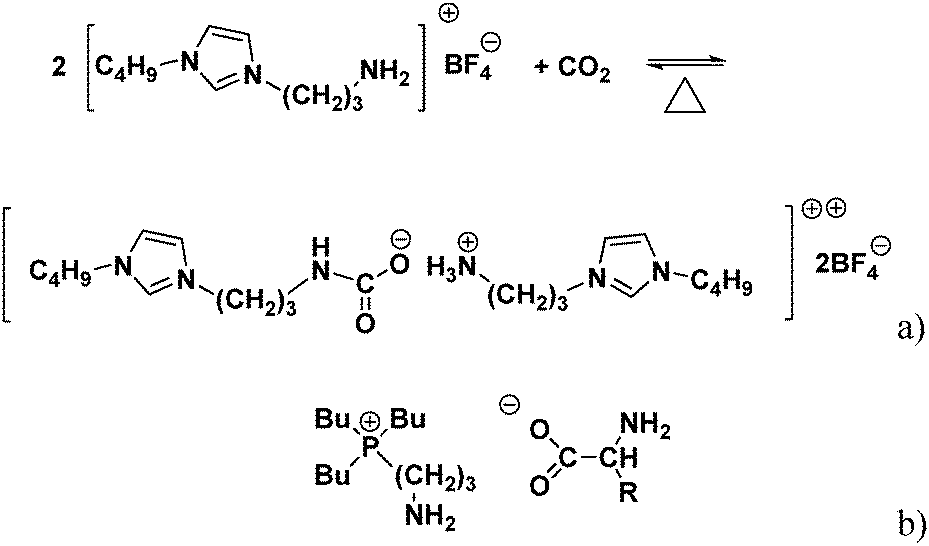 | ||
| Fig. 1 (a) Proposed reaction mechanism for uptake of CO2 by carbamate formation in the reaction between [NH2p-bim][BF4] and CO2.4 (b) Structure of the dual amino-functionalized phosphonium ILs ([aP4443][AA]. [AA] = [Ala], [Arg], [Asn], [Asp], [Cys], [Gln], [Glu], [Gly], [His], [Ile], [Leu], [Lys], [Met], [Phe], [Pro], [Ser], [Thr], [Trp], [Tyr], and [Val]).5,6 | ||
Amino acid-based ILs have potential for use in CO2 capture because they possess amine functional groups and Zhang et al.5,6 developed a series of single and dual amino-functionalized phosphonium ILs for CO2 capture (Fig. 1b). Because their viscosities were also high, they were supported on porous silica gel where they exhibited stoichiometric uptake of 0.5 mol and 1.0 mol CO2 per mole IL.5,6 Gurkan et al., also used amino acid-based ILs and observed stoichiometric CO2 uptake.7
Recently, Wang et al., developed an efficient CO2 capture method based on anion-functionalized protic ILs (PILs). The results showed that these ILs had higher CO2 capacities than the above mentioned ILs. Moreover, CO2 absorption into, and release from these IL could be repeatedly cycled 25 times with high absorption capacity.8–12 Nonetheless, these functionalized ILs were not easy to synthesize and purify.
While many ILs described for CO2 absorption over the last 5 years have exhibited excellent capacity on a molar basis, usually indicating completion of carbamate formation, many of these compounds do not compare well on a weight/weight (w/w) or a weight/volume (w/v) basis because of the high molecular weight of the IL involved. Significant in this respect, the recent work of Vijayraghavan has demonstrated that the loading capacity of the industry standard monoethanolamine can be matched by inexpensive ILs based on small protic ions such as (N,N-dimethyl)aminoethylammonium formate.13
Supported IL membranes (SILMs) consist of two phases, a supporting porous membrane and an IL phase. The potential of SILMs for gas separation has been investigated by several researchers.14–21 Bara et al., investigated SILMs based on [C2mim][NTf2], [C2mim][OTf], and [C2mim][N(CN)2] reporting CO2 permeabilities in the range of 350–1000 barriers and ideal CO2/N2 selectivities of 15–61.22 Iarikov et al., studied SILMs based on ammonium, imidazolium, pyridinium, pyrrolidinium, and phosphonium ILs obtaining a CO2/CH4 selectivity of 5–30.23 Yoo et al., obtained an O2/CH4 selectivity of 26 using imidazolium-based ILs in a Nafion membrane.17 However, one of the issues with this approach is that “blow-out” of the IL component through the pores of the support limits the pressure differential that can be maintained.
Although functionalized ILs can increase the capacity of CO2 absorption by incorporating an amine or other functional group in the structure of the ILs, the synthesis of amine-functionalized ILs requires several steps that make them relatively expensive to produce. In 2008, Noble et al., put forward the idea of mixing ILs and alkanolamines for CO2 capture and indicated that such mixtures were capable of capturing 0.5 mole of CO2 per mole of dissolved alkanolamine.24 This method has two advantages: (1) the desirable properties of both the IL and the alkanolamines may be incorporated and (2) energy can be saved during the regeneration processes without affecting their absorption performance.25–27 In general, IL-based mixtures such as these represent a platform technology with broad industrial applicability wherein the properties of the mixture can be modulated to take best advantage of the individual components.
Anderson et al., reported the first example of the use of conventional ILs for SO2 capture. The results showed that 85 mol% of SO2 could dissolve in [C6mim][NTf2] and [hmpy][NTf2] at 25 °C and pressures up to 4 bar by simple physical absorption.28 Huang et al., found that some ILs could absorb large amounts of SO2 gas corresponding to molar ratios of SO2 to ILs of 1.18, 1.27, 1.33, 1.50, 1.60, (in wt%: 20.4%, 40.0%, 40.1%, 19.2%, and 20.1%) for [TMG][NTf2], [TMG][BF4], [C4mim][NTf2], [C4mim][BF4], and [TMGB2][NTf2], respectively, after being saturated with SO2 gas at 1 bar and 20 °C.29 Hong et al., developed a series of ether-functionalized imidazolium methanesulfonates (Fig. 2) where at least 2 moles of SO2 per mole of IL could be dissolved at 30 °C and atmospheric pressure.30
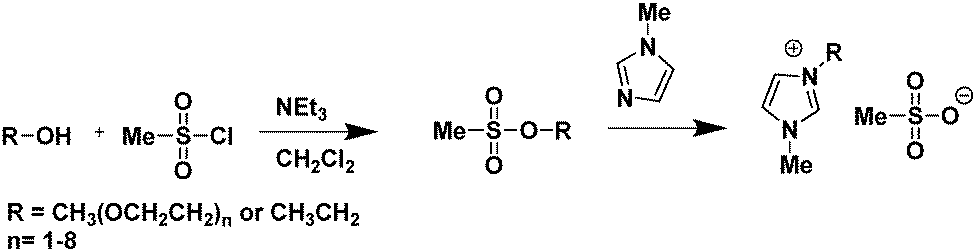 | ||
| Fig. 2 Synthesis scheme of the ether-functionalized imidazolium methanesulfonate ILs.30 | ||
Wu et al., first used 1,1,3,3-tetramethylguanidinium lactate for the removal of SO2, and found that the mole ratio of SO2 to IL could reach 1.7 at 40 °C and 1.2 bar of SO2.31 They also investigated the influence of H2O and O2 on the desulfurization performance of the ILs and their physical properties.32–35 Zhang et al., reported new ILs based on 1,1,3,3-tetramethylguanidinium phenol, 2,2,2-trifluoroethanol, and imidazolate. The viscosities of these ILs are much lower than that of other ILs used for absorbing SO2, and these ILs have high absorption capacities of SO2.36
Recently, Wang et al., synthesised a series of azole-based ILs using a phosphonium cation (e.g., [P66614][Tetr], [P66614][Im], and [P44410][Tetr]) which exhibited very high absorption capacities for SO2. They also combined ether-functionalized cations and tetrazolate anions to form new ILs that physically and chemically absorb SO2 with high absorption capacities: 5.0 moles of SO2 per mole of IL at 20 °C and 1 bar.37–39 These functionalized ILs have high absorption capacities of SO2 even when the partial pressure of SO2 is very low and as a result are more efficient than conventional ILs when used in real flue gas with low SO2 concentrations.
ILs have some advantages for capturing CO2/SO2 from flue gas or natural gas. Their negligible vapour pressure is one of the main factors that make them superior to conventional solvents, however, if this technology is to be commercialized, there are several problems to be resolved. These include (i) the price of the ILs should be low enough for commercial use; (ii) the long-term chemical and thermal stability of ILs must be quantified; (iii) pilot scale plants should be set up to assess the feasibility of ILs for capturing CO2/SO2; and (iv) biodegradability and toxicity of candidate ILs should be evaluated thoroughly to prevent environmental pollution.
2. Enabling the new: fuel cells
In efforts to shift to other, more sustainable fuels, the use of ILs as anhydrous proton conductors has significant implications for the further development of proton exchange membrane fuel cells (PEMFC). These devices react hydrogen and oxygen, directly converting chemical energy into electrical energy with only water as the by-product. Thus, these are potentially very clean sources of energy if, for example, the hydrogen is produced from solar water splitting. However, there remain a number of economic and technical barriers that must be overcome before the PEMFC can be widely used for either mobile or stationary applications.40,41At the heart of a traditional PEMFC is a proton conducting polymer electrolyte membrane such as Nafion®, and the conductivities of these membranes rely on maintaining a sufficient hydration level. This limits their operating temperature to <80 °C. However, at these temperatures the Pt catalyst is readily poisoned by CO, which can be present in the gas stream. The tolerance of the electrocatalysts to CO poisoning is significantly improved at operating temperatures >120 °C.42–44 Such temperatures also improve the kinetics of the oxygen reduction reaction and are more suitable for electric vehicle applications (reducing the heat management requirements). Increasing the temperature of operation requires either, (i) modification of the traditional perfluorosulfonic acid membranes so that hydration is not required to achieve sufficient conductivity, (ii) the development of new proton-conducting membranes, or (iii) use of a less volatile proton conducting material as a replacement for water.45–47
ILs that contain a mobile proton (protic ILs, PILs) can exhibit fast proton conduction and also facile hydrogen oxidation reactions (HOR) and oxygen reduction reactions (ORR).43,48,49 Sometimes referred to as Brønsted acid–base ILs, PILs are formed by classic acid–base neutralization reactions. The free energy change associated with the proton transfer process has a strong influence on the extent of PIL formation and its thermal stability.50–52
To ensure high ionic character (i.e., negligible amounts of acid and base starting material), and thus low vapour pressure and high thermal stability, a large proton energy gap is required. In some cases this can be approximated by the difference in the pKaaq values of the component acid and base in aqueous solutions. However, in the case of tertiary amine-based PILs it would appear that the IL environment is very different from aqueous and these estimations fail dramatically.52 Decomposition temperatures are observed to increase with increasing acidity of the acid used:53 with ΔpKaaq of 4–10 proton transfer is almost complete,52,54 and with ΔpKaaq > 20 decomposition of the PIL can occur prior to the volatilization of the neutral acid or base species,54 akin to the aprotic IL decomposition behaviour. In some cases higher conductivity PILs may be achieved by use of a non-stoichiometric mixture of acid and bases.52,55,56 The PILs can also be either organic or inorganic (e.g., ammonium salt mixtures), both of which are promising for fuel cell applications.43,50
One of the most important observations from studies on both organic and inorganic PILs is the correlation between the open circuit voltage (OCV) of H2/O2 fuel cells and the ΔpKaaq of the acid/base pair of the electrolyte.49,57 The OCV increases with ΔpKaaq until it reaches the theoretical value (1.15 V) at ΔpKaaq = 14–17, where it decreases with further increases in ΔpKaaq.49,57 The lower OCV at low ΔpKaaq has been attributed to the presence of neutral species (as a result of incomplete proton transfer) while the voltage decrease at higher ΔpKaaq may be due to decreased proton activities (because of a stronger N–H bond) and an increase in the viscosity of the PIL.57 This is clearly an important area of future study to enable the choice or design of new PILs for fuel cell applications. Also advantageous would be a direct method for quantifying the free energy of proton transfer in anhydrous Brønsted acid–base systems rather than relying on aqueous parameters.
Following their first demonstration of organic PILs as fuel cell electrolytes,48 the screening of >100 candidates by the Watanabe group has identified [NH122][OTf] as the most promising thus far.43 With good thermal stability (Tdecomp = 360 °C) and high ionic conductivity (10 mS cm−1 at room temperature) this PIL can produce an OCV of 1.03 V in a H2/O2 fuel cell at 150 °C.53,58
[NH122][OTf] has also been incorporated into a sulfonated polyimide membrane and used in a fuel cell without humidification to produce maximum current densities of 400 mA cm−2 and powers of 100 mW cm−2.58,59 The permanent incorporation of PILs into a membrane material, e.g., through their incorporation into a polymer, an ionogel, or the synthesis of polymerised ILs,46,60 is important for the longevity of the fuel cell. The proton conductivity in these materials can be improved by optimizing the polymer morphology and chemistry, the PIL content, and the use of nano-fillers.41,46,61
One strategy to overcoming the possible volatility of PILs for fuel cell applications is the use of di-cations with linked protic and aprotic groups.62 These novel materials are liquid at room temperature and exhibit significant hydrogen bonding but display no mass loss after being held under vacuum overnight. They can also be prepared using combinations of anions with different Brønsted basicity, which may be pertinent to the idea of creating a range of energetically accessible proton energy levels to allow better proton conduction.63
An alternative to PILs is the use of proton-conducting organic ionic plastic crystals (OIPCs),61,64 which can display good H+ diffusivities upon addition of acid. The plasticity of these materials, as a result of disorder within the solid state matrix, enables good ionic conductivity. Research has predominantly focused on [Chol][H2PO4] doped with phosphoric acid, triflic acid, or HNTf264–66 which can exhibit high ionic conductivity and proton activity. The incorporation of cellulose acetate as a support for these solid-state electrolytes improves the mechanical strength while still retaining the desired OIPC properties, such as good thermal stability (up to 200 °C) and significant proton reduction currents in the CV analysis (Fig. 3).67 Future work in this area is concentrating on increasing the OCV of the H2/O2 fuel cell and exploration of other OIPCs to improve device performance under anhydrous conditions.
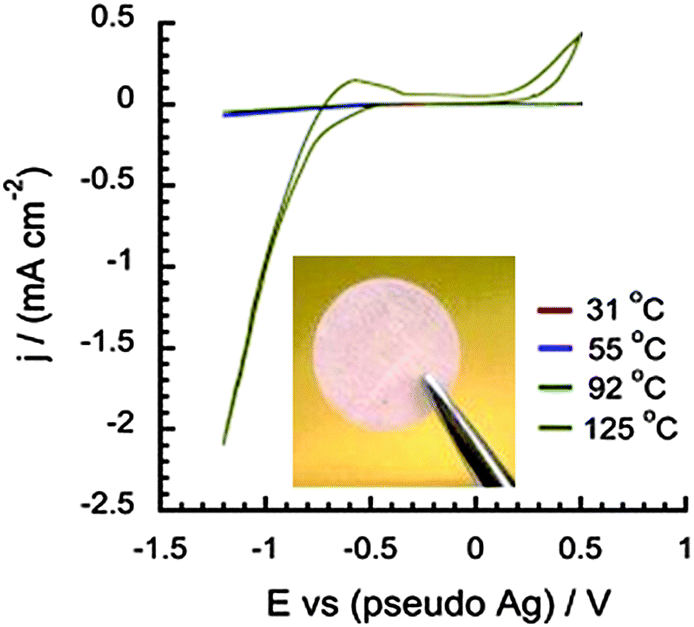 | ||
| Fig. 3 The appearance of the acid-containing plastic crystal supported on cellulose acetate membrane, and the cyclic voltammogram for 4% HNTf2 + [Chol][H2PO4] membrane. Reproduced from ref. 67 with permission from Elsevier. | ||
Another challenge in the use of PILs in fuel cells is the need for a catalyst layer to function effectively with the anhydrous proton conductor. Significant research into this area is required to overcome problems with absorption onto the catalyst and to facilitate a move away from Pt-based systems.68 Judicious choice of the PIL cation may help maintain electrocatalytic activity, e.g., Ke et al.,69 achieved current densities of >600 mA cm−2 at 140 °C using [NH111][H2PO4] on a polypropylene matrix, whereas pyridine and imidazole-based ILs poisoned the Pt/C catalyst.
Encapsulating nanoparticles of a catalyst within a medium of high O2 solubility, such as a hydrophobic PIL, can also lead to significant increases in the kinetics of the oxygen reduction reaction.70 Finally, the use of new in situ NMR techniques to study the chemical processes occurring at the electrode and catalyst surfaces during fuel cell operation can provide valuable insights into these critical processes.71
These studies are clearly opening up an important new avenue for medium temperature fuel cell development and application. With increasing scope for such fuel cells to be used as part of a renewable energy storage cycle, this regime of fuel cell operation is likely to be become of increasing importance and the impact of ILs may be significant.
3. Enabling the new: dye-sensitized solar cells
The non-volatility of ILs is a primary motivation for their use in dye-sensitized solar cells (DSSCs), as a strategy to improve long-term device stability. However, at present the use of ILs in DSSCs results in a reduction in efficiency to ca. 2/3 of those with molecular-solvent based electrolytes. This is primarily a result of mass transport limitations of the redox couple arising from the higher viscosity of the ILs studied, although diffusion of the I−/I3− redox couple can be aided by the presence of a Grotthus-type exchange mechanism at high triiodide concentrations.72 The formulation of IL-based electrolytes for DSSCs generally involves combination of the redox active component (e.g., an imidazolium iodide salt) with a more fluid IL to enhance transport. However, the concentration of triiodide used in ILs is higher than in molecular solvents, to minimize diffusion limitations, and this increases the dark current, lowers the open circuit voltage and reduces the efficiency of the device.At present, the most widely used IL system is a ternary melt of imidazolium iodides and [C2mim][B(CN)4] which gives efficiencies of 8.2% and good stability.73 However, it would be advantageous to find a more available alternative to this toxic anion. [C2mim][N(CN)2] can yield 8.4% efficiency in a DSSC,74 but the long-term stability is lower.73 In addition to good chemical stability and redox couple diffusivity, for use in a DSSC the IL-redox electrolyte must also allow fast electron donation by the I− to regenerate the dye, but slow charge recombination between the I3− and the injected electron at the photoanode. The addition of lithium salts and nitrogen heterocyclic bases to the IL electrolyte allows modification of the surface charges of the TiO2 photoanode and adjustment of the conduction band to optimize the OCV and current density of the device.74–77
The most widely-used photosensitiser is N719 (cis-diisothiocyanato-bis(2,2′-bipyridyl-4,4′-dicarboxylato) ruthenium(II) bis(tetrabutylammonium).75 However, the Z907 dye ([Ru(H2dcbpy)(dnbpy)(NCS)2], where H2dcbpy is 4,4′-dicarboxylic acid-2,2′-bipyridine and dnbpy is 4,4′-dinonyl-2,2′-bipyridine), can offer better performance with IL electrolytes because the long hydrocarbon chains increase the separation between the triiodide and the photoanode, reducing unwanted back electron transfer.78 Use of a porphyrin dye with long hydrophobic alkyl chains provides a similar barrier layer and achieves efficiencies of 4.9% with [C2mim][B(CN)4)]-based IL electrolytes, which is only ca. 1% lower than with the acetonitrile-based electrolyte.79
Extensive research has focused on developing new dyes to improve affordability, stability, incident photon-to-current conversion efficiency (IPCE), and to allow the use of thinner TiO2 films.80,81 For example, organic dyes can be cheaper and have higher extinction coefficients than Ru-based species, while the design of dyes with bulky structures that decrease the proximity of the redox species to the TiO2 surface may be particularly beneficial for the use of one-electron outer-sphere redox couples such as the CoII/III species.81,82 The CoII/III(bpy)3 couple can yield an unprecedented 12% photovoltaic efficiency when used in molecular solvents.82
To address solubility limitations of this couple in ILs, the ligands can be functionalized with imidazolium groups.83 Used in [C2mim][SCN], the device performance of this new couple was relatively poor (1.7%) but in a binary IL (with 1-propyl-3-methylimidazolium iodide) power conversion efficiencies of 7.37% at 1 sun were achieved.83 The significant increase in efficiency upon addition of the iodide salt has been attributed to its role in regenerating the Co2+ species after electron donation to the dye.83 A new sulfide/polysulfide couple was also used in the presence of an iodide salt, giving efficiencies of 6.4% in [C2mim][SCN].84 Modification of the counter anion of the CoII/III couple is another possible strategy to enable higher redox couple concentrations in ILs, while alternatives to the [PF6]− anion, which is easily hydrolysed, may also provide better stability.85
To eliminate any problems of leakage of the electrolyte from the DSSC, the IL may be incorporated into polymer gels, or physically gelled via the addition of inorganic nanoparticles.80,86,87 However, to achieve optimum device efficiency the diffusion rate of the redox couple must be maintained. Organic ionic plastic crystals (OIPCs) can also be used as solid-state electrolytes, allowing I−/I3− diffusion rates sufficient to support efficiencies of >5%.86,88,89 The nature of the cation and anion of the OIPC can have a significant influence on the device efficiency, which is a reflection of both the rates of diffusion of the redox couple through the different electrolytes and also of the influence of the anion – particularly the basic dicyanamide, [N(CN)2]− – on the energy levels of the TiO2,88 as observed in analogous IL systems.90 OIPCs also perform well at elevated temperatures and with accelerated ageing.91 Work in this area is now focused on strategies for further enhancing the diffusion rates of the redox couple.
In a DSSC, efficiency can be lost over time through electrolyte or redox species loss/degradation, deactivation of the electrocatalyst, corrosion of internal current collectors (e.g., Ag wires), and dye desorption or degradation.92 Stability is ideally assessed by accelerated aging at 60 °C under 1 sun illumination for at least 1000 h, under which conditions the standard [C2mim][B(CN)4]-based DSSCs on glass substrates are similar to those using a 3-methoxypropionitrile (MPN) electrolyte (retaining >90% of initial efficiency).73,92,93 However, with thermal cycling between −45 °C and +85 °C, under 1 sun illumination, DSSCs using this IL show slightly better stability (17% loss in relative efficiency) compared to MPN-based devices (22% loss), albeit from a lower initial efficiency.92
The long-term stability of DSSCs on plastic substrates requires an electrolyte that does not permeate the substrate and thus ILs are of particular interest in this context. These flexible DSSCs have the advantage of being lighter weight, more suitable for a range of applications and offer the possibility of large scale, low cost production via roll-to-roll printing.94 In the counter electrode role, alternatives to thermally deposited platinum are also required to enable lower cost flexible devices and the use of plastic substrates. However, relatively few of the counter electrode materials available have been utilized with IL electrolytes.95 CoS electrochemically deposited onto ITO/PEN films can produce 6.5% efficient DSSCs with a [C2mim][B(CN)]4-based electrolyte and a glass working electrode.96 Poly(3,4-ethylenedioxythiophene) (PEDOT) can also be electrochemically deposited onto ITO/PEN films using very short (5 s) deposition times, which is pertinent to the development of roll-to-roll processes, yielding 5.7% efficient devices with a [C2mim][B(CN)]4 electrolyte.97 PEDOT can give lower charge transfer resistances than Pt when used with I−/I3− in ILs, where the ability to make thicker films may be an advantage given the need for high triiodide concentrations.86,98 Materials such as PEDOT and gold can also give better performance than Pt with the CoII/III redox couple,99 but these have yet to be utilized with an IL electrolyte.
Looking to the future, improvements in efficiency, stability and cost are all required for the wide-spread uptake of DSSC technologies. Ionic liquids clearly have a leading role as low volatility electrolytes. To this end, the development of efficient and stable metal-free photosensitisers and electrocatalysts that operate efficiently with the IL electrolytes is an important goal. Similarly, eliminating the need for transparent conducting oxide (TCO) layers on one or both substrates would be highly beneficial for commercialisation. For flexible DSSCs on plastic substrates there is an additional need for semiconductor and blocking layers that can be processed at low (<150 °C) temperatures. Overcoming these material and engineering challenges will be extremely valuable in realising the full promise of DSSC technologies.
4. Enabling the new: thermoelectrochemical cells
While the possibilities for harvesting solar energy are widely recognized and this is a relatively mature field of research, utilization of the plethora of low-level (<200 °C) waste heat sources through the direct conversion of thermal to electrical energy has not been widely explored. Semiconductor-based thermoelectrics have shown significant progress in recent years, with nano-dimensional materials engineering enabling the long awaited figure of merit increases.100–102 However, these devices remain relatively expensive and more suitable for harvesting high temperature waste heat.Thermoelectrochemical cells (TECs) utilize an alternative design, incorporating a redox couple within an electrolyte. Here, it is the temperature dependence of the electrochemical redox potential that generates the potential difference across the device when a temperature gradient exists.103 Until recently, electrolytes tested in these devices have been limited primarily to aqueous-based systems,104 which restrict the operating temperatures and long term stability. On the other hand, the thermal stability and relatively low thermal conductivity of many ILs makes them ideal for harvesting waste heat in the 100–200 °C range. This increases both the variety of possible applications and the power outputs that can be achieved by increasing ΔT.
Adopting the nomenclature from the semiconductor-based thermoelectric field, the magnitude of the change in electrode potential with temperature is given by the Seebeck coefficient, Se. The Seebeck coefficient of a redox electrolyte system is related to the entropy change associated with the redox process, and is thus influenced by both the nature of the redox couple and the surrounding solvent.85,105,106 Therefore, an additional motivation for the use of ILs in TECs is the possibility of increasing the magnitude of this entropy change through the different solvation environments afforded by ILs.107
Migita and co-workers have demonstrated the linear relationship between the Seebeck coefficient and the charge density of a series of Fe and Cr complexes in [C4mpyr][NTf2].106 The highest value obtained in this IL was for the Fe(CN)63−/4− couple, at −1.49 mV K−1, which is similar to the aqueous value. This, and a recent report by Yamato et al.108 on a range of Fe, Ru, and Ni redox couples in different ILs, highlights the importance of the strength of the redox couple-IL interactions in determining Se.
To enable the comparison of different electrolytes with respect to their suitability for use in a thermoelectrochemical cell, a modified figure of merit has been proposed.109 This takes into account the diffusion rate of the redox couple, rather than the electrical conductivity that is of importance in semiconductor thermoelectrics, and is therefore more applicable to devices using a liquid redox electrolyte. The top performing I−/I3− based electrolyte tested used [C2mim][BF4], which produced power densities up to 29 mW m−2 in unoptimized devices operating with a hot side at 130 °C.109
Taking inspiration from the DSSC work discussed above, the first use of a CoII/III(bpy)3 redox couple in thermoelectrochemical cells achieved Seebeck coefficients of up to 1.88 mV K−1 using IL electrolytes, and 2.2 mV K−1 with MPN.85 This unprecedented Se value is believed to be primarily a result of the high/low spin transition upon electron transfer in the redox couple. Characterization of the power and current vs. potential outputs for thermoelectrochemical cells demonstrated a strong dependence on the nature of the IL used, with maximum power outputs of 183 mW m−2 with [C2mim][B(CN)4] (Fig. 4).85 This was further increased to 240 mW m−2 upon deposition of a rough layer of Pt-black onto the Pt disk electrodes.
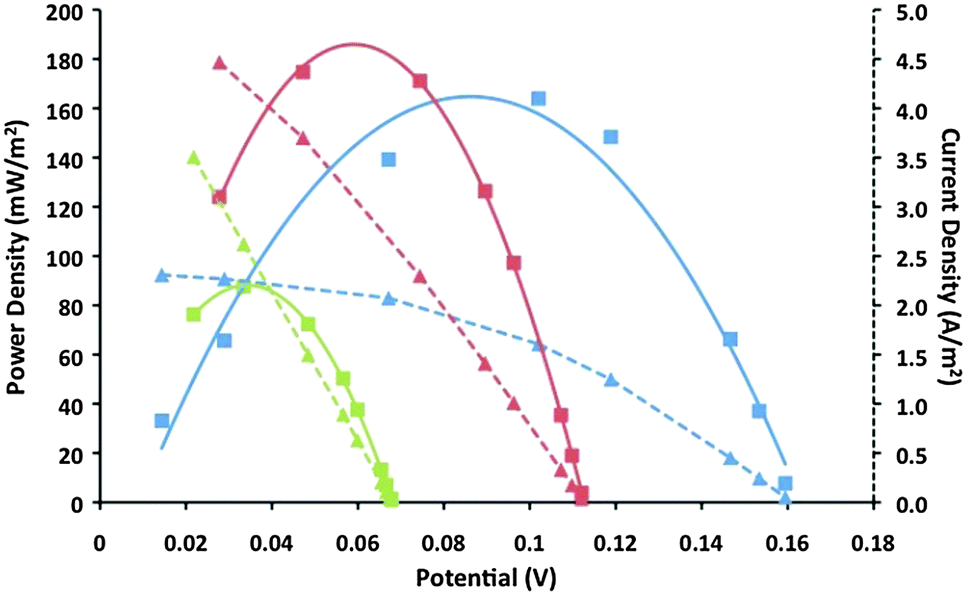 | ||
Fig. 4 Power vs. potential (solid line) and current vs. potential (dotted line) plots from thermoelectrochemical devices utilizing platinum electrodes and 0.1 M CoII/III(bpy)3(NTf2)2/3 in [C2mim][B(CN)4] at three temperature differences: Thot (°C)/Tcold (°C):  130/30 (blue); 130/30 (blue);  130/60 (red); 130/60 (red);  130/90 (green). Reproduced from ref. 85 with permission of the RSC. 130/90 (green). Reproduced from ref. 85 with permission of the RSC. | ||
In these IL-based thermoelectrochemical cells the output power is primarily limited by diffusion of the Co redox couple, with the MPN electrolyte producing up to 522 mW m−2. Thus, further improvements of device efficiency will result from optimization of device design (electrode spacing, cell orientation, etc.) and increasing the operating temperature. In parallel, further investigation of the Seebeck coefficient of different redox couple/IL systems and more fundamental understanding of how to maximize the redox entropy will lead to further advances in this promising new area.
5. Enabling the new: ionic liquids in energy storage – lithium, sodium, and magnesium batteries
ILs have a very clear set of advantageous properties to offer the battery world, especially given recent instability issues with battery packs in both aircraft and vehicles.110 The lack of flammability of IL-based electrolytes, even in mixtures with other solvents, may offer the prospect of improvements in these devices that are needed to develop large scale energy storage in various aspects of transportation technology, as well as renewable energy storage. The ability of a number of IL families such as the pyrrolidinium cation and its relatives, along with fluorinated anions such as [NTf2]−, to support lithium and more recently sodium electrochemistry is well known.3 However, commercial applications have been slow to develop due in part to the cost of the IL and also due to their relatively poor transport properties that translate into relatively poor charge/discharge characteristics. Charge rate is particularly important to most applications, including hybrid vehicles, electric vehicles, and renewable energy storage. The problem becomes particularly acute at sub-ambient temperatures.In regard to cost and charge rate, two recent advances are of note. The first development demonstrated that potentially low cost anions containing only carbon and nitrogen could support stable lithium cycling.111 Emphasizing once again the importance of the solid-electrolyte interface (SEI) layer in generating a stable interface, the dicyanamide ILs proved capable of high efficiency lithium cycling only in the presence of an optimum, but small (∼250 ppm), amount of water in the electrolyte, this presumably serving to provide some of the reactions necessary to create a stable SEI layer. While this nitrile-based IL may ultimately not be the ideal electrolyte for safe large-scale use, it demonstrates the possibility of non-fluoride containing electrolytes and the importance of SEI generating additives in developing the ideal low cost electrolyte.
A second important development in recent work involved the demonstration of high-rate charging of an IL-based Li/LiCoO2 cell at rates not possible in traditional carbonate electrolytes.112 The key to this breakthrough was the discovery of very high lithium salt content IL electrolytes which are probably more appropriately thought of as metal-complex IL electrolytes. In this case the electrolytes consisted of an equimolar mixture of [Li][FSI] and [C3mpyr][FSI], the Li+ probably forming a complex ion species of the stoichiometry [Li][FSI]2−. The key to their high rate charging effect appears in the charging transient on the initial stages of charging the battery (Fig. 5). The traditional carbonate electrolyte produces a large charging transient associated with polarization of the electrolyte, exceeding the allowed maximum voltage of the cell. On the other hand the IL electrolyte shows no such transient even at a 5 C (i.e., a 12 minute) charge rate, because of the very high concentration of both Li+ and [FSI]− ions in the electrolyte.
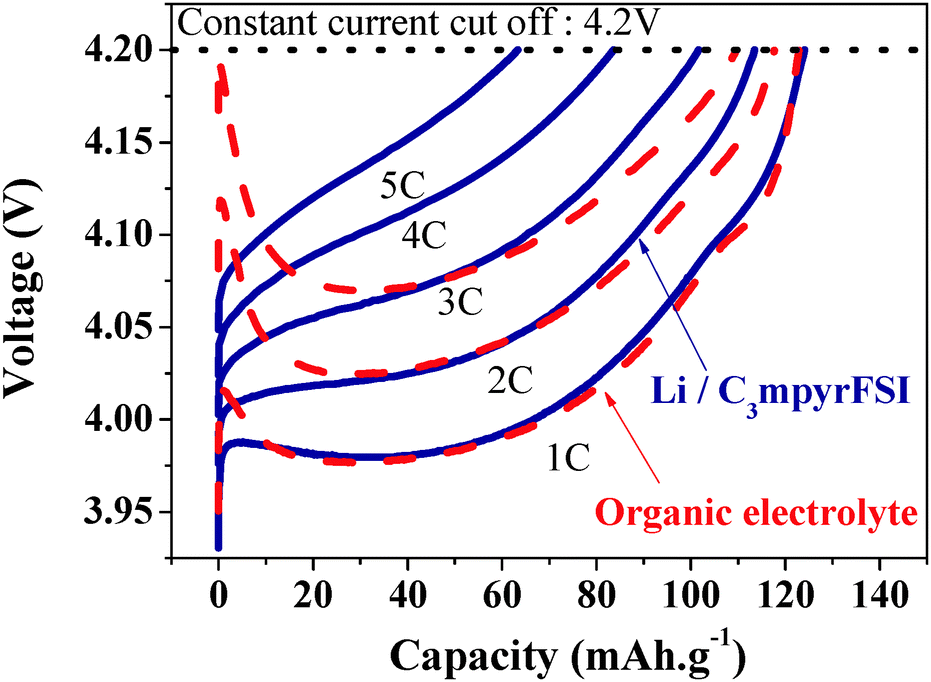 | ||
| Fig. 5 Charge cycles of an equimolar mixture of [Li][FSI] and [C3mpyr][FSI] in a Li|LiCoO2 cell; dashed lines = organic electrolyte, solid lines = the Li+/[C3mpyr][FSI] electrolyte. Redrawn from ref. 112. | ||
Related to this, ILs are currently being investigated for use with high voltage cathode materials. A number of inorganic materials of lithium intercalation potential >4.5 V vs. Li/Li+ are potentially available, but limited both practically, and even in respect to research investigations, by the lack of sufficiently stable electrolytes.113 What is required here is very high oxidative stability in both anion and cation of the IL. The lack of overpotential transients observed in high Li+ salt electrolytes discussed above is of assistance in this respect.
Similarly, Li–air concepts are under investigation that utilize the special properties of ILs.114,115 In these devices, the necessary isolation of the anode from the air and moisture exposed cathode is achieved using a hydrophobic IL and the stability of the IL towards the O2− radical anion was induced by incorporation of ether functional groups into the alkyl chains of the cation.
It is important to note here that ILs have also become of utility in “ionothermal” synthesis reactions (see Materials section below) in the preparation of the lithium cathode materials themselves.116 In this context the IL environment offers unique solubility properties that create the potential for lower temperature processing. Unique metastable phases also become stable and thereby this approach offers access to materials otherwise difficult to produce.
Returning to the issue of cost, recently work has begun to focus on the intrinsic cost of lithium materials and the impact of this on large scale energy devices that are required, for example for wind-mill and solar array energy storage.117 Thus ambient temperature sodium-based battery technology has become of intense interest. Since sodium electrochemistry bears many features in common with lithium, it is not surprising that the electrolytes investigated for sodium cells have borrowed heavily from lithium battery know-how, including IL electrolyte studies. Sodium cells based on [NTf2]− and [FSI]− have shown initial success and are the focus of considerable further attention.118–121
While research into sodium intercalation into anodic materials such as graphite is in its infancy, the greater reactivity of sodium metal compared to lithium reminds us that the creation of a stable SEI layer will be of potentially even greater importance to these devices. As above, the addition of small amounts of SEI forming additives will be one of the key strategies in the search for very stable SEI forming electrolytes for Na-based cells.
In a similar vein of striving for low cost on a $/MJ scale, there are efforts to develop Mg-based battery systems and, again, these are making use of the potentially advantageous properties of ILs to provide a stable SEI layer against a reactive interface.122,123 A controlled amount of water is the key to the operation of the Mg electrolyte and a range of phosphonium ILs including [P66614][Cl] have been studied in this regard.124 This IL has an upper solubility for water around 8%, this being a convenient saturation point that ensures that the electrolyte cannot absorb more than this level on exposure to the atmosphere. However, more data on the saturation points as a function of temperature for this and other hydrophobic ILs is needed.
Being considerably heavier than Li, a Mg-based battery is most likely to emerge in combination with an air electrode in order to improve the overall cell energy and power density. The challenge then includes the development of a reversible air electrode, i.e., a dual oxygen reduction electrode/water (or peroxide) oxidation electrode, where the required catalysts are combined in a single material or single electrode. Interest in the oxygen reduction electrode operating in an IL electrolyte (with or without water being present in the electrolyte) has been growing in recent years, partly driven by fuel cell concepts as well as air batteries.125–128 Pozo-Gonzalo et al.,129 have demonstrated a facile four-electron reduction of oxygen to water in [P66614][Cl] and investigated the important role of a weak proton source in determining the outcome of the reaction. Where such a proton source is absent a reversible O2/O2˙− couple (the one electron reduction reaction) is observed instead in [P66614][Cl]125 and this also has the potential to be used in energy storage applications. Similarly, oxidation of solute amounts of water in an IL electrolyte has been studied in recent years and this is discussed further below.
Clearly, cost is a critical barrier to be overcome in order for the advantageous properties of ILs to find their way into large scale battery application. However, if breakthrough performance can be enabled by an IL electrolyte, such as in the use of high voltage cathode materials, this may be less of a challenge. In this respect a greater understanding, both practically and theoretically, of the oxidative stability and the factors determining it would be of tremendous assistance to progress in this field.
Common to many of the electrochemical device applications discussed in the sections above is the requirement for enhanced transport of the target species through the IL electrolyte. The development of more fluid ILs will go some way to facilitate this, and there is significant scope for optimising transport properties through judicious choice of binary IL or IL–solvent combinations. However, for many applications the advantages of solid state electrolytes are significant, e.g., for improved safety and stability. Thus, research into IL-based polymer electrolyte systems, or organic ionic plastic crystal electrolytes, is of equal importance. Achieving high transport rates in these different electrolyte materials requires more fundamental understanding of the speciation and transport mechanism of the target ions (I−/I3−, Li+, Na+, Mg+, H+, CoII/IIIetc.) in the ILs, and how this can be optimised by anion and cation design. Similarly, more detailed studies of the structure and electrochemical processes at the IL/electrode interphase is key, to improve efficiency (lower the overpotential), eliminate catalyst poisoning or improve the SEI layer, and also facilitate the move towards lower cost materials and devices.
6. Enabling the new: ionic liquids in hydrogen generation by water splitting
Water electrolysis (otherwise known as water splitting) has become of intense interest as a means of producing hydrogen as a fuel.130 The process involves simultaneous water oxidation and water reduction on the anode and cathode of the cell, respectively. The water oxidation reaction, producing 4 electrons and 4 protons, is intrinsically complex and represents the largest fundamental source of inefficiency in this process; a huge effort has therefore focused on discovering and understanding new catalysts for this reaction. Izgorodin et al.,131–133 investigated the use of the classic PIL, ethyl ammonium nitrate, as an electrolyte medium for the electrodeposition of a MnOx (x ∼ 1.8) birnessite-type catalyst layer for this purpose via the reaction: Mn(II) + 2H2O ↔ MnO2 + 4H+ + 2e−. The IL represents a convenient way to access elevated temperatures, around 140 °C, for this electrodeposition reaction; the reduced vapour pressure of the solute quantity of water involved in the IL allows operation at these temperatures without the need for a pressure vessel. The catalysts obtained proved to represent some of the best ever observed in the manganese oxide family of water oxidation materials. The reasons for this high activity are still under investigation.Using an IL-based electrolyte as a medium for the water oxidation reaction has also been investigated recently. The MnOx catalyst described above was found133 to produce very high water oxidation activity in hydrated IL buffer134 electrolytes based on butylammonium sulfate and water. In a low overpotential range the product was shown to be hydrogen peroxide via a 2 electron pathway, rather than the full 4 electron oxidation to oxygen. It was shown that the hydrogen peroxide could be separately decomposed into oxygen via a simple catalyst pathway. This low overpotential oxidation is extremely valuable in terms of achieving a high energy efficiency reversible air electrode.
These relatively preliminary observations suggest that further investigation of ILs as electrolytes in water splitting, and the related challenge of CO2 reduction, may provide the basis for significant advances in these fields. In particular, a deeper understanding of water activity in ionic liquid mixtures, protic and aprotic, would be of great value.
7. Enabling the new: organic salts as phase change materials
Finally, in the context of energy applications it is important to mention the role of ILs in addressing the growing need for thermal energy storage materials and working fluids for solar thermal applications. Phase change materials have been investigated recently based on the solid–liquid phase change in organic salts. There is some interest in materials that have a sub-100 °C melting point, but these are of relatively limited application. The temperature range between 100 °C and 200 °C is of considerably greater interest and is a temperature range where other materials are much more limited. What is needed here is a melting point in the desired temperature region, coupled most critically with a high enthalpy of fusion. Recent work135 has shown that the strong hydrogen bonded lattice formed by the guandinium cation with a range of anions produces a competitively high enthalpy of fusion for these materials. The development of ILs as working fluids for solar thermal applications either as heat transfer fluids or as working fluids in Organic Rankine Engines is still in its infancy, however the design flexibility offered by organic salts offers considerable potential for important breakthroughs in these areas. For example, the exceptional thermal stability offered by the tetra-arylphosphonium cation family of ILs136 provides an important direction for future work in the heat transfer field.ILs in materials preparation
In 2000, the synthesis of SiO2 aerogel using [C2mim][NTf2] opened a new path for the synthesis of inorganic materials.137 ILs have since received much attention in materials synthesis due to their unique properties in this context including, in many cases, their weakly coordinating molecular ions and their high-temperature stability. Many new inorganic materials and their unique morphologies have been summarized in a number of excellent recent review articles.138,139 The range of these materials has been very broad, including metal structures, non-metal elements, silicas, organosilicas, metal oxides, chalcogenides, metal salts, open-framework structures, IL-functionalized materials, and supported ILs.140 The ILs have been used as solvents, structural templates, precursors, stabilizers, and scavenging agents. Here we will focus on a number of intensely studied areas including molecular sieves, metal–organic frameworks (MOFs), polyoxometalates (POMs), reduced graphene oxides (rGOs), and recently described photocatalysts.8. Ionothermal synthesis of open-framework structures
 | ||
| Fig. 6 A [001] perspective view of the framework structure of aluminophosphate molecular sieve DNL-1 (Al yellow, P green, and O red). The bridging oxygen atoms in the framework and all hydrogen atoms have been omitted for clarity. Reproduced from ref. 144 with permission from Wiley-VCH. | ||
Similarly, using either 1,2-ethylenediamine or 1,6-hexanediamine as SDA in [C2mim][Cl], two new 2D layered aluminophosphates [Al3P4O16][NH3CH2CH2NH3]0.5[C6N2H11]2 and [Al3P4O16][NH3(CH2)6NH3][NH3(CH2)6NH2]0.5[C6N2H11]0.5[H2O] were synthesised ionothermally.142 It appears that the amines and the IL cations act cooperatively as co-templates in the process.
Tian et al., also reported the synthesis of permeable aluminophosphate molecular sieve membranes (Fig. 7) on porous alumina disks by a substrate-surface conversion.147 In the in situ process, the porous alumina substrate acted as both support and Al source. By adjusting the type of IL and organic amines in the initial solution, different types of membranes could be synthesised.
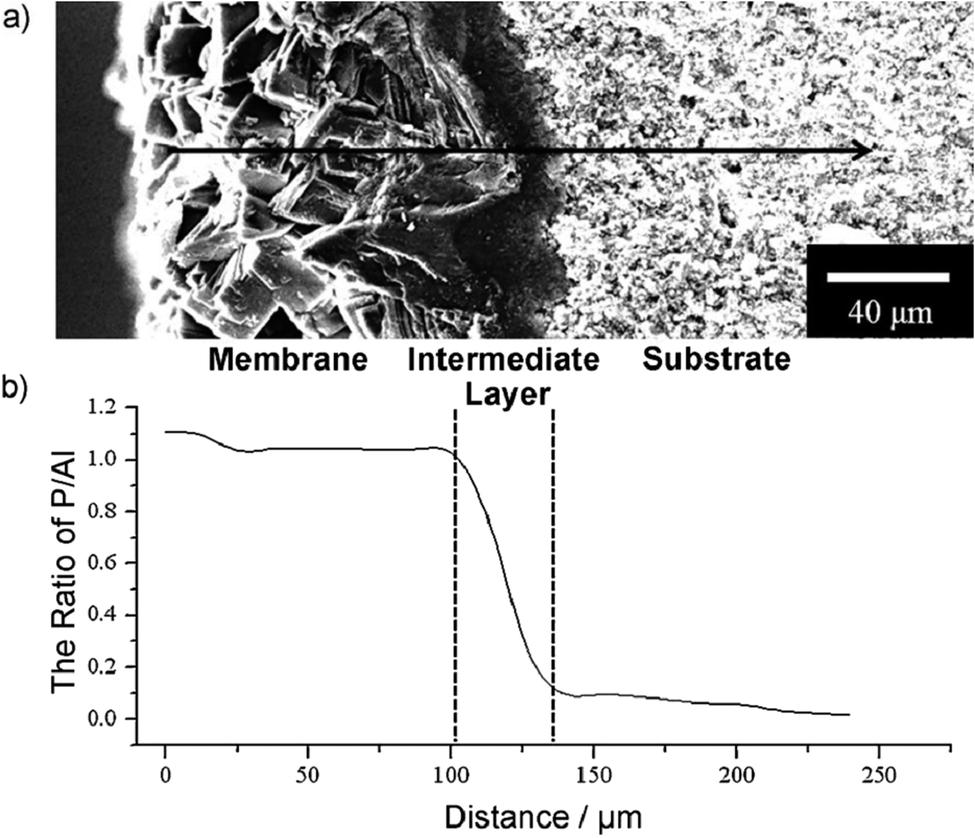 | ||
| Fig. 7 (a) Cross-section SEM image of chabasite (CHA) membrane. The arrow represents the path of the EDX line scan; (b) P/Al ratio along the path of the EDX line scan. Reproduced from ref. 147 with permission from Wiley-VCH. | ||
With the advantages of the ILs, ionothermal synthesis can be a highly-efficient, safe, environmental-friendly synthetic strategy for preparation of crystalline materials. During the process, the IL may direct the crystal growth, and template the pore formation with new structures. However, this approach is immature, and needs much more exploration of the versatile structures and special properties of the ILs.
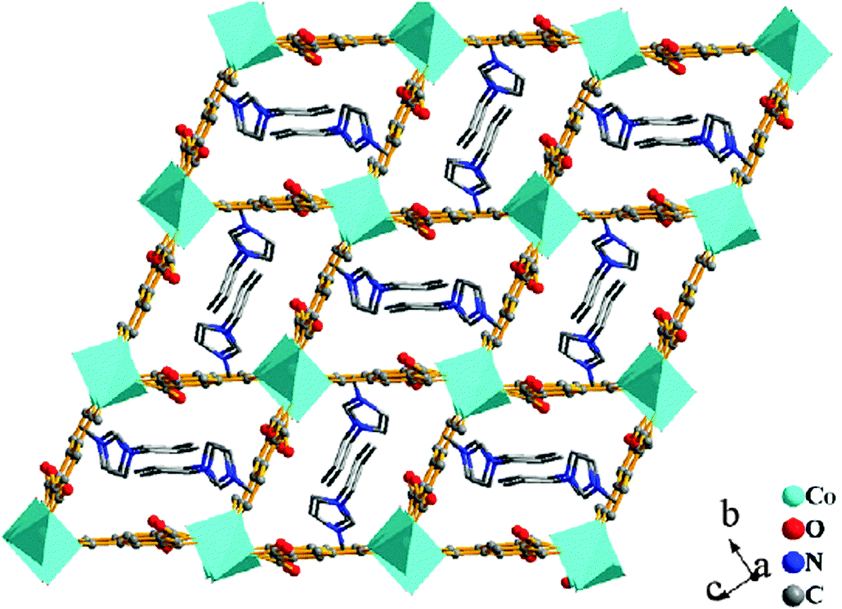 | ||
| Fig. 8 The 3D [Co2(BTC)2(H2O)2]n2n− framework with the [C3mim]+ cations residing in the 1D channels. Reproduced from ref. 150 with permission from Elsevier. | ||
In an example demonstrating the different roles of ILs in ionothermal synthesis, Zhai et al., synthesised a novel 3D ferroelectric MOF using IL as both the solvent and structure directing agent.151 The asymmetric nature of the [C2mim]+ cations was a key factor in the formation of helical chains, which further led to the non-centrosymmetric packing arrangement. Parise et al., synthesised five, novel 2D formate-bridged magnetic MOFs based on Co, Ni, and Mn ionothermally ([NH3C2H4OH]2[M(CHO2)4], M = Co, Ni, Mn and [NH3C3H6OH][M(CHO2)3(H2O)], M = Co, Mn).152 Here, the ILs 2-hydroxyethylammonium formate, and 1-hydroxy-3-propylammonium formate served the roles of solvent, organocation template source, and ligand source.
Huang et al., created F-centered Cd3F MOFs via an in situ ionothermal oxidation and hydrolysis process (Fig. 9).153 This work addressed the application of an IL ([C4mim][BF4]) with four functions, including solvent, structure-directing agent, fluoride source, and catalyst promoter.
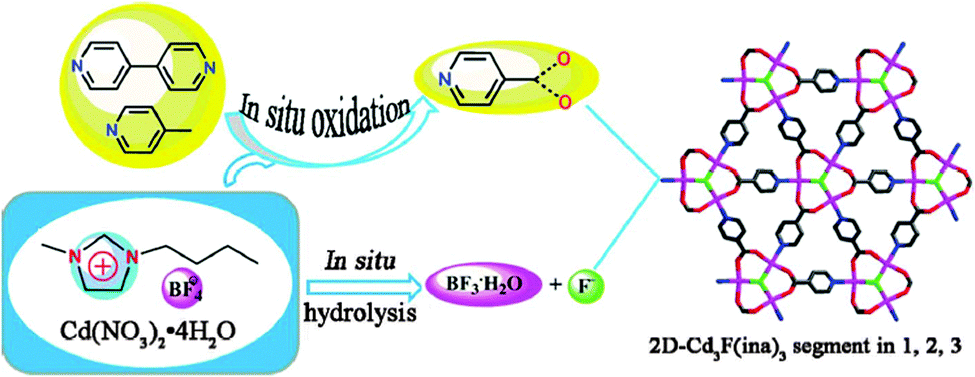 | ||
| Fig. 9 The formation routes of F-centered Cd3F MOFs via in situ oxidation and hydrolysis. Reproduced from ref. 153 with permission from the RSC. | ||
The successful preparation of new MOF structures by ionothermal synthesis demonstrates that the method is a promising one for synthesizing new kinds of open-framework materials. Further investigation of these systems may also yield many classes of MOF compounds with interesting features and useful properties.
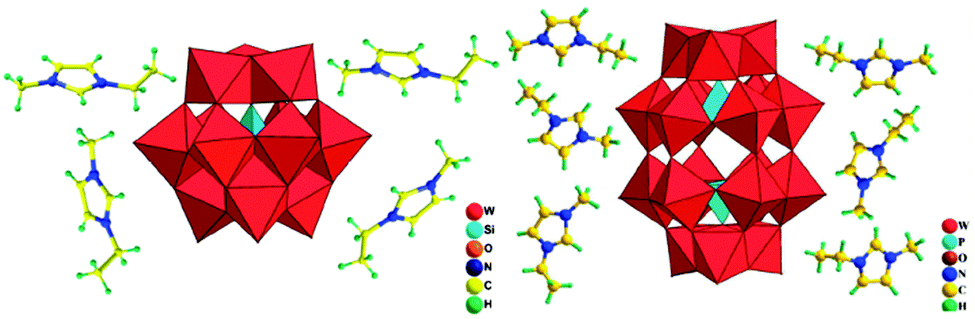 | ||
| Fig. 10 Polyhedral and ball-and-stick representation of [C2mim]4[SiW12O40] (left) and [C2mim]6[P2W18O62]·4H2O (right). Reproduced from ref. 162 with permission from Taylor & Francis. | ||
During the synthetic processes described above, the ILs act as both solvents and charge compensating species. Noticeably, owing to the unique characteristics of the ILs, ionothermal synthesis opens up new routes to new POM-based aggregates. We anticipate much new chemistry based on these exciting solids.
9. ILs for synthesizing reduced graphene oxides (rGOs) and exfoliating graphene
Graphene and its derivatives, such as graphene oxide (GO) and reduced graphene oxide (rGO), have attracted increasing research interest in both scientific studies and technological development such as batteries, supercapacitors, fuel cells, photovoltaic devices, and biosensors.163,164 As one of the most attractive techniques for larger scale and low-cost production of rGO, new methods have been developed to reduce GO that has been homogeneously exfoliated in ILs.165–167,176,177 For example, Yoon et al., developed a facile and scalable method to rapidly produce rGO by IL-assisted microwave chemistry (Fig. 11).168 Due to its thermal stability upon microwave radiation and high dielectric constant, [C2mim][NTf2] was chosen as the IL.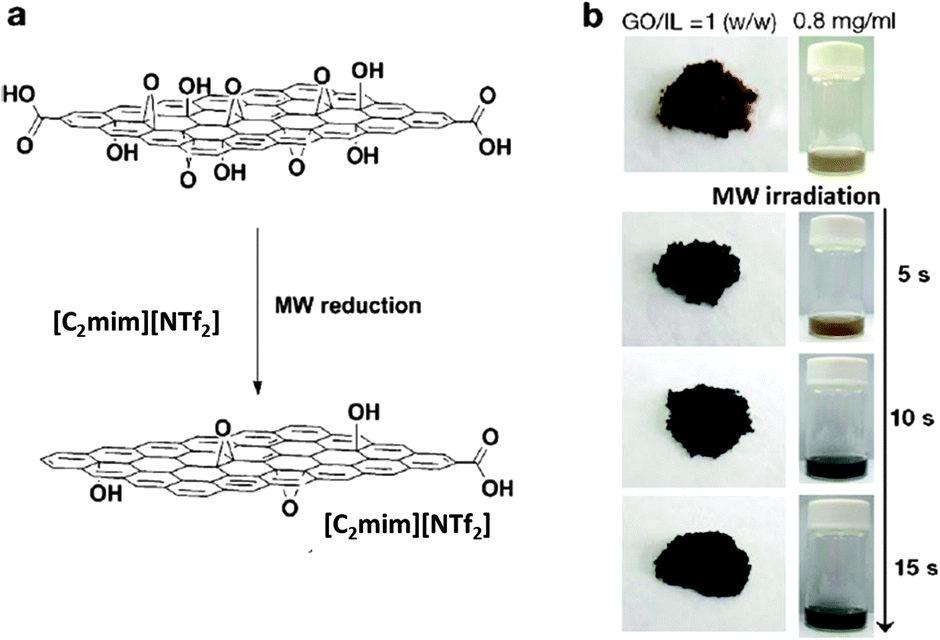 | ||
| Fig. 11 (a) Schematic representation of IL-assisted microwave reduction of GO and (b) photographs of prepared reduced graphene oxide. Reproduced from ref. 168 with permission from the RSC. | ||
A similar microwave-assisted ionothermal approach carried out at 200 °C and atmospheric pressure has also been demonstrated169 (Fig. 12). Clearly, ILs have an important role to play in the synthesis of rGO and further investigations of the effect of the IL on the formation of different rGO structures, offering different functional properties, are much needed.
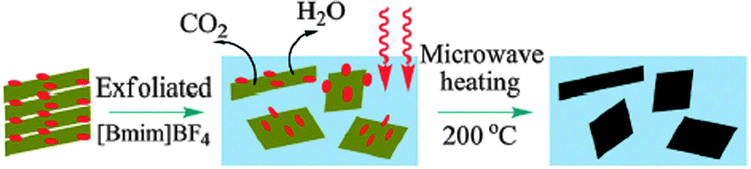 | ||
| Fig. 12 Schematic illustration of reaction processes of microwave assisted thermal treatment to produce graphene. Reproduced from ref. 169 with permission from the RSC. | ||
In an interesting unique application of ILs, it has been reported that certain ILs can exfoliate graphene sheets directly from graphite.170–172 This could allow the production and use of pristine graphene sheets from direct exfoliation of graphite rather than producing GO which would then have to be reduced. It is believed that the combination of the unique properties of ILs and graphene will help to speed up the utilization of graphene materials in existing areas and to open up new applications.
10. ILs with multifunctional roles for synthesizing photocatalysts
Photocatalytic technology offers a potentially sustainable approach to industrial oxidation processes and has been widely investigated for environmental pollution treatment and energy applications. Hierarchical semiconductor nanostructures such as nanorods, nanowires, and nanotubes are receiving much attention because of their unusual properties in this regard.173,174 Recently, novel methods to prepare photocatalysts with high activity and stability have been developed using various IL processes.175,176ILs have been used as templates to prepare titanium oxides.177–180 Liu et al.,176 used [C4mim][BF4] as solvent and titanium isopropoxide (TTIP) as precursor to produce uniform anatase TiO2 nanocrystals. This method could also be extended to the synthesis of other metal oxides. Rutile TiO2 nano-flowers (TNFs) comprising bunches of aligned nanorods with uniform size and shape were synthesised via a hydrothermal method in [(carboxymethyl)mim][HSO4] as illustrated in Fig. 13.180 Most of the structures displayed microspheres containing nanorods of 250 nm diameter.
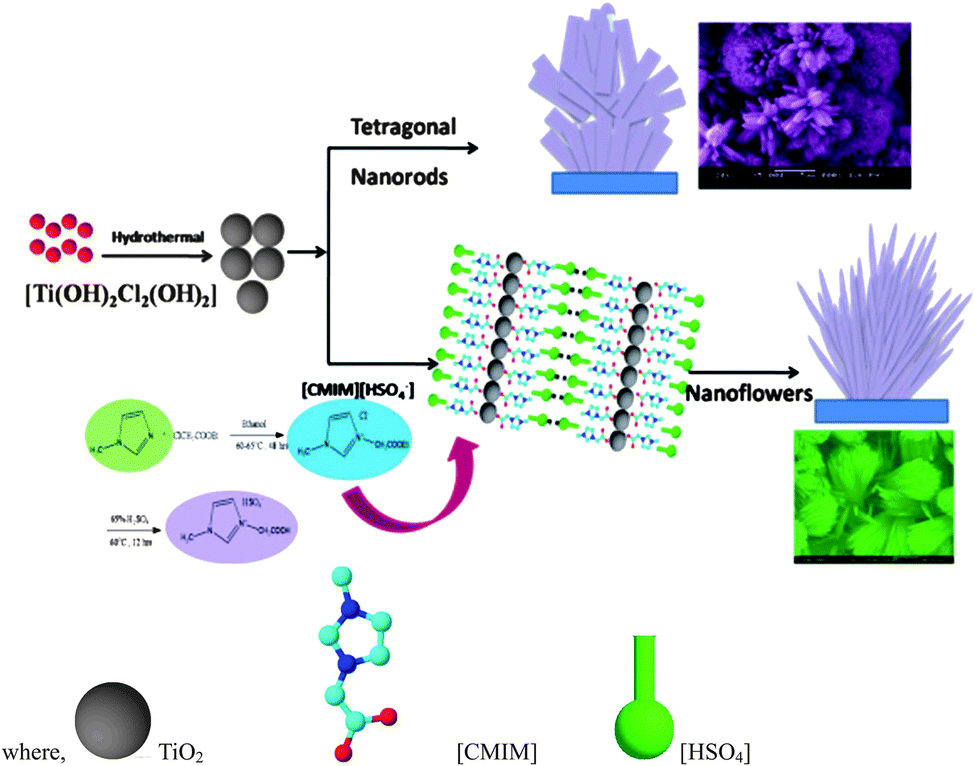 | ||
| Fig. 13 Schematic illustration for the growth process of TiO2. Reproduced from ref. 180 with permission from the RSC. | ||
Huang et al.,181 reported a facile one-pot approach to uniform BiOBr hollow microspheres (HMSs), with a diameter of 1–2 mm and shell thickness of 100 nm (Fig. 14), in a mixture of 2-methoxyethanol and the IL [C16mim][Br]. The band gap of the prepared BiOBr HMSs was estimated to be 2.7 eV, and they displayed highly efficient photocatalytic activity in the degradation of Rhodamine B (RhB) dye and reduction of CrVI under visible-light irradiation. Other BiOBr hierarchical nano- and microstructures have been synthesised through solvothermal processes in the presence of [C16mim][Br].182 In [C4mim][I],183 the IL not only acts as solvent and template, but also acts as an I source for the fabrication of BiOI hollow microspheres. Li et al., also used a [C16mim][Cl]/[C16mim][Br] mixture for the preparation of BiOCl/BiOBr composite photocatalysts;184 the photocatalytic activity of the BiOCl/BiOBr in the degradation of RhB under visible light irradiation was higher than that of either BiOCl or BiOBr alone.
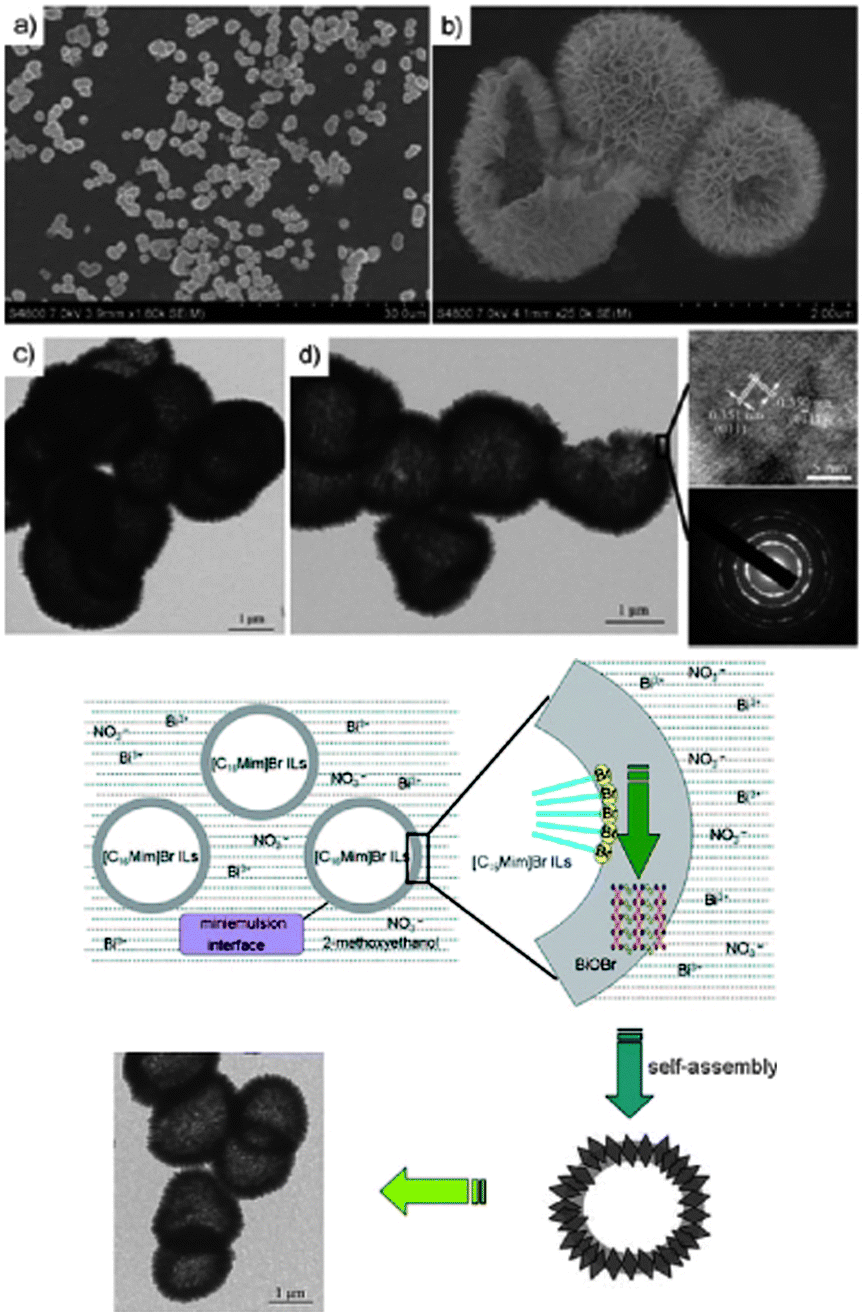 | ||
| Fig. 14 SEM and HRTEM images of BiOBr HMSs (top) and a plausible schematic formation process of the BiOBr HMSs (bottom). Reproduced from ref. 181 with permission from Wiley-VCH. | ||
The studies demonstrated that the IL could prevent the aggregation of the nanocrystals to some extent, and modulate the nanocrystal formation. Thus the ionothermal method opens a new way for controlled synthesis of nanocrystals and will facilitate the exploration of the phase, shape, and size effect of nanocrystals in various applications.
In this brief review, we have surveyed several synthetic strategies involving ILs for inorganic materials including molecular sieves, metal–organic frameworks (MOFs), polyoxometalates (POMs), reduced graphene oxides (rGOs), and certain photocatalysts. In the majority of cases, the ILs were used as media, working as neutral solvent, structure directing agent, and/or charge compensating species. However, despite the remarkable progress made in the field, many challenges, as well as opportunities still remain. One of the most significant challenges is the lack of a series of general methods for synthesizing inorganic materials with controllable sizes, compositions, and shapes. At present, there are still many questions that need to be answered such as the nature of the interaction between the ILs and inorganic species, reactants, and products during the formation process of materials; the mechanism of the self-assembly processes in ILs; and the influence of ILs on the morphology.138 Thus, more spectroscopic and in situ experiments, as well as theoretical modelling are needed to understand the formation process of the special morphologies. Structure–activity relationships between the physicochemical properties of ILs and the activity of the inorganic materials also remain to be established.
Ionic liquids for and as pharmaceuticals
Ionic liquids were once studied as ‘green’ solvents to help address the growing concern in the pharmaceutical industry about the environmental impact of making these specialty chemicals.185,186 However, the often unknown toxicity and safety profiles of ILs (or any new chemical) were inconsistent with the tightly regulated active pharmaceutical ingredient manufacturing practices and ILs were almost summarily dismissed in the context of pharma.187 Unfortunately, this backlash of ‘ILs are bad for pharma’ persists today despite the many opportunities where ILs might make a positive impact in industries that make and use biologically active compounds (e.g., pharmaceuticals, agrochemicals, nutraceuticals, etc.).188,189 Here we will briefly explore the growing potential for ILs to make a positive impact in the isolation, delivery, use, or form of biologically active compounds.As the study of ILs became more widespread and the understanding of their nature grew, the recognition that ILs are customizable and their functions could be designed to fit a given property or application led to many new ILs based on cations and anions never before associated with ILs. That is, the application, or need, drove the search for specific ILs and not vice versa. While green chemistry and sustainability drove the IL community to less and less toxic and more biodegradable ions, another class of ions, those which are biologically active by design, but also just as likely to form ILs, were waiting to be exploited.
Currently, the costs associated with delivering to market new pharmaceuticals, herbicides, biocides, or any other biologically active ingredients is very high. These costs are not only associated with the expenses related to patent protection of particular molecules and the approval by the appropriate agencies to enter the market, but also because of requirements (i) to test new compounds for many years before approval, (ii) to develop the synthetic procedures and build new production plants, and (iii) to cover the costs associated with failed experiments, failed trials, and any other losses that may come from the fact that not all previously investigated compounds end-up getting to the market. Thus much effort is being put into finding new active compounds, new delivery technologies, and new methods for the controlled-release of biologically-active substances.190 In these contexts, ILs have, perhaps, much to offer.
11. Aspects of drug delivery
McCrary et al.,199 investigated tuning IL hydrophilic–lipophilic balance (HLB) for improving water solubility of amphoceritin B (Amp B) and itraconazole. In order to solubilize those hard to dissolve APIs, functionality providing HLB was chosen in the IL's ions independently. Acetates were selected as target ILs to dissolve Amp B by tuning cation hydrophobicity and polyethylene glycol (PEG)-based ILs with a hydrophilic cation and tunable hydrophobicity in the anion were selected to dissolve itraconazole. As a result it was shown that Amp B (30 mg mL−1) and itraconazole (5 mg mL−1) could be dissolved into specific ILs. It was also reported that the introduction of such API-IL solutions into water resulted in achieving aqueous solubility at high concentrations (Amp B, 0.10 mg mL−1; itraconazole, 0.25 mg mL−1). This work suggested that with proper knowledge of the structural requirements of the APIs and of IL chemistry, many drugs could be dissolved and effectively solubilized in water.
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 times by dissolution in [C4mim][PF6] compared to water.200
000 times by dissolution in [C4mim][PF6] compared to water.200
In related work, Smith et al., investigated the solubilities of paracetamol and ibuprofen in [C4mim][PF6] and [C6mim][PF6].201 Similarly, Jaitely et al., reported the solubilization of APIs in water immiscible ILs based on the [PF6]− anion.202 This paper explored the IL–water partition coefficients of sucrose, penicillin V potassium, dexametasone, progesterone, and dehydro-epiandrosterone and compared the data to octanol–water coefficients. The authors concluded that as a result of the water immiscibility of the IL, the release of sucrose and dexametasone from IL reservoirs into water can be prolonged over 48 h. Of course as one can expect the use of PF6−-based ILs, generally considered as toxic due to possible evolution of HF by slow hydrolysis in water,203 may not be very practical. However this proof of concept opens new possibilities and exploration paths for the search of other less toxic or potentially harmfully hydrophobic ILs as carriers for APIs.
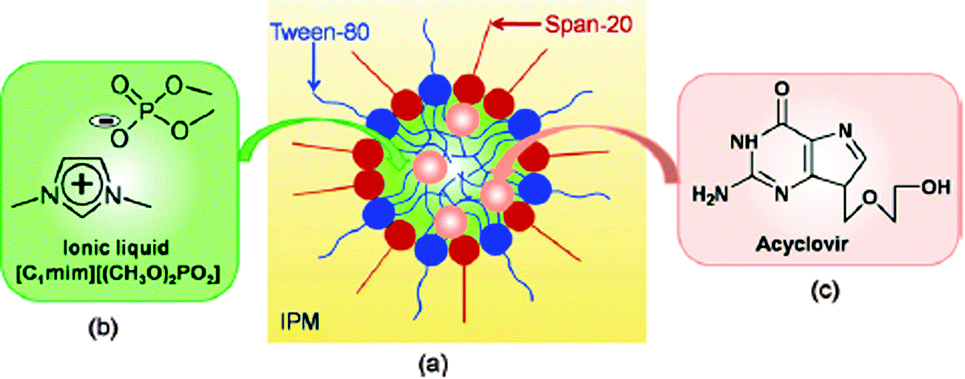 | ||
| Fig. 15 (a) Schematic representation of IL-in-oil (IL/O) microemulsions containing drug molecules, (b) chemical structure of the IL, and (c) acyclovir. Reproduced from ref. 204 with permission of Elsevier. | ||
Another example of using ILs to increase the solubility of sparingly soluble drugs and to enhance their topical and transdermal delivery, was presented recently by Dobler et al.205 In their work, the authors reported the preparation of oil-in-water (O/W) and water-in-oil (W/O) emulsions containing hydrophilic [C6mim][Cl] and hydrophobic [C4mim][PF6] incorporated into the emulsion, resulting in stable formulations. As suggested by the authors, and confirmed by preservative efficacy tests, such systems could be used as preservatives.
12. ILs for extraction of medicinal natural ingredients
Ionic liquid-based extraction of natural products from plants is now being investigated as an alternative to conventional solvent extraction, supercritical fluid extraction, and cloud point extraction, where cost, energy, or environmental impact might be improved. For example, Marrucho et al., recently published interesting survey studies regarding the use of various imidazolium- and cholinium-based ILs in the extraction of saponins and polyphenols from tea and mate.206 The results obtained indicated that high concentration of saponins could be achieved using cholinium chloride and K3PO4 following the recovery of saponins in water. The authors therefore suggested that it is possible to tune the IL affinity for a specific solvent and considerably increase the extraction efficiency of saponins and phenols.There are now many literature examples of utilization of ILs in the extraction of medicinal compounds from medicinal plants with three main approaches of the most interest: (i) use of just ILs as solvent in the ultrasound-assisted extraction of natural products; (ii) microwave-assisted IL extraction of natural products from plants; and (iii) reactive dissolution of biomass in order to extract natural products. We will briefly discuss each in turn.
Salvia miltiorrhiza bunge (SMB) also known as red sage, is used in treatment of cardiovascular and cerebrovascular diseases in Chinese traditional medicine. Bi et al., successfully extracted three active components of SMB, cryptotanshinone, tanshinone I, and tanshinone II utilizing ultrasound-assisted aqueous IL extraction techniques.208 The efficiency of this method proved to be a few times higher than using other solvent systems. Moreover, in order to separate the products from the IL, the metathesis reaction to convert the IL from [C8mim][Cl] to [C8mim][PF6] was performed in situ allowing phase separation and product isolation from the IL.
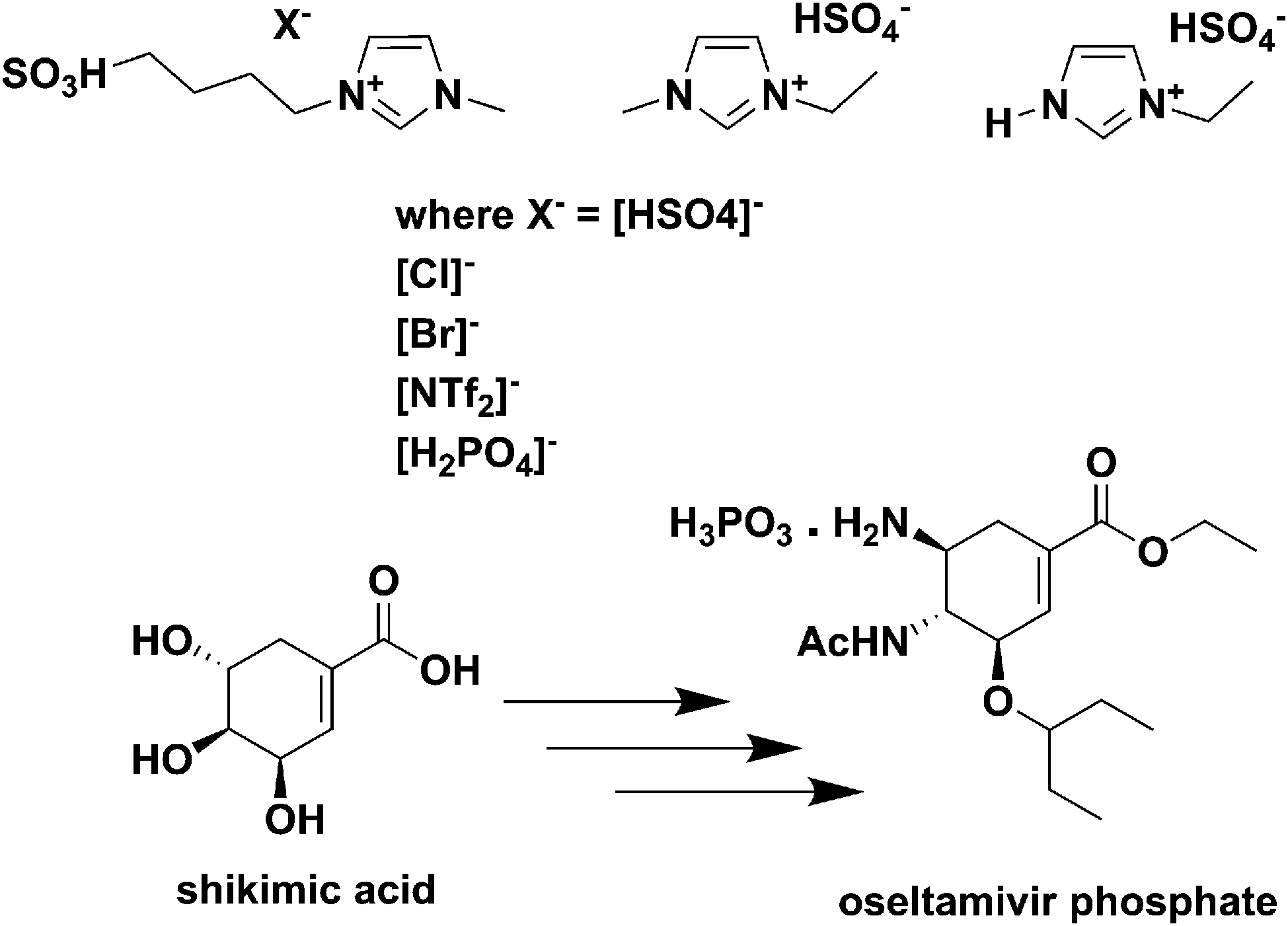 | ||
| Fig. 16 Acidic ILs used for the reactive dissolution of star anise and formation of shikimic acid.213 | ||
Bica et al., reported the use of ILs such as [C2mim][OAc] in the extraction of the pharmaceutically active triterpene betulin (up to 31 wt%) from birch bark with very high extraction yield and purity. Moreover, the reported recovery of the IL via azeotropic distillation of EtOH–H2O proved that this extraction method could be successfully used at large scale (Fig. 17).214 In a similar fashion, [C4mim][Cl] was used in microwave-assisted pretreatment of biomass in order to destroy cell walls of Cynanchum paniculatum before paeonol extraction by solvents such as water, methanol, and ethanol.215
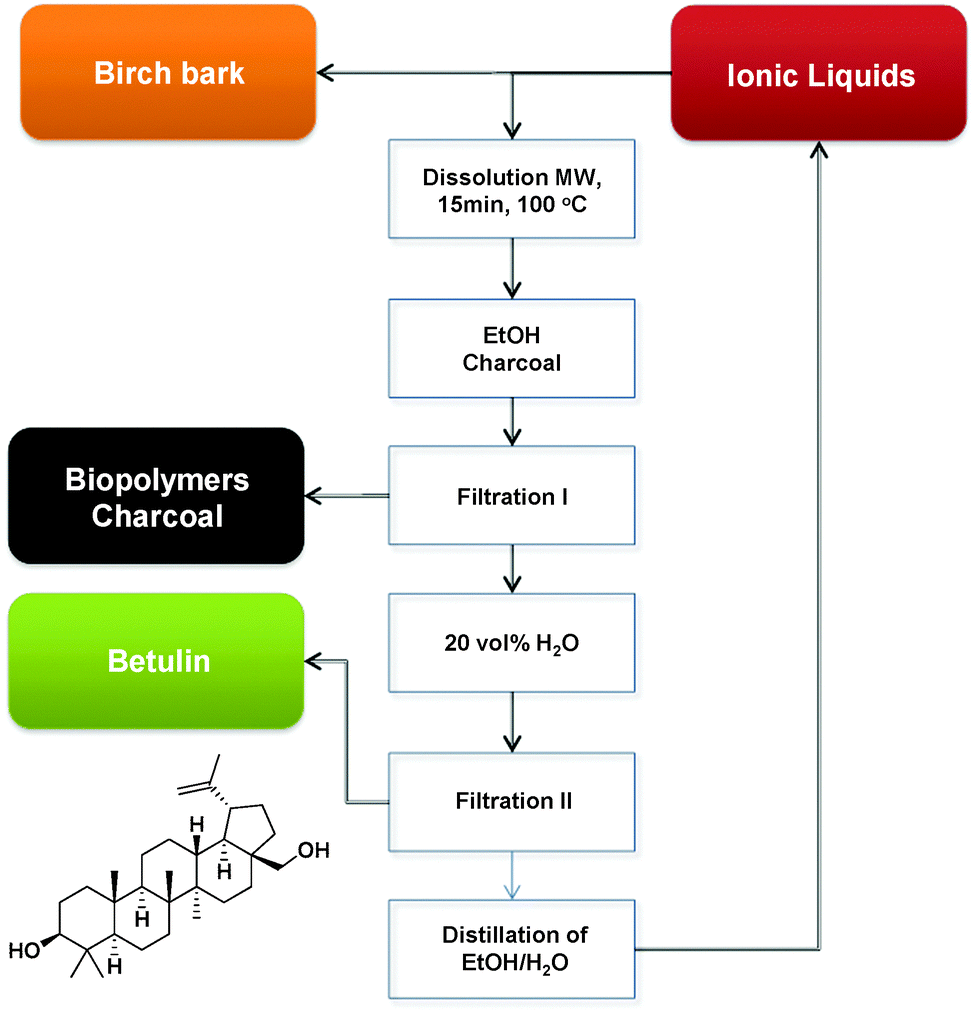 | ||
| Fig. 17 Scaled up betulin isolation process (MW: microwave heating).214 | ||
Chowdhury et al., demonstrated the use of the distillable IL dimethylammonium dimethylcarbamate (DIMCARB) in the extraction of tannins from plant sources such as catechu (Acacia Catechu) and myrobolan (Terminalia Chebula).216 On hydrolysis, these tannins are a source of pharmaceutically active compounds such as ellagic acid and catechin. The DIMCARB IL has a buffering action in the process, deprotonating the tannins to facilitate extraction. The relatively low distillation point of the DIMCARB also allows efficient separation of the products from the IL.
13. Ionic liquids as active pharmaceutical ingredients
In 1998, Davis et al., reported the synthesis of an alkylated derivative of miconazole (an antifungal drug) which was combined with [PF6]− to form the IL alkylmiconazolium hexafluorophosphate.217 In 2004, Davis and Hoffman et al.,218 reported the formation of ILs from non-nutritive sweeteners combining the anions of saccharin and acesulfame with organic cations. In both cases the emphasis was on using safer chemicals or natural products of known activity in the formation of ILs, which might negate the rather negative perception of some ILs as being toxic, rather than what the IL form might bring to the use of the active ions.This changed in 2007,219 when some of us asked the question, “Why not let the biological activity of an ion be the primary IL attribute that was being developed?” Designing active pharmaceutical ingredients (APIs) as ILs rather than as crystalline materials, could be another tool to help resolve current problems related to polymorphism, solubility, delivery, release rates, handling, and efficacy.
The first active pharmaceutical ingredient ILs included the synthesis and analysis of lidocainium docusate, prepared in a simple synthesis from lidocaine hydrochloride (a local surface anesthetic) and sodium docusate (an emollient) resulting in a hydrophobic IL. This API-IL in comparison to the parent salts, exhibited modified solubility, increased thermal stability, and improved efficacy in topical analgesia. The improvement in topical analgesia was attributed to synergistic effects of combining the pharmaceutical ions into a single compound.
In another publication, Stoimenovski et al.189 reported IL forms of the common antimuscarinic drug propantheline bromide, which suffers from the presence of different polymorphs.220 After an anion exchange reaction to form the acesulfamate derivative (a calorie-free artificial sweetener) a new IL was obtained, propantheline acesulfamate. Also Ferraz et al., reported recently on the successful synthesis and analysis of ampicillin-based ILs (Fig. 18), where the ampicillin anion was paired with various organic cations including cholinium and hexadecylpyridinium.221,222
 | ||
| Fig. 18 Schematic synthetic procedure for the preparation of ampicillin-based ILs.221 | ||
While the initial focus in the study of API-ILs was to improve the properties of one of the ions, the logical next step in using ILs as a platform for development of new (or old) APIs was incorporation of two pharmaceutical functions in the same compound but separated into the individual ions. Such an approach would not only allow for the formation of new interesting forms of drugs, but also introduce new and unique properties in comparison to current solid pharmaceutical forms.
There are indeed many biologically active ions which might form dual functional ILs. For example, from the many classes of available functional ions, reports can be found on using cations with antibacterial, local anaesthetic, anticholinergic, or antifungal properties, and combining them with anions which are antibiotics, NSAIDs, vitamins, emollients or anti-acne agents.223,224
Rogers et al., reported the synthesis of dual functional IL forms of aspirin by pairing pharmaceutically-active cations (antibacterial, analgesic, local anaesthetics, and antiarrhythmic) and acetylsalicylic acid or its metabolite salicylic acid (Fig. 19).225 The authors discussed the physical properties of the obtained salts in comparison to the parent compounds. With the exception of tramadolium and hexetidinium salts, all obtained salicylates were found to be low melting. Comparison of the acetylsalicylate salts and the corresponding salicylate salts revealed a significant reduction in phase transition temperatures for the acetylsalicylates, likely caused by the absence of inter- or intramolecular hydrogen bonds in the acetylsalicylate anion.
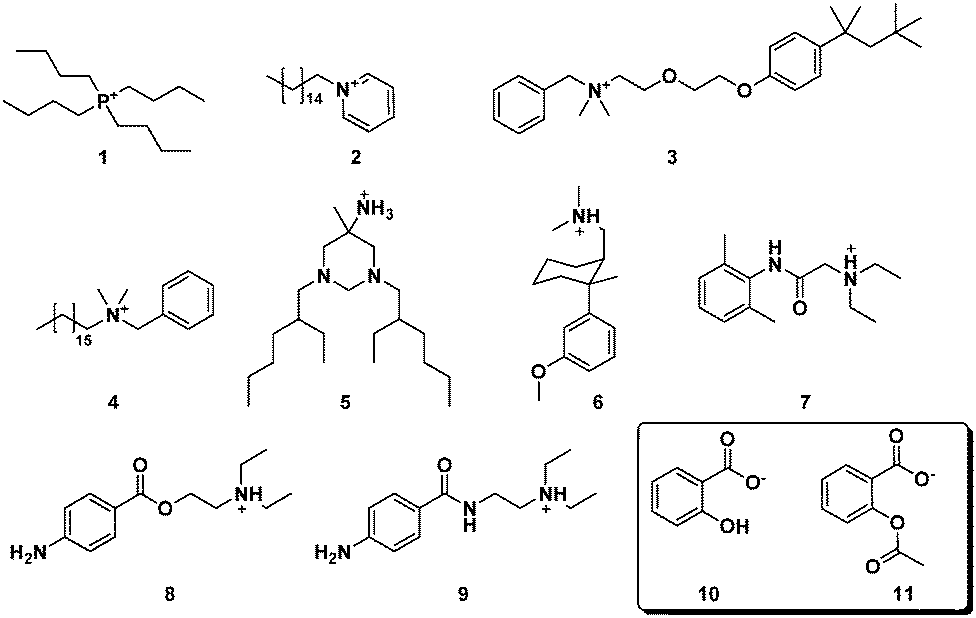 | ||
| Fig. 19 Antibacterial (1, 2, 3, 4, 5), analgesic (6), local anesthetic (7, 8) and antiarrhythmic (9) cations used in combination with the salicylate (10) and acetylsalicylate anion (11).225 | ||
Interestingly, the addition of excess protic acid or excess base (not necessarily of the parent compounds) to an existing salt can lead to the liquefaction of low melting salts through the formation of oligomeric ions with confused hydrogen bonding (i.e., strong complexes between ionized and unionized species).228 This ‘oligomeric approach’ was presented by Bica et al.,229 as both a liquefaction strategy (lowering melting points to below body or room temperatures) and a strategy to allow the two pharmaceutically active ions to be dosed at different ratios. This is exemplified in Fig. 20 with lidocaine and salicylic acid.230
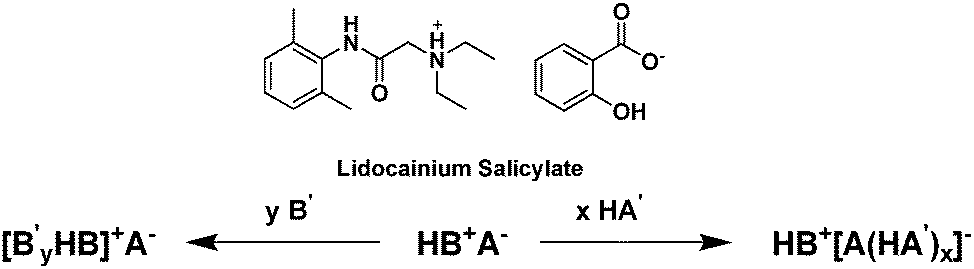 | ||
| Fig. 20 Oligomeric approach as both a liquefaction strategy and a strategy to allow the two pharmaceutically active ions to be dosed at different ratios.230 | ||
It is also important to note when studying protic ionic liquids (whether pharmaceuticals or not), that just because a liquid is obtained when an acid or base are mixed (even when both are solid), does not necessarily mean that a salt has been formed. In a study of different compositions of local anaesthetic lidocaine combined with various fatty acids, spectroscopic and thermal studies revealed the formation of deep eutectics which likely exist at the boundary between simple mixtures and partially ionized salts. By changing the molar composition of lidocaine or the fatty acids, low melting, single fraction mixtures were obtained. For example, the mixtures of lidocaine with decanoic and oleic acids led to liquids which only exhibited glass transitions below −40 °C (Fig. 21).229 The deep eutectic behaviour was the result of very strong hydrogen bonds between the lidocaine and acid. These are similar to the types of interactions being used to design co-crystals of APIs and hence the terminology ‘liquid co-crystal’ was adopted.229,231,232
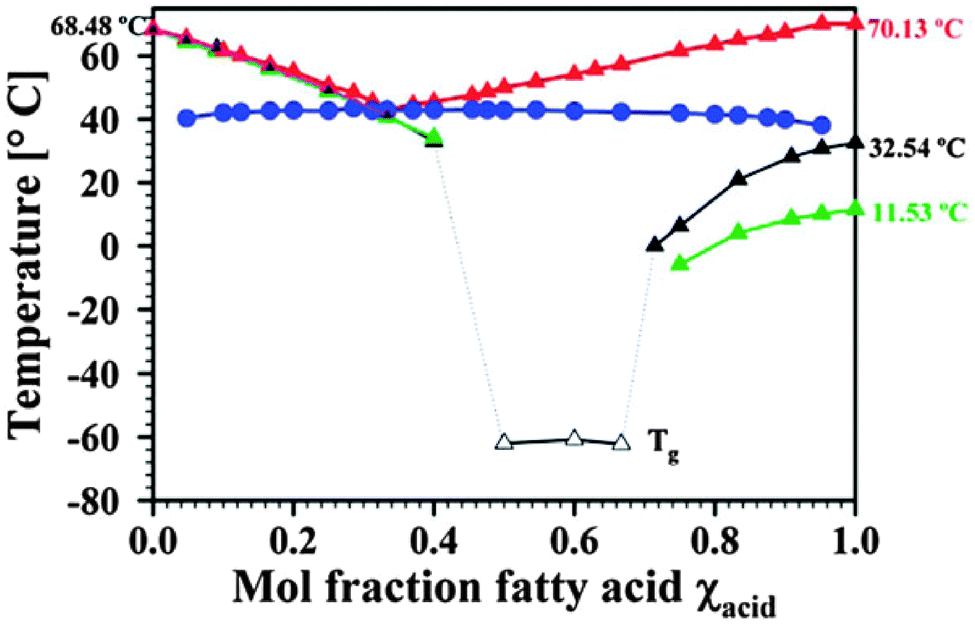 | ||
| Fig. 21 Transition temperatures for lidocaine:stearic acid (liquidus line red; solidus line blue), lidocaine:decanoic acid (black), and lidocaine:oleic acid (green). Reproduced from ref. 229 with permission from the RSC. | ||
Another interesting approach to liquidity in API-ILs was presented by Lovejoy et al.,233 where by combining cationic pharmaceutical Lewis acids with ZnCl2, it was possible to obtain liquid forms. Due to the oligomeric forms of zinc chloride anions, the melting points of the parent pharmaceutical chloride salt could be depressed. The authors prepared a series of new API-ILs based on hematropine, ethabutol, ranitidine, and benzethonium. Moreover, it was shown that metal halide-based API-ILs often possess better shelf life.
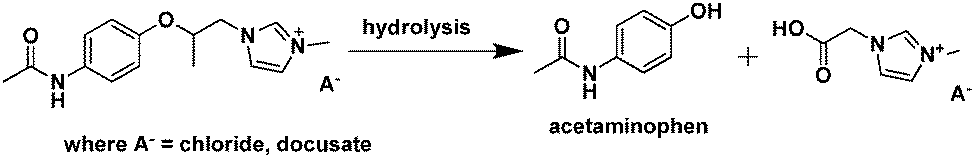 | ||
| Fig. 22 Hydrolysis of the IL prodrug 1-(2-(4-acetamidophenoxy)-2-oxoethyl)-3-methyl-1H-imidazol-3-ium docusate.236 | ||
14. Antimicrobial ionic liquids
Quaternary ammonium compounds (QACs) have been staple cations for the IL field for many years, as has the use of QACs for disinfection (e.g., common household detergents contain large amounts of various QACs). However, because the initial focus on ILs was as green, non-toxic solvents, the use of antimicrobial ILs took some time to develop. Some of the early examples involved use of protonated 1-alkylimidazolium and 1-alkoxymethylimidazolium lactate-based ILs as antibacterial agents.237 As the minimum inhibitory concentration (MIC) and minimum biocidal concentrations (MBC) were established it was noted that the antimicrobial activity of these ILs was strongly related to the length of the alkyl substituent, where an alkyl chain length below pentyl or pentyloxymethyl resulted in inactivity of the salt toward microorganisms.Following the concept of using ILs as antiseptics, disinfectants, or antifouling reagents, in recent years a number of publications describing antimicrobial activity of imidazolium, pyridinium, and other quaternary ammonium ILs have been published. One such example describes the preparation and biological activity of a series of ILs composed of imidazolium or pyridinium-based cations and β-lactam antibiotic anions.238 Many of the prepared salts exhibited excellent antimicrobial properties, as shown by the MIC and MBC values against Escherichia coli O157:H7, Klebsiella pneumoniae, Staphylococcus aureus, and Enterococcus faecium. Moreover, when comparing prepared ampicillin ILs (Fig. 23) to parent compounds, sodium ampicillin, and quaternary halides, it was found that in most cases the antibacterial activity of the products was higher than that of the starting materials. Moreover, when normalized for ampicillin content, it was concluded that most of the prepared ILs had much greater antimicrobial activity (up to 43 times higher) than sodium ampicillin alone.
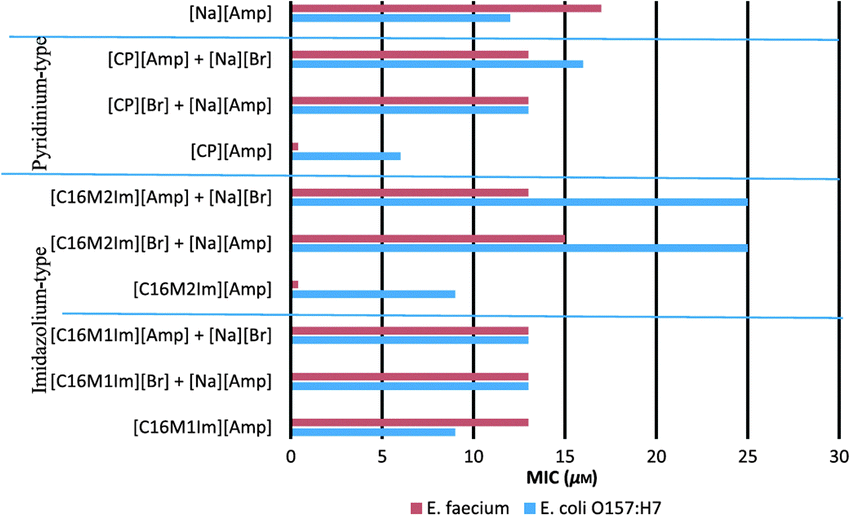 | ||
| Fig. 23 Comparison of MIC between ampicillin ILs and their starting materials tested in combination. Reproduced from ref. 238 with permission from John Wiley & Sons. | ||
Similarly, in recent studies by Carson et al.239 and Busetti et al.,240 the authors reported the synthesis and biological properties of a series of 1-alkyl-3-methylimidazolium chloride and 1-alkylquinolinium ILs (Fig. 24), which possessed broad antimicrobial activity against various Gram positive and Gram negative bacteria, and fungi, with clear dependency between this activity and the alkyl chain length. As it was described by the authors, the microbial biofilms, which protect microorganisms from antiseptics, disinfectants, and antibiotics, can be broken down by long alkyl chain-containing ILs.
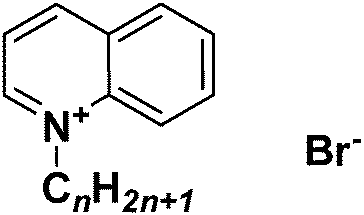 | ||
| Fig. 24 Generic structure of alkylquinolinium salts ([Cnquin][Br]).240 | ||
Surface decontamination and sterilization is also one of the big challenges for clinical environments as bacteria such as Staphylococcus aureus (MRSA) are very difficult to eliminate from the surfaces of medical devices, which are mostly made of various polymers and plastics. In a recent report by Choi et al., the authors presented the preparation of two dual functional ILs 1-ethylpyridinium docusate and tributyl(2-hydroxyethyl)phosphonium docusate, which were used as both a plasticising additive for production of polymers and at the same time antimicrobial and antibiofilm-forming agents to a range of antibiotic resistant bacteria.241 As suggested by the authors such an approach could be adopted in the future for the preparation of multifunctional additives for medical grade polymers.
Antimicrobials also have an important application in corrosion inhibition, since many metal corrosion processes, for example in pipelines and ship hulls, are initiated by microbial action. In this context Seter et al.,242,243 have recently described and tested a number of novel dual-active salts, some of which are ILs, which are designed to be active against a range of bacteria commonly found to be associated with biofouling in the marine environment, including Citrobacter freundii. For example, cetrimonium nalidixate was prepared as a novel dual-active and shown to combat both biofilm formation and microbiologically influenced corrosion.
It is apparent from the literature results recently reported, and from knowledge in the fields of disinfectants and other antimicrobial agents, that the use of ILs (in many cases QACs) as new forms of antimicrobial agents is well justified. Moreover, due to the fact that the disinfectants and other antimicrobial agents, especially for use on surfaces do not necessarily have to go through rigorous biological testing procedures as is the case for new pharmaceutical agents, it can be expected that in this particular field in the near future we may observe the first commercial uses of biologically-active ILs.
15. Agricultural chemical-based ionic liquids
The use of biologically-active ILs is not limited to pharmaceutical compounds. Several agrochemicals are also amenable to being made into ILs. As the human population grows, better methods for plant protection are needed in order to facilitate growth and protect crops from unwanted diseases and other stress factors. One of the biggest challenges in plant protection and plant growth is the presence of weeds that coexist with the crops in the same field and strongly compete for water, minerals, and light. The use of herbicides can help in protecting crops, but often the herbicides can have an adverse effect on non-weed plants and animals as well.244Recent studies on herbicidal ILs have concentrated on the synthesis of various derivatives of commonly known herbicides in order to moderate the solubility of the herbicide, improve its efficacy, and to reduce its drift into the environment. Pernak et al., presented the synthesis and characterization of a group of herbicidal ILs based on anionic derivatives of 2,4-D,245,246 MCPA,245 and others.247 Use of ILs based on the 2,4-D anion in combination with cations such as alkyldi(2-hydroxyethyl)methylammonium, dialkyldimethylammonium, and alkyltrimethylammonium, resulted in formation of salts with much higher activity against broad-leaf weeds when tested in the field compared to commonly used dimethylammonium salts of 2,4-D. Moreover, the thermal stabilities of the ILs were often higher than of the parent herbicide and showed substantially lower water solubility (which can be advantageous in terms of slow release and lack of washing-out effects). The authors also speculated that due to the nonvolatile nature of the ILs, these new herbicides may be safer to the operators and neighbouring plants.245
Similarly, when analysing the biological activity of MCPA-based ILs, which were composed of plant growth regulator cations (e.g., ammonium, phosphonium, pyridinium, imidazolium, morpholinium, and piperidinium) and the MCPA anion, higher biological activity was observed for ILs along with higher thermal stability and substantially lower solubility in water. The authors speculated that the acute toxicity of such ILs could be controlled by appropriate selection of cation types.248
Also recently, Pernak and Rogers reported their work on a group of new dicamba (3,6-dichloro-2-methoxybenzoic acid)-based herbicidal salts through pairing the dicamba anion with quaternary tetraalkyl- or alkoxyammonium, piperidinium, imidazolium, pyridinium, morpholinium, quinolinium, and phosphonium cations. These salts were prepared to improve the efficacy of this widely known herbicide used to protect maize, grassland, and other cultures. Growth chamber and field test data suggested that IL forms of dicamba offer substantially increased efficacy which would allow less to be applied in the field.249
Another application where the IL concept is applied to modify the properties of biologically-active substances was recently presented by Smiglak et al.,250 where the authors focused on the chemical modification of plant resistance inducer benzo[1,2,3]thiadiazole-7-carboxylate (BTH) in order to produce its anionic and cationic form and later to convert it to an IL. The counter ions used for pairing with the BTH anion were cholinium and decylbutyldimethylammonium. In both cases it was possible to obtain salts that were soluble in water, in comparison with non-water soluble BTH. Moreover, use of long alkyl chain quaternary ammonium cations brings antibacterial properties to these new salts.
Another application for the use of dual functional ILs is in the preparation of novel antifungal agents. Bica et al.,251 reported hydrophobic fungicide forms of the actives thiabendazole and imazalil with increased rain persistence and activity against potato tuber diseases. Analysing for example, the activity of thiabendazolium docusate at various concentrations, it was found that the activity of the antifungal agent was retained with EC50 values similar to those obtained with neutral thiabendazole (Fig. 25). In light of the possible applications of ILs as herbicidal agents, it is important to remember that such compounds in use will be exposed to the environment. Thus, it is important to be aware of the issues related to biodegradability, eco-toxicity, and to possible accumulation in soils of various herbicidal ILs. Ongoing research focusing on these aspects of ILs is currently well represented in the literature with, for example, the work of Scammells and Singer,252–255 and Stolte.256,257
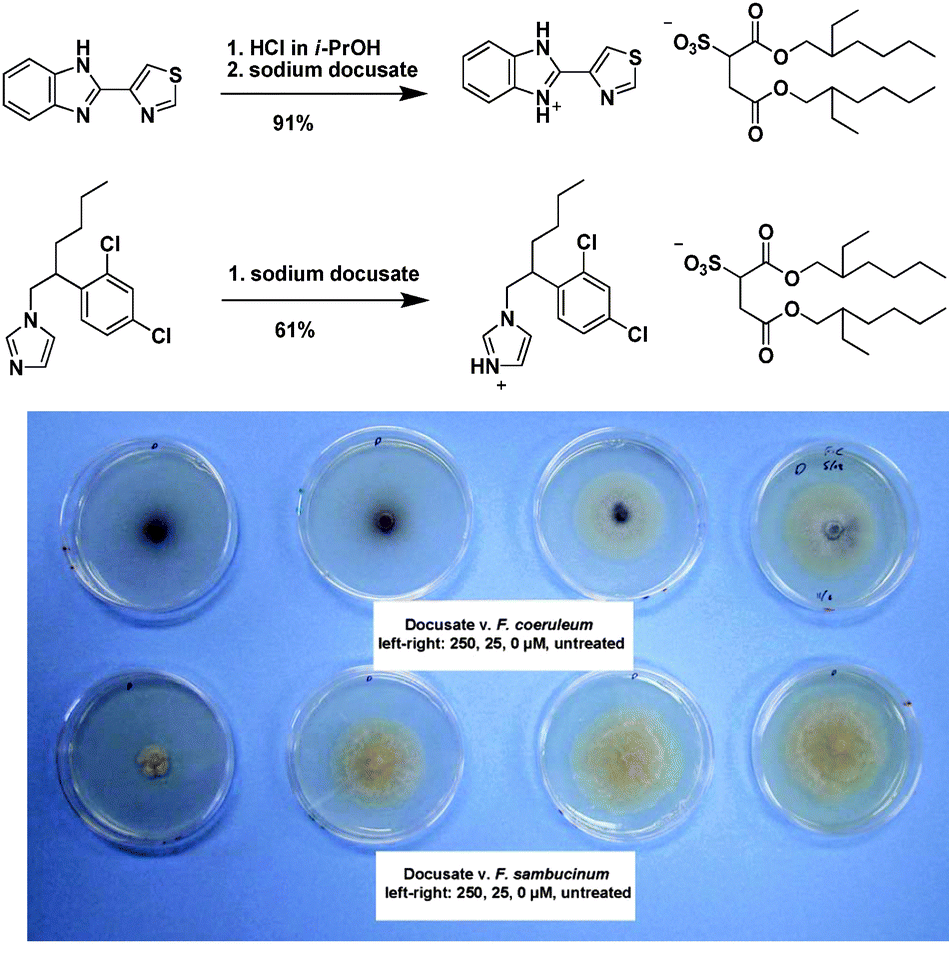 | ||
| Fig. 25 In vitro activity of thiabendazolium docusate against F. coeruleum and F. sambucinum. Reproduced from ref. 251 with permission from the RSC. | ||
Conclusions
Here we could only touch the surface of a few areas of modern ILs research. As knowledge in the field grows from both experiment and theory, imaginative uses of ILs continue to arise. As IL researchers become more adept at describing their findings to new research communities outside their own disciplines, exploitation of the property sets inherent in ILs become apparent in areas no one had anticipated. We fully expect this trend to continue.Key aspects requiring further development within the field itself that will support advances in both existing and new applications in our view include: (i) a greater focus and understanding of mixtures of ILs, both with molecular species and also other ILs,258 (ii) a deeper understanding of thermodynamic activities in ILs, including solutes such as Li+ and H2O and the main component ions, especially in regard to proton activities, (iii) investigations of ion-induced structure at the nanometer level and its influence on physical and chemical properties; structure that may persist for long times and even after dilution into solvents, (iv) transport properties and their link to structure (and thereby to ionicity) both in the bulk liquid, as well as in confined states such as mesoporous catalyst and electrode structures; (v) biological activity including synergistic effects of IL ions in neutral, charged, and partially ionized forms; and (vi) environmental and economic impacts of using ILs in large and small scale industrial processes. We ask the research community to continue these investigations and others yet to be anticipated as the field looks forward to the exciting developments that will emerge.
Abbreviations
Cations
| [Allmim]+ | 1-Allyl-3-methylimidazolium |
| [BA]+ | (Benzyl)dimethylalkylammonium |
| [(carboxymethyl)mim]+ | 1-Carboxymethyl-3-methylimidazolium |
| [C10mim]+ | 1-Decyl-3-methylimidazolium |
| [C16mim]+ | 1-Hexadecyl-3-methylimidazolium |
| [C1mim]+ | 1,3-Dimethylimidazolium |
| [C2mim]+ | 1-Ethyl-3-methylimidazolium |
| [C2OHmim]+ | 1-Ethanol-3-methylimidazolium |
| [C3mim]+ | 1-Propyl-3-methylimidazolium |
| [C4mim]+ | 1-Butyl-3-methylimidazolium |
| [C4mpyr]+ | 1-Butyl-1-methylpyrrolidinium |
| [C6mim]+ | 1-Hexyl-3-methylimidazolium |
| [C8mim]+ | 1-Octyl-3-methylimidazolium |
| [Chol]+ | Cholinium |
| [DDA]+ | Didecyldimethylammonium |
| [hmpy]+ | 1-n-Hexyl-3-methylpyridinium |
| [N1122(OH)]+ | Ethyl(2-hydroxyethyl)dimethylammonium |
| [N2222]+ | Tetraethylammonium |
| [N4444]+ | Tetrabutylammonium |
| [aP4443]+ | Tributyl-3-aminopropylphosphonium |
| [NH111]+ | Trimethylammonium |
| [NH122]+ | Diethylmethylammonium |
| [NH2p-bim]+ | 1-Butyl-3-propylamine-imidazolium |
| [P44410]+ | Tributyldecylphosphonium |
| [P66614]+ | Trihexyltetradecylphosphonium |
| [TMG]+ | 1,1,3,3-Tetramethylguanidinium |
| [TMGB2]+ | 1,1,3,3-Tetramethyl-1,1-dibutylguanidinium |
Anions
| [B(CN)4]− | Tetracyanoborate |
| [BF4]− | Tetrafluoroborate |
| [Br]− | Bromide |
| [Cl]− | Chloride |
| [DMP]− | Dimethyl phosphate |
| [FSI]− | Bis(fluorosulfonyl)imide |
| [HSO4]− | Hydrogensulfate |
| [H2PO4]− | Dihydrogen phosphate |
| [I]− | Iodide |
| [Im]− | Imidazolate |
| [N(CN)2]− | Dicyanamide |
| [NO3]− | Nitrate |
| [NTf2]− | Bis((Trifluoromethane)sulfonyl)imide |
| [OAc]− | Acetate |
| [OTf]− | Trifluoromethanesulfonate |
| [PF6]− | Hexafluorophosphate |
| [SCN]− | Thiocyanate |
| [Tetr]− | Tetrazolate |
Neutrals
| 2,4-D | 2,4-Dichlorophenoxyacetic acid |
| Amp B | Amphoceritin B |
| H2BDC | 1,4-Benzenedicarboxylic acid |
| H3BTC | 1,3,5-Benzenetricarboxylic acid |
| DIMCARB | Dimethylammonium dimethylcarbamate |
| HDA | 1,6-Hexanediamine |
| HNTf2 | Bis((trifluoromethane)sulfonyl)imide acid |
| ITO/PEN | Indium tin oxide/polyethylene naphthalate |
| MCPA | 2-Methyl-4-chlorophenoxyacetic acid |
| MPN | 3-Methoxypropionitrile |
| PEDOT | Poly(3,4-ethylenedioxythiophene) |
| PEG | Polyethylene glycol |
| TTIP | Titanium isopropoxide |
Miscellaneous
| API | Active pharmaceutical ingredient |
| BAIL | Brønsted acidic ionic liquid |
| CHA | Chabasite |
| DSSC | Dye-sensitized solar cell |
| GO | Graphene oxide |
| HLB | Hydrophilic–lipophilic balance |
| HMS | Hollow microsphere |
| HOR | Hydrogen oxidation reaction |
| IPCE | Incident photon-to-current conversion efficiency |
| MOF | Metal–organic frameworks |
| NSAID | Non-steroidal anti-inflammatory drug |
| OCV | Open circuit voltage |
| OIPC | Organic ionic plastic crystal |
| ORR | Oxygen reduction reaction |
| PEMFC | Proton exchange membrane fuel cell |
| PIL | Protic ionic liquids |
| POM | Polyoxometalate |
| QAC | Quaternary ammonium compound |
| rGO | Reduced graphene oxide |
| SDA | Structure directing agent |
| SEI | Solid-electrolyte interface |
Acknowledgements
M. Smiglak gratefully acknowledges funding from the National Science Center of Poland (project SONATA, grant number 2011/03/D/ST5/06200 and OPUS, grant number 2012/05/B/ST5/00375), the Foundation for Polish Science (project HOMING PLUS, grant number 2012-5/13), and a fellowship from the Polish Ministry of Higher Education and Science. J. M. Pringle and D. R. MacFarlane gratefully acknowledge funding from the Australian Research Council through its Centre of Excellence program, and also for fellowship support for D. R. MacFarlane (Australian Laureate Fellowship). S. Zhang, X. Lu, L. Han, and H. Gao gratefully acknowledge the financial support from the international scientific and technological cooperation and exchange projects (2014DFA61670), the National Natural Science Foundation of China (grant number 21210006), and the Beijing Municipal Natural Science Foundation (grant number 2131005). R. D. Rogers thanks the U.S. Department of Energy, Office of Nuclear Energy (Award # DE-NE0000672), the U.S. National Science Foundation Alabama EPSCoR Research Infrastructure Improvement Project (EPS-0814103), the U.S. Air Force Office of Scientific Research (agreement number FA9550-10-1-0521), Monsanto, 525 Solutions, Reliance, and the Novartis-Massachusetts Institute of Technology (MIT) Center for Continuous Manufacturing (CCM) for current support of ionic liquids projects.References
- http://pubs.rsc.org/en/journals/articlecollectionlanding?sercode=cc&themeid=d3759160-edca-4baf-871d-b8873930c974, last accessed on Feb. 16, 2014.
- J. M. Pandolfi, S. R. Connolly, D. J. Marshall and A. L. Cohen, Science, 2011, 333, 418–422 CrossRef CAS PubMed.
- D. R. MacFarlane, N. Tachikawa, M. Forsyth, J. M. Pringle, P. C. Howlett, G. D. Elliott, J. H. Davis, M. Watanabe, P. Simon and C. A. Angell, Energy Environ. Sci., 2014, 7, 232–250 CAS.
- E. D. Bates, R. D. Mayton, I. Ntai and J. H. Davis, J. Am. Chem. Soc., 2002, 124, 926–927 CrossRef CAS PubMed.
- J. Zhang, S. Zhang, K. Dong, Y. Zhang, Y. Shen and X. Lv, Chem. – Eur. J., 2006, 12, 4021–4026 CrossRef CAS PubMed.
- Y. Zhang, S. Zhang, X. Lu, Q. Zhou, W. Fan and X. Zhang, Chem. – Eur. J., 2009, 15, 3003–3011 CrossRef CAS PubMed.
- B. E. Gurkan, J. C. de la Fuente, E. M. Mindrup, L. E. Ficke, B. F. Goodrich, E. A. Price, W. F. Schneider and J. F. Brennecke, J. Am. Chem. Soc., 2010, 132, 2116–2117 CrossRef CAS PubMed.
- C. Wang, H. Luo, X. Luo, H. Li and S. Dai, Green Chem., 2010, 12, 2019–2023 RSC.
- C. Wang, S. M. Mahurin, H. Luo, G. A. Baker, H. Li and S. Dai, Green Chem., 2010, 12, 870–874 RSC.
- C. Wang, X. Luo, H. Luo, D.-e. Jiang, H. Li and S. Dai, Angew. Chem., Int. Ed., 2011, 50, 4918–4922 CrossRef CAS PubMed.
- C. Wang, Y. Guo, X. Zhu, G. Cui, H. Li and S. Dai, Chem. Commun., 2012, 48, 6526–6528 RSC.
- C. Wang, H. Luo, H. Li, X. Zhu, B. Yu and S. Dai, Chem. – Eur. J., 2012, 18, 2153–2160 CrossRef CAS PubMed.
- R. Vijayraghavan, S. J. Pas, E. I. Izgorodina and D. R. MacFarlane, Phys. Chem. Chem. Phys., 2013, 15, 19994–19999 RSC.
- J. Ilconich, C. Myers, H. Pennline and D. Luebke, J. Membr. Sci., 2007, 298, 41–47 CrossRef CAS PubMed.
- J. E. Bara, C. J. Gabriel, E. S. Hatakeyama, T. K. Carlisle, S. Lessmann, R. D. Noble and D. L. Gin, J. Membr. Sci., 2008, 321, 3–7 CrossRef CAS PubMed.
- P. Scovazzo, D. Havard, M. McShea, S. Mixon and D. Morgan, J. Membr. Sci., 2009, 327, 41–48 CrossRef CAS PubMed.
- S. Yoo, J. Won, S. W. Kang, Y. S. Kang and S. Nagase, J. Membr. Sci., 2010, 363, 72–79 CrossRef CAS PubMed.
- J. Albo, E. Santos, L. A. Neves, S. P. Simeonov, C. A. M. Afonso, J. G. Crespo and A. Irabien, Sep. Purif. Technol., 2012, 97, 26–33 CrossRef CAS PubMed.
- L. A. Neves, C. Afonso, I. M. Coelhoso and J. G. Crespo, Sep. Purif. Technol., 2012, 97, 34–41 CrossRef CAS PubMed.
- T. K. Carlisle, E. F. Wiesenauer, G. D. Nicodemus, D. L. Gin and R. D. Noble, Ind. Eng. Chem. Res., 2013, 52, 1023–1032 CrossRef CAS.
- L. C. Tome, D. Mecerreyes, C. S. R. Freire, L. P. N. Rebelo and I. M. Marrucho, J. Membr. Sci., 2013, 428, 260–266 CrossRef CAS PubMed.
- J. E. Bara, D. E. Camper, D. L. Gin and R. D. Noble, Acc. Chem. Res., 2010, 43, 152–159 CrossRef CAS PubMed.
- D. D. Iarikov, P. Hacarlioglu and S. T. Oyama, Chem. Eng. J., 2011, 166, 401–406 CrossRef CAS PubMed.
- D. Camper, J. E. Bara, D. L. Gin and R. D. Noble, Ind. Eng. Chem. Res., 2008, 47, 8496–8498 CrossRef CAS.
- A. Ahmady, M. A. Hashim and M. K. Aroua, J. Chem. Eng. Data, 2010, 55, 5733–5738 CrossRef CAS.
- N. A. Sairi, R. Yusoff, Y. Alias and M. K. Aroua, Fluid Phase Equilib., 2011, 300, 89–94 CrossRef CAS PubMed.
- M. M. Taib and T. Murugesan, Chem. Eng. J., 2012, 181, 56–62 CrossRef PubMed.
- J. L. Anderson, J. K. Dixon, E. J. Maginn and J. F. Brennecke, J. Phys. Chem. B, 2006, 110, 15059–15062 CrossRef CAS PubMed.
- J. Huang, A. Riisager, P. Wasserscheid and R. Fehrmann, Chem. Commun., 2006, 4027–4029 RSC.
- S. Y. Hong, J. Im, J. Palgunadi, S. D. Lee, J. S. Lee, H. S. Kim, M. Cheong and K. D. Jung, Energy Environ. Sci., 2011, 4, 1802–1806 CAS.
- W. Z. Wu, B. X. Han, H. X. Gao, Z. M. Liu, T. Jiang and J. Huang, Angew. Chem., Int. Ed., 2004, 43, 2415–2417 CrossRef CAS PubMed.
- S. Tian, Y. Hou, W. Wu, S. Ren and C. Zhang, RSC Adv., 2013, 3, 3572–3577 RSC.
- M. Jin, Y. Hou, W. Wu, S. Ren, S. Tian, L. Xiao and Z. Lei, J. Phys. Chem. B, 2011, 115, 6585–6591 CrossRef CAS PubMed.
- S. Ren, Y. Hou, W. Wu and M. Jin, Ind. Eng. Chem. Res., 2011, 50, 998–1002 CrossRef CAS.
- S. Ren, Y. Hou, W. Wu, Q. Liu, Y. Xiao and X. Chen, J. Phys. Chem. B, 2010, 114, 2175–2179 CrossRef CAS PubMed.
- Y. Shang, H. Li, S. Zhang, H. Xu, Z. Wang, L. Zhang and J. Zhang, Chem. Eng. J., 2011, 175, 324–329 CAS.
- C. Wang, J. Zheng, G. Cui, X. Luo, Y. Guo and H. Li, Chem. Commun., 2013, 49, 1166–1168 RSC.
- G. Cui, C. Wang, J. Zheng, Y. Guo, X. Luo and H. Li, Chem. Commun., 2012, 48, 2633–2635 RSC.
- C. Wang, G. Cui, X. Luo, Y. Xu, H. Li and S. Dai, J. Am. Chem. Soc., 2011, 133, 11916–11919 CrossRef CAS PubMed.
- F. de Bruijn, Green Chem., 2005, 7, 132–150 RSC.
- R. Devanathan, Energy Environ. Sci., 2008, 1, 101–119 CAS.
- S. M. Haile, Acta Mater., 2003, 51, 5981–6000 CrossRef CAS PubMed.
- S.-Y. Lee, A. Ogawa, M. Kanno, H. Nakamoto, T. Yasuda and M. Watanabe, J. Am. Chem. Soc., 2010, 132, 9764–9773 CrossRef CAS PubMed.
- Q. Li, R. He, J. O. Jensen and N. J. Bjerrum, Chem. Mater., 2003, 15, 4896–4915 CrossRef CAS.
- S. Yi, F. Zhang, W. Li, C. Huang, H. Zhang and M. Pan, J. Membr. Sci., 2011, 366, 349–355 CrossRef CAS PubMed.
- Y.-S. Ye, J. Rick and B.-J. Hwang, J. Mater. Chem. A, 2013, 1, 2719–2743 CAS.
- F. Lu, X. Gao, X. Yan, H. Gao, L. Shi, H. Jia and L. Zheng, ACS Appl. Mater. Interfaces, 2013, 5, 7626–7632 CAS.
- M. A. B. H. Susan, A. Noda, S. Mitsushima and M. Watanabe, Chem. Commun., 2003, 938–939 RSC.
- J.-P. Belieres, D. Gervasio and C. A. Angell, Chem. Commun., 2006, 4799–4801 RSC.
- J.-P. Belieres and C. A. Angell, J. Phys. Chem. B, 2007, 111, 4926–4937 CrossRef CAS PubMed.
- M. S. Miran, H. Kinoshita, T. Yasuda, M. A. B. H. Susan and M. Watanabe, Phys. Chem. Chem. Phys., 2012, 14, 5178–5186 RSC.
- J. Stoimenovski, E. I. Izgorodina and D. R. MacFarlane, Phys. Chem. Chem. Phys., 2010, 12, 10341–10347 RSC.
- H. Nakamoto and M. Watanabe, Chem. Commun., 2007, 2539–2541 RSC.
- M. Yoshizawa, W. Xu and C. A. Angell, J. Am. Chem. Soc., 2003, 125, 15411–15419 CrossRef CAS PubMed.
- A. Noda, M. A. B. H. Susan, K. Kudo, S. Mitsushima, K. Hayamizu and M. Watanabe, J. Phys. Chem. B, 2003, 107, 4024–4033 CrossRef CAS.
- T. L. Greaves and C. J. Drummond, Chem. Rev., 2008, 108, 206–237 CrossRef CAS PubMed.
- M. S. Miran, T. Yasuda, M. A. B. H. Susan, K. Dokko and M. Watanabe, RSC Adv., 2013, 3, 4141–4144 RSC.
- T. Yasuda, S.-i. Nakamura, Y. Honda, K. Kinugawa, S.-Y. Lee and M. Watanabe, ACS Appl. Mater. Interfaces, 2012, 4, 1783–1790 CAS.
- T. Yasuda and M. Watanabe, MRS Bull., 2013, 38, 560–566 CrossRef CAS.
- H. Ohno and M. Yoshizawa-Fujita, Electrochemical Aspects of ILs, 2nd edn, 2011, pp. 419–431 Search PubMed.
- U. A. Rana, M. Forsyth, D. R. MacFarlane and J. M. Pringle, Electrochim. Acta, 2012, 84, 213–222 CrossRef CAS PubMed.
- A. Mirjafari, L. N. Pham, J. R. McCabe, N. Mobarrez, E. A. Salter, A. Wierzbicki, K. N. West, R. E. Sykora and J. H. Davis, RSC Adv., 2013, 3, 337–340 RSC.
- J. A. Bautista-Martinez, L. Tang, J. P. Belieres, R. Zeller, C. A. Angell and C. Friesen, J. Phys. Chem. C, 2009, 113, 12586–12593 CAS.
- M. Yoshizawa-Fujita, K. Fujita, M. Forsyth and D. R. MacFarlane, Electrochem. Commun., 2007, 9, 1202–1205 CrossRef CAS PubMed.
- U. A. Rana, P. M. Bayley, R. Vijayaraghavan, P. Howlett, D. R. MacFarlane and M. Forsyth, Phys. Chem. Chem. Phys., 2010, 12, 11291–11298 RSC.
- U. A. Rana, R. Vijayaraghavan, R. MacFarlane Douglas and M. Forsyth, J. Mater. Chem., 2012, 22, 2965–2974 RSC.
- U. A. Rana, I. Shakir, R. Vijayaraghavan, D. R. MacFarlane, M. Watanabe and M. Forsyth, Electrochim. Acta, 2013, 111, 41–48 CrossRef CAS PubMed.
- S. Zhang, Y. Shao, G. Yin and Y. Lin, J. Mater. Chem. A, 2013, 1, 4631–4641 CAS.
- C. Ke, J. Li, X. Li, Z. Shao and B. Yi, RSC Adv., 2012, 2, 8953–8956 RSC.
- J. Snyder, K. Livi and J. Erlebacher, Adv. Funct. Mater., 2013, 23, 5494–5501 CrossRef CAS.
- F. Blanc, M. Leskes and C. P. Grey, Acc. Chem. Res., 2013, 46, 1952–1963 CrossRef CAS PubMed.
- N. Papageorgiou, Y. Athanassov, M. Armand, P. Bonhote, H. Pettersson, A. Azam and M. Graetzel, J. Electrochem. Soc., 1996, 143, 3099–3108 CrossRef CAS PubMed.
- Y. Bai, Y. Cao, J. Zhang, M. Wang, R. Li, P. Wang, S. M. Zakeeruddin and M. Gratzel, Nat. Mater., 2008, 7, 626–630 CrossRef CAS PubMed.
- Y. Bai, J. Zhang, Y. Wang, M. Zhang and P. Wang, Langmuir, 2011, 27, 4749–4755 CrossRef CAS PubMed.
- M. K. Nazeeruddin, A. Kay, I. Rodicio, R. Humphry-Baker, E. Mueller, P. Liska, N. Vlachopoulos and M. Graetzel, J. Am. Chem. Soc., 1993, 115, 6382–6390 CrossRef CAS.
- C. A. Kelly, F. Farzad, D. W. Thompson, J. M. Stipkala and G. J. Meyer, Langmuir, 1999, 15, 7047–7054 CrossRef CAS.
- Z. Yu, M. Gorlov, G. Boschloo and L. Kloo, J. Phys. Chem. C, 2010, 114, 22330–22337 CAS.
- P. Wang, S. M. Zakeeruddin, J.-E. Moser and M. Graetzel, J. Phys. Chem. B, 2003, 107, 13280–13285 CrossRef CAS.
- V. Armel, J. M. Pringle, M. Forsyth, D. R. MacFarlane, D. L. Officer and P. Wagner, Chem. Commun., 2010, 46, 3146–3148 RSC.
- S. M. Zakeeruddin and M. Graetzel, Adv. Funct. Mater., 2009, 19, 2187–2202 CrossRef CAS.
- L. M. Peter, J. Phys. Chem. Lett., 2011, 2, 1861–1867 CrossRef CAS.
- A. Yella, H.-W. Lee, H. N. Tsao, C. Yi, A. K. Chandiran, M. K. Nazeeruddin, E. W.-G. Diau, C.-Y. Yeh, S. M. Zakeeruddin and M. Graetzel, Science, 2011, 334, 629–634 CrossRef CAS PubMed.
- D. Xu, H. Zhang, X. Chen and F. Yan, J. Mater. Chem. A, 2013, 1, 11933–11941 CAS.
- J. Liu, X. Yang, J. Cong, L. Kloo and L. Sun, Phys. Chem. Chem. Phys., 2012, 14, 11592–11595 RSC.
- T. J. Abraham, D. R. MacFarlane and J. M. Pringle, Energy Environ. Sci., 2013, 6, 2639–2645 CAS.
- J. M. Pringle and V. Armel, Int. Rev. Phys. Chem., 2011, 30, 371–407 CrossRef CAS.
- J. Nei de Freitas, A. F. Nogueira and M.-A. De Paoli, J. Mater. Chem., 2009, 19, 5279–5294 RSC.
- V. Armel, M. Forsyth, D. R. MacFarlane and J. M. Pringle, Energy Environ. Sci., 2011, 4, 2234–2239 CAS.
- Q. Li, X. Chen, J. Zhao, L. Qiu, Y. Zhang, B. Sun and F. Yan, J. Mater. Chem., 2012, 22, 6674–6679 RSC.
- M. Zhang, J. Zhang, Y. Bai, Y. Wang, M. Su and P. Wang, Phys. Chem. Chem. Phys., 2011, 13, 3788–3794 RSC.
- Q. Li, J. Zhao, B. Sun, B. Lin, L. Qiu, Y. Zhang, X. Chen, J. Lu and F. Yan, Adv. Mater., 2012, 24, 945–950 CrossRef CAS PubMed.
- R. Harikisun and H. Desilvestro, Sol. Energy, 2011, 85, 1179–1188 CrossRef CAS PubMed.
- D. Kuang, C. Klein, Z. Zhang, S. Ito, J.-E. Moser, S. M. Zakeeruddin and M. Gratzel, Small, 2007, 3, 2094–2102 CrossRef CAS PubMed.
- T. Miyasaka, J. Phys. Chem. Lett., 2011, 2, 262–269 CrossRef CAS.
- M. Wu and T. Ma, ChemSusChem, 2012, 5, 1343–1357 CrossRef CAS PubMed.
- M. Wang, A. M. Anghel, B. Marsan, N.-L. Cevey Ha, N. Pootrakulchote, S. M. Zakeeruddin and M. Gratzel, J. Am. Chem. Soc., 2009, 131, 15976–15977 CrossRef CAS PubMed.
- J. M. Pringle, V. Armel and D. R. MacFarlane, Chem. Commun., 2010, 46, 5367–5369 RSC.
- Y. Saito, W. Kubo, T. Kitamura, Y. Wada and S. Yanagida, J. Photochem. Photobiol., A, 2004, 164, 153–157 CrossRef CAS PubMed.
- S. Carli, E. Busatto, S. Caramori, R. Boaretto, R. Argazzi, C. J. Timpson and C. A. Bignozzi, J. Phys. Chem. C, 2013, 117, 5142–5153 CAS.
- T. M. Tritt, Annual Reviews in Materials Research, 2011, 41, 433–448 CrossRef CAS.
- J. R. Szczech, J. M. Higgins and S. Jin, J. Mater. Chem., 2011, 21, 4037–4055 RSC.
- K. Biswas, J. He, I. D. Blum, C.-I. Wu, T. P. Hogan, D. N. Seidman, V. P. Dravid and M. G. Kanatzidis, Nature, 2012, 489, 414–418 CrossRef CAS PubMed.
- H. L. Chum and R. A. Osteryoung, Sol. Energy Res. Inst., Golden, CO, USA, 1981, p. 223 Search PubMed.
- R. C. Hu, B. A. Cola, N. Haram, J. N. Barisci, S. Lee, S. Stoughton, G. Wallace, C. Too, M. Thomas, A. Gestos, M. E. dela Cruz, J. P. Ferraris, A. A. Zakhidov and R. H. Baughman, Nano Lett., 2010, 10, 838–846 CrossRef CAS PubMed.
- J. T. Hupp and M. J. Weaver, Inorg. Chem., 1984, 23, 3639–3644 CrossRef CAS.
- T. Migita, N. Tachikawa, Y. Katayama and T. Miura, Electrochemistry, 2009, 77, 639–641 CrossRef CAS PubMed.
- A. J. DeBethune, T. S. Licht and N. Swendeman, J. Electrochem. Soc., 1959, 106, 616–625 CrossRef CAS PubMed.
- Y. Yamato, Y. Katayama and T. Miura, J. Electrochem. Soc., 2013, 160, H309–H314 CrossRef CAS PubMed.
- T. J. Abraham, D. R. Mac Farlane, R. H. Baughman, L. Jin, N. Li and J. M. Pringle, Electrochim. Acta, 2013, 113, 87–93 CrossRef CAS PubMed.
- http://www.boeing.com/787-media-resource/docs/fct-031514-787%20battery%20timeline.pdf, last accessed on Feb. 13, 2014.
- H. Yoon, G. H. Lane, Y. Shekibi, P. C. Howlett, M. Forsyth, A. S. Best and D. R. MacFarlane, Energy Environ. Sci., 2013, 6, 979–986 CAS.
- H. Yoon, P. C. Howlett, A. S. Best, M. Forsyth and D. R. MacFarlane, J. Electrochem. Soc., 2013, 160, A1629–A1637 CrossRef CAS PubMed.
- J. B. Goodenough and K. S. Park, J. Am. Chem. Soc., 2013, 135, 1167–1176 CrossRef CAS PubMed.
- S. Higashi, Y. Kato, K. Takechi, H. Nakamoto, F. Mizuno, H. Nishikoori, H. Iba and T. Asaoka, J. Power Sources, 2013, 240, 14–17 CrossRef CAS PubMed.
- F. Mizuno, K. Takechi, S. Higashi, T. Shiga, T. Shiotsuki, N. Takazawa, Y. Sakurabayashi, S. Okazaki, I. Nitta, T. Kodama, H. Nakamoto, H. Nishikoori, S. Nakanishi, Y. Kotani and H. Iba, J. Power Sources, 2013, 228, 47–56 CrossRef CAS PubMed.
- N. Recham, J.-N. Chotard, L. Dupont, C. Delacourt, W. Walker, M. Armand and J.-M. Tarascon, Nat. Mater., 2009, 9, 68–74 CrossRef PubMed.
- B. Dunn, H. Kamath and J.-M. Tarascon, Science, 2011, 334, 928–935 CrossRef CAS PubMed.
- V. A. Nikitina, A. Nazet, T. Sonnleitner and R. Buchner, J. Chem. Eng. Data, 2012, 57, 3019–3025 CrossRef CAS.
- M. Egashira, T. Tanaka, N. Yoshimoto and M. Morita, Electrochemistry, 2012, 80, 755–758 CrossRef CAS PubMed.
- (a) S. A. M. Noor, P. C. Howlett, D. R. MacFarlane and M. Forsyth, Electrochim. Acta, 2013, 114, 766–771 CrossRef PubMed; (b) H. Yoon, H. Zhu, A. Hervault, M. Armand, D. R. MacFarlane and M. Forsyth, Phys. Chem. Chem. Phys., 2014 10.1039/C4CP00365A.
- C. Ding, T. Nohira, K. Kuroda, R. Hagiwara, A. Fukunaga, S. Sakai, K. Nitta and S. Inazawa, J. Power Sources, 2013, 238, 296–300 CrossRef CAS PubMed.
- T. Kakibe, J. Y. Hishii, N. Yoshimoto, M. Egashira and M. Morita, J. Power Sources, 2012, 203, 195–200 CrossRef CAS PubMed.
- T. Khoo, P. C. Howlett, M. Tsagouria, D. R. MacFarlane and M. Forsyth, Electrochim. Acta, 2011, 58, 583–588 CrossRef CAS PubMed.
- T. Khoo, A. Somers, A. A. J. Torriero, D. R. MacFarlane, P. C. Howlett and M. Forsyth, Electrochim. Acta, 2013, 87, 701–708 CrossRef CAS PubMed.
- C. Pozo-Gonzalo, A. A. J. Torriero, M. Forsyth, D. R. MacFarlane and P. C. Howlett, J. Phys. Chem. Lett., 2013, 1834–1837 CrossRef CAS.
- E. E. Switzer, R. Zeller, Q. Chen, K. Sieradzki, D. A. Buttry and C. Friesen, J. Phys. Chem. C, 2013, 117, 8683–8690 CAS.
- S. Monaco, A. M. Arangio, F. Soavi, M. Mastragostino, E. Paillard and S. Passerini, Electrochim. Acta, 2012, 83, 94–104 CrossRef CAS PubMed.
- A. Rene, D. Hauchard, C. Lagrost and P. Hapiot, J. Phys. Chem. B, 2009, 113, 2826–2831 CrossRef CAS PubMed.
- C. Pozo-Gonzalo, N. Byrne, P. C. Howlett, D. R. MacFarlane and M. Forsyth, Electrochem. Commun., 2014, 38, 24–27 CrossRef CAS PubMed.
- T. Hisatomi, J. Kubota and K. Domen, Chem. Soc. Rev., 2014 10.1039/C3CS60378D.
- F. L. Zhou, A. Izgorodin, R. K. Hocking, L. Spiccia and D. R. MacFarlane, Adv. Energy Mater., 2012, 2, 1013–1021 CrossRef CAS.
- A. Izgorodin and D. R. MacFarlane, Patent application, WO2013138845A1 2013 Search PubMed.
- A. Izgorodin, E. Izgorodina and D. R. MacFarlane, Energy Environ. Sci., 2012, 5, 9496–9501 CAS.
- D. R. MacFarlane, R. Vijayaraghavan, H. N. Ha, A. Izgorodin, K. D. Weaver and G. D. Elliott, Chem. Commun., 2010, 46, 7703–7705 RSC.
- R. Vijayraghavan, U. A. Rana, G. D. Elliott and D. R. MacFarlane, Energy Technol., 2013, 1, 609–612 CrossRef CAS.
- C. G. Cassity, A. Mirjafari, N. Mobarrez, K. J. Strickland, R. A. O'Brien and J. H. Davis, Jr, Chem. Commun., 2013, 49, 7590–7592 RSC.
- S. Dai, Y. H. Ju, H. J. Gao, J. S. Lin, S. J. Pennycook and C. E. Barnesc, Chem. Commun., 2000, 243–244 RSC.
- Z. Ma, J. Yu and S. Dai, Adv. Mater., 2010, 22, 261–285 CrossRef CAS PubMed.
- D. Freudenmann, S. Wolf, M. Wolff and C. Feldmann, Angew. Chem., Int. Ed., 2011, 50, 11050–11060 CrossRef CAS PubMed.
- E. Ahmed, J. Breternitz, M. F. Groh and M. Ruck, CrystEngComm, 2012, 14, 4874–4885 RSC.
- E. R. Cooper, C. D. Andrews, P. S. Wheatley, P. B. Webb, P. Wormald and R. E. Morris, Nature, 2004, 430, 1012–1016 CrossRef CAS PubMed.
- Y. Wei, B. Marler, L. Zhang, Z. Tian, H. Graetsch and H. Gies, Dalton Trans., 2012, 41, 12408–12415 RSC.
- E. R. Parnham, E. A. Drylie, P. S. Wheatley, A. M. Slawin and R. E. Morris, Angew. Chem., Int. Ed., 2006, 45, 4962–4966 CrossRef CAS PubMed.
- Y. Wei, Z. Tian, H. Gies, R. Xu, H. Ma, R. Pei, W. Zhang, Y. Xu, L. Wang, K. Li, B. Wang, G. Wen and L. Lin, Angew. Chem., Int. Ed., 2010, 49, 5367–5370 CrossRef CAS PubMed.
- E. A. Drylie, D. S. Wragg, E. R. Parnham, P. S. Wheatley, A. M. Slawin, J. E. Warren and R. E. Morris, Angew. Chem., Int. Ed., 2007, 46, 7839–7843 CrossRef PubMed.
- E. R. Parnham and R. E. Morris, J. Mater. Chem., 2006, 16, 3682–3684 RSC.
- K. Li, Z. Tian, X. Li, R. Xu, Y. Xu, L. Wang, H. Ma, B. Wang and L. Lin, Angew. Chem., Int. Ed., 2012, 51, 4397–4400 CrossRef CAS PubMed.
- S. M. Chen, J. Zhang, T. Wu, P. Y. Feng and X. H. Bu, J. Am. Chem. Soc., 2009, 131, 16027–16029 CrossRef CAS PubMed.
- C. Wang, Z. Xie, K. E. DeKrafft and W. Lin, J. Am. Chem. Soc., 2011, 133, 13445–13454 CrossRef CAS PubMed.
- Y. L. Wang, N. Zhang, Q. Y. Liu, X. Yang, H. Bai, L. Y. Duan and H. Y. Liu, Inorg. Chem. Commun., 2011, 14, 380–383 CrossRef CAS PubMed.
- W. J. Ji, Q. G. Zhai, S. N. Li, Y. C. Jiang and M. C. Hu, Chem. Commun., 2011, 47, 3834–3836 RSC.
- P. J. Calderone, P. M. Forster, L. A. Borkowski, S. J. Teat, M. Feygenson, M. C. Aronson and J. B. Parise, Inorg. Chem., 2011, 50, 2159–2167 CrossRef CAS PubMed.
- Z. L. Xie, M. L. Feng, B. Tan and X. Y. Huang, CrystEngComm, 2012, 14, 4894–4901 RSC.
- B. Tan, Z. L. Xie, M. L. Feng, B. Hu, Z. F. Wu and X. Y. Huang, Dalton Trans., 2012, 41, 10576–10584 RSC.
- W. J. Ji, Q. G. Zhai, S. N. Li, Y. C. Jiang and M. C. Hu, Inorg. Chem. Commun., 2012, 24, 209 CrossRef CAS PubMed.
- A. Dolbecq, E. Dumas, C. R. Mayer and P. Mialane, Chem. Rev., 2010, 110, 6009–6048 CrossRef CAS PubMed.
- E. Ahmed and M. Ruck, Angew. Chem., Int. Ed., 2012, 51, 308–309 CrossRef CAS PubMed.
- F. H. Aidoudi, D. Aldous, R. J. Goff, A. M. Z. Slawin, J. P. Attfield, R. E. Morris and P. Lightfoot, Nat. Chem., 2011, 3, 801–806 CrossRef CAS PubMed.
- H. Fu, C. Qin, Y. Lu, Z. M. Zhang, Y. G. Li, Z. M. Su, W. L. Li and E. B. Wang, Angew. Chem., Int. Ed., 2012, 51, 7985–7989 CrossRef CAS PubMed.
- N. Zou, W. Chen, Y. Li, W. Liu and E. Wang, Inorg. Chem. Commun., 2008, 11, 1367–1370 CrossRef CAS PubMed.
- A. S. Pakhomova and S. V. Krivovichev, Inorg. Chem. Commun., 2010, 13, 1463–1465 CrossRef CAS PubMed.
- W. Liu, H. Tan, W. Chen, Y. Li and E. Wang, J. Coord. Chem., 2010, 63, 1833–1843 CrossRef CAS.
- A. K. Geim, Science, 2009, 324, 1530–1534 CrossRef CAS PubMed.
- C. N. Rao, A. K. Sood, K. S. Subrahmanyam and A. Govindaraj, Angew. Chem., Int. Ed., 2009, 48, 7752–7777 CrossRef CAS PubMed.
- M. Tunckol, J. Durand and P. Serp, Carbon, 2012, 50, 4303–4334 CrossRef CAS PubMed.
- J. Y. Liu, H. Y. Chang, Q. D. Truong and Y. C. Ling, J. Mater. Chem. C, 2013, 1, 1713–1716 RSC.
- Y. Fu, J. Zhang, H. Liu, W. C. Hiscox and Y. Gu, J. Mater. Chem. A, 2013, 1, 2663–2674 CAS.
- T. Kim, H. C. Kang, T. T. Tung, J. D. Lee, H. Kim, W. S. Yang, H. G. Yoon and K. S. Suh, RSC Adv., 2012, 2, 8808–8812 RSC.
- B. Wang, X. Wang, W. Lou and J. Hao, New J. Chem., 2012, 36, 1684–1690 RSC.
- E. K. Choi, I. Y. Jeon, S. Y. Bae, H. J. Lee, H. S. Shin, L. Dai and J. B. Baek, Chem. Commun., 2010, 46, 6320–6322 RSC.
- P. D. McCrary, P. A. Beasley, S. A. Alaniz, C. G. Griggs, R. M. Frazier and R. D. Rogers, Angew. Chem., Int. Ed., 2012, 51, 9784–9787 CrossRef CAS PubMed.
- R. M. Frazier, D. T. Daly, S. K. Spear and R. D. Rogers, PCT Int. Appl., WO, 2010065346, A1, 2010 Search PubMed.
- Z. Zeng, Z. Yin, X. Huang, H. Li, Q. He, G. Lu, F. Boey and H. Zhang, Angew. Chem., Int. Ed., 2011, 50, 11093–11097 CrossRef CAS PubMed.
- H. Zhang, G. Duan, Y. Li, X. Xu, Z. Dai and W. Cai, Cryst. Growth Des., 2012, 12, 2646–2652 CAS.
- J. L. Mi, C. Clausen, M. Bremholm, N. Lock, K. M. Ø. Jensen, M. Christensen and B. B. Iversen, Cryst. Growth Des., 2012, 12, 6092–6097 CAS.
- K. L. Ding, Z. J. Miao, Z. M. Liu, Z. F. Zhang, B. X. Han, G. M. An, S. D. Miao and Y. Xie, J. Am. Chem. Soc., 2007, 129, 6362–6363 CrossRef CAS PubMed.
- K. Ding, Z. Miao, B. Hu, G. An, Z. Sun, B. Han and Z. Liu, Langmuir, 2010, 26, 5129–5134 CrossRef CAS PubMed.
- H. Li, J. Qu, Q. Cui, H. Xu, H. Luo, M. Chi, R. A. Meisner, W. Wang and S. Dai, J. Mater. Chem., 2011, 21, 9487–9490 RSC.
- C. Wesse, L. Zhao, S. Urban, R. Ostermann, I. Djerdj, B. M. Smarsly, L. Chen, Y. S. Hu and S. Sallard, Chem. – Eur. J., 2011, 17, 775–779 CrossRef PubMed.
- S. S. Mali, C. A. Betty, P. N. Bhosale, R. S. Devan, Y. R. Ma, S. S. Kolekar and P. S. Patil, CrystEngComm, 2012, 14, 1920–1924 RSC.
- H. Cheng, B. Huang, Z. Wang, X. Qin, X. Zhang and Y. Dai, Chem. – Eur. J., 2011, 17, 8039–8043 CrossRef CAS PubMed.
- J. Xia, S. Yin, H. Li, H. Xu, L. Xu and Y. Xu, Dalton Trans., 2011, 40, 5249–5258 RSC.
- J. Xia, S. Yin, H. Li, H. Xu, Y. Yan and Q. Zhang, Langmuir, 2011, 27, 1200–1206 CrossRef CAS PubMed.
- J. Zhang, J. Xia, S. Yin, H. Li, H. Xu, M. He, L. Huang and Q. Zhang, Colloids Surf., A, 2013, 420, 89–95 CrossRef CAS PubMed.
- K. Seddon, Chem. Eng., 2002, 730, 33–35 CAS.
- M. C. Uzagare and M. Salunkhe, Asian Chem. Lett., 2004, 8, 269–287 CAS.
- R. Freer and A. Curzons, in Green Industrial Applications of Ionic Liquids, ed. R. D. Rogers, K. R. Seddon and S. Volkov, NATO Science Series II. Mathematics, Physics and Chemistry, Kluwer, Dordrecht, 2003, vol. 92, pp. 129–136 Search PubMed.
- J. S. Shamshina, P. S. Barber and R. D. Rogers, Expert Opin. Drug Delivery, 2013, 10, 1367–1381 CrossRef CAS PubMed.
- J. Stoimenovski, D. R. MacFarlane, K. Bica and R. D. Rogers, Pharm. Res., 2010, 27, 521–526 CrossRef CAS PubMed.
- M. M. Oniruzzaman and M. G. Oto, J. Chem. Eng. Jpn., 2011, 44, 370–381 CrossRef.
- K. Fujita, D. R. MacFarlane and M. Forsyth, Chem. Commun., 2005, 4804–4806 RSC.
- K. Fujita, M. Forsyth, D. R. MacFarlane, R. W. Reid and G. D. Elliott, Biotechnol. Bioeng., 2006, 94, 1209–1213 CrossRef CAS PubMed.
- V. Kumar, V. S. Parmar and S. V. Malhotra, Tetrahedron Lett., 2007, 48, 809–812 CrossRef CAS PubMed.
- Y. Fukaya, K. Hayashi, M. Wada and H. Ohno, Green Chem., 2008, 10, 44–46 RSC.
- M. Moniruzzaman, Y. Tahara, M. Tamura, N. Kamiya and M. Goto, Chem. Commun., 2010, 47, 1452–1454 RSC.
- R. P. Swatloski, S. K. Spear, J. D. Holbrey and R. D. Rogers, J. Am. Chem. Soc., 2002, 124, 4974–4975 CrossRef CAS PubMed.
- M. C. Uzagare, Y. S. Sanghvi and M. M. Salunkhe, Green Chem., 2003, 5, 370–372 RSC.
- C. I. Melo, R. Bogel-Lukasik, M. Nunes da Ponte and E. Bogel-Lukasik, Fluid Phase Equilib., 2013, 338, 209–216 CrossRef CAS PubMed.
- P. D. McCrary, P. A. Beasley, G. Gurau, A. Narita, P. S. Barber, A. Cojocaru and R. D. Rogers, New J. Chem., 2013, 37, 2196–2202 RSC.
- H. Mizuuchi, V. Jaitely, S. Murdan and A. T. Florence, Eur. J. Pharm. Sci., 2008, 33, 326–331 CrossRef CAS PubMed.
- K. B. Smith, R. H. Bridson and G. A. Leeke, J. Chem. Eng. Data, 2011, 56, 2039–2043 CrossRef CAS.
- V. Jaitely, A. Karatas and A. T. Florence, Int. J. Pharm., 2008, 354, 168–173 CrossRef CAS PubMed.
- R. P. Swatloski, J. D. Holbrey and R. D. Rogers, Green Chem., 2003, 5, 361–363 RSC.
- M. Moniruzzaman, M. Tamura, Y. Tahara, N. Kamiya and M. Goto, Int. J. Pharm., 2010, 400, 243–250 CrossRef CAS PubMed.
- D. Dobler, T. Schmidts, I. Klingenhoefer and F. Runkel, Int. J. Pharm., 2013, 441, 620–627 CrossRef CAS PubMed.
- B. D. Ribeiro, M. A. Z. Coelho, L. P. N. Rebelo and I. M. Marrucho, Ind. Eng. Chem. Res., 2013, 52, 12146–12153 CrossRef CAS.
- Y. Sun, Z. Liu, J. Wang, S. Yang, B. Li and N. Xu, Ultrason. Sonochem., 2013, 20, 180–186 CrossRef CAS PubMed.
- W. Bi, M. Tian and K. H. Row, Talanta, 2011, 85, 701–706 CrossRef CAS PubMed.
- Y. Qin, X. Lu, N. Sun and R. D. Rogers, Green Chem., 2010, 12, 968 RSC.
- X. Liu, Y. Wang, J. Kong, C. Nie and X. Lin, Anal. Methods, 2012, 4, 1012–1018 RSC.
- Y. Yuan, Y. Wang, R. Xu, M. Huang and H. Zeng, Analyst, 2011, 136, 2294–2305 RSC.
- H. Zeng, Y. Wang, J. Kong, C. Nie and Y. Yuan, Talanta, 2010, 83, 582–590 CrossRef CAS PubMed.
- A. K. Ressmann, P. Gaertner and K. Bica, Green Chem., 2011, 13, 1442–1447 RSC.
- A. K. Ressmann, K. Strassl, P. Gaertner, B. Zhao, L. Greiner and K. Bica, Green Chem., 2012, 14, 940 RSC.
- R. Jin, L. Fan and X. An, Sep. Purif. Technol., 2012, 83, 45–49 CrossRef CAS PubMed.
- S. A. Chowdhury, R. Vijayaraghavan and D. R. MacFarlane, Green Chem., 2010, 12, 1023–1028 RSC.
- J. H. Davis, K. J. Forrester and T. Merrigan, Tetrahedron Lett., 1998, 39, 8955–8958 CrossRef CAS.
- E. B. Carter, S. L. Culver, P. A. Fox, R. D. Goode, I. Ntai, M. D. Tickell, R. K. Traylor, N. W. Hoffman and J. H. Davis, Chem. Commun., 2004, 630–631 RSC.
- W. L. Hough, M. Smiglak, H. Rodríguez, R. P. Swatloski, S. K. Spear, D. T. Daly, J. Pernak, J. E. Grisel, R. D. Carliss, M. D. Soutullo, J. H. Davis and R. D. Rogers, New J. Chem., 2007, 31, 1429 RSC.
- L. Borka and J. K. Haleblian, Acta Pharm. Jugosl., 1990, 40, 71–94 CAS.
- R. Ferraz, L. C. Branco, I. M. Marrucho, J. M. M. Araújo, L. P. N. Rebelo, M. N. da Ponte, C. Prudêncio, J. P. Noronha and Ž. Petrovski, MedChemComm, 2012, 3, 494 RSC.
- C. Florindo, J. M. M. Araujo, F. Alves, C. Matos, R. Ferraz, C. Prudencio, J. P. Noronha, Z. Petrovski, L. Branco, L. P. N. Rebelo and I. M. Marrucho, Int. J. Pharm., 2013, 456, 553–559 CrossRef CAS PubMed.
- W. L. Hough-Troutman, M. Smiglak, S. Griffin, W. Matthew Reichert, I. Mirska, J. Jodynis-Liebert, T. Adamska, J. Nawrot, M. Stasiewicz, R. D. Rogers and J. Pernak, New J. Chem., 2009, 33, 26 RSC.
- J. Cybulski, A. Wisniewska, A. Kulig-Adamiak, Z. Dabrowski, T. Praczyk, A. Michalczyk, F. Walkiewicz, K. Materna and J. Pernak, Tetrahedron Lett., 2011, 52, 1325–1328 CrossRef CAS PubMed.
- K. Bica, C. Rijksen, M. Nieuwenhuyzen and R. D. Rogers, Phys. Chem. Chem. Phys., 2010, 12, 2011–2017 RSC.
- J. Stoimenovski, P. M. Dean, E. I. Izgorodina and D. R. MacFarlane, Faraday Discuss., 2012, 154, 335 RSC.
- J. Stoimenovski and D. R. MacFarlane, Chem. Commun., 2011, 47, 11429–11431 RSC.
- K. M. Johansson, E. I. Izgorodina, M. Forsyth, D. R. MacFarlane and K. R. Seddon, Phys. Chem. Chem. Phys., 2008, 10, 2972–2978 RSC.
- K. Bica, J. Shamshina, W. L. Hough, D. R. MacFarlane and R. D. Rogers, Chem. Commun., 2011, 47, 2267–2269 RSC.
- K. Bica and R. D. Rogers, Chem. Commun., 2010, 46, 1215–1217 RSC.
- S. Aitipamula, R. Banerjee, A. K. Bansal, K. Biradha, M. L. Cheney, A. Roy Choudhury, G. R. Desiraju, A. G. Dikundwar, R. Dubey, N. Duggirala, P. P. Ghogale, S. Ghosh, P. K. Goswami, N. R. Goud, R. R. K. R. Jetti, P. Karpinski, P. Kaushik, D. Kumar, V. Kumar, B. Moulton, A. Mukherjee, G. Mukherjee, A. S. Myerson, V. Puri, A. Ramanan, T. Rajamannar, C. M. Reddy, N. Rodriguez-Hornedo, R. D. Rogers, T. N. G. Row, P. Sanphui, N. Shan, G. Shete, A. Singh, C. C. Sun, J. A. Swift, R. Thaimattam, T. S. Thakur, R. K. Thaper, S. P. Thomas, S. Tothadi, V. R. Vangala, N. Variankaval, P. Vishweshwar, D. R. Weyna and M. J. Zaworotko, Cryst. Growth Des., 2012, 12, 2147–2152 CAS.
- S. P. Kelley, A. Narita, J. D. Holbrey, K. D. Green, W. M. Reichert and R. D. Rogers, Cryst. Growth Des., 2013, 13, 965–975 CAS.
- K. S. Lovejoy, G. M. Purdy, C. A. Corley, J. S. Wilkes, A. T. Koppisch and S. Del Rico E., Abstracts of Papers, 243rd ACS National Meeting & Exposition, San Diego, CA, United States, March 25–29, 2012, American Chemical Society, 2012, p. IEC-291.
- M. Hacker, W. S. MesserII and K. A. Bachmann, Pharmacology: Principles and Practice, 2009, p. 216 Search PubMed.
- R. D. Rogers, O. A. Cojocaru and J. L. Shamshina, Chim. Oggi, 2013, 5, 24–29 Search PubMed.
- O. A. Cojocaru, K. Bica, G. Gurau, A. Narita, P. D. McCrary, J. L. Shamshina, P. S. Barber and R. D. Rogers, MedChemComm, 2013, 4, 559–563 RSC.
- J. Pernak, I. Goc and I. Mirska, Green Chem., 2004, 6, 323–329 RSC.
- M. R. Cole, M. Li, B. El-Zahab, M. E. Janes, D. Hayes and I. M. Warner, Chem. Biol. Drug Des., 2011, 78, 33–41 CAS.
- L. Carson, P. K. W. Chau, M. J. Earle, M. A. Gilea, B. F. Gilmore, S. P. Gorman, M. T. McCann and K. R. Seddon, Green Chem., 2009, 11, 492–497 RSC.
- A. Busetti, D. E. Crawford, M. J. Earle, M. A. Gilea, B. F. Gilmore, S. P. Gorman, G. Laverty, A. F. Lowry, M. McLaughlin and K. R. Seddon, Green Chem., 2010, 12, 420–425 RSC.
- S. Y. Choi, H. Rodríguez, A. Mirjafari, D. F. Gilpin, S. McGrath, K. R. Malcolm, M. M. Tunney, R. D. Rogers and T. McNally, Green Chem., 2011, 13, 1527 RSC.
- M. Seter, M. J. Thomson, J. Stoimenovski, D. R. MacFarlane and M. Forsyth, Chem. Commun., 2012, 48, 5983–5985 RSC.
- M. Seter, M. J. Thomson, A. Chong, D. R. MacFarlane and M. Forsyth, Aust. J. Chem., 2013, 66, 921–929 CrossRef CAS.
- http://purdueturftips.blogspot.com/2012/01/imprelis-herbicide-injury-2011-ppdl.html, last accessed on Jan. 22, 2014.
- J. Pernak, A. Syguda, K. Materna, E. Janus, P. Kardasz and T. Praczyk, Tetrahedron, 2012, 68, 4267–4273 CrossRef CAS PubMed.
- T. Praczyk, P. Kardasz, E. Jakubiak, A. Syguda, K. Materna and J. Pernak, Weed Sci., 2012, 60, 189–192 CrossRef CAS PubMed.
- J. Pernak, J. Shamshina, P. Tadeusz, A. Syguda, D. Janiszewska, M. Smiglak, G. Gurau, D. T. Daly and R. D. Rogers, US20130109572, 2013.
- J. Pernak, A. Syguda, D. Janiszewska, K. Materna and T. Praczyk, Tetrahedron, 2011, 67, 4838–4844 CrossRef CAS PubMed.
- O. A. Cojocaru, J. L. Shamshina, G. Gurau, A. Syguda, T. Praczyk, J. Pernak and R. D. Rogers, Green Chem., 2013, 15, 2110–2120 RSC.
- (a) P. Lewandowski, R. Kukawka, H. Pospieszny and M. Smiglak, New J. Chem., 2014, 38, 1372–1375 RSC; (b) M. Smiglak, R. Kukawka, P. Lewandowski and H. Pospieszny, Tetrahedron Lett., 2014 DOI:10.1016/j.tetlet.2014.04.108.
- K. Bica, L. R. Cooke, P. Nugent, C. Rijksen and R. D. Rogers, Green Chem., 2011, 13, 2344 RSC.
- L. Ford, J. R. Harjani, F. Atefi, M. T. Garcia, R. D. Singer and P. J. Scammells, Green Chem., 2010, 12, 1783–1789 RSC.
- F. Atefi, M. T. Garcia, R. D. Singer and P. J. Scammells, Green Chem., 2009, 11, 1595–1604 RSC.
- J. R. Harjani, J. Farrell, M. T. Garcia, R. D. Singer and P. J. Scammells, Green Chem., 2009, 11, 821–829 RSC.
- J. R. Harjani, R. D. Singer, M. T. Garcia and P. J. Scammells, Green Chem., 2009, 11, 83–90 RSC.
- S. Steudte, P. Stepnowski, C.-W. Cho, J. Thöming and S. Stolte, Chem. Commun., 2012, 48, 9382–9384 RSC.
- J. Neumann, S. Steudte, C.-W. Cho, J. Thöming and S. Stolte, Green Chem., 2014, 16, 2174–2184 RSC.
- G. Chatel, J. F. B. Pereira, V. Debetti, H. Wang and R. D. Rogers, Green Chem., 2014, 16, 2051–2083 RSC.
| This journal is © The Royal Society of Chemistry 2014 |