 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Ditopic bis-terdentate cyclometallating ligands and their highly luminescent dinuclear iridium(III) complexes†
Pierre-Henri
Lanoë
a,
Chi Ming
Tong
a,
Ross W.
Harrington
b,
Michael R.
Probert
b,
William
Clegg
b,
J. A. Gareth
Williams
*c and
Valery N.
Kozhevnikov
*a
aDepartment of Applied Sciences, Northumbria University, Newcastle upon Tyne, NE1 8ST, UK. E-mail: valery.kozhevnikov@northumbria.ac.uk
bSchool of Chemistry, Newcastle University, Newcastle upon Tyne, NE1 7RU, UK. E-mail: bill.clegg@ncl.ac.uk
cDepartment of Chemistry, University of Durham, South Road, Durham, DH1 3LE, UK. E-mail: j.a.g.williams@durham.ac.uk
First published on 1st April 2014
Abstract
A new family of bridged cyclometallating ligands is reported, which incorporate two terdentate N∧C∧N-coordinating binding sites linked via pyrazine, pyrimidine or pyridazine units. Dinuclear Ir(III) complexes of one ligand have been prepared and crystallographically characterised; they display intense red phosphorescence.
Multimetallic complexes formed by rigid ditopic ligands have become well-known in contemporary coordination chemistry. For example, polypyridine-type bridging ligands have underpinned the development of metallosupramolecular chemistry, leading to a wide variety of functional materials and unusual forms of chirality.1 Amongst the many useful properties of polymetallic complexes are their increased extinction coefficients compared to mononuclear analogues and, frequently, their lower-energy absorption maxima.2 Such improved light-harvesting features are particularly attractive in areas such as photoinduced electron- and energy-transfer for solar energy conversion and in photocatalysis.3 Meanwhile, for luminescence, the presence of additional metal centres may facilitate spin–orbit coupling (SOC) pathways and augment radiative rate constants, increasing the efficiency of phosphorescence from triplet states, as required for use in organic light-emitting diodes.4 For example, efficient red emitters have been prepared using bis-cyclometallating ligands that bring together two N∧C-coordinating sites to bind simultaneously to Pt(II) and Ir(III) ions.5
It is widely recognised that terdentate ligands can offer structural advantages over bidentate ligands, including the absence of chirality in some cases and greater rigidity.6 However, the classic N∧N∧N-binding ligand 2,2′:6′,2′′-terpyridine, despite having been used to generate a plethora of structures with interesting geometries, rarely leads to systems that are strongly luminescent at room temperature, owing in part to the poor bite angle that leads to efficient non-radiative decay.7 In contrast, the isoelectronic N∧C∧N-binding ligand 1,3-bis(2-pyridyl)-benzene (dpyb)8 and its derivatives have been used to prepare highly luminescent complexes of metals such as Ir(III), Rh(III) and Pt(II).9–11 The strong ligand field in combination with high rigidity inhibits non-radiative decay, whilst efficient spin–orbit coupling pathways promote triplet phosphorescence.12 Although such complexes have been incorporated as units into multinuclear assemblies,13 the few examples reported to date fall into the class of supramolecular system where the individual units largely retain their original excited state properties, and energy transfer occurs between them. In this contribution, we describe a new family of rigid, bridged cyclometallating proligands that feature potential ditopic N∧C∧N–N∧C∧N coordination, and report on the preparation and luminescent properties of mono- and dinuclear Ir(III) complexes of one such ligand.
The synthetic strategy used to prepare the ligands is summarised in Scheme 1, and is based on a combination of boronic acid synthesis via ortho-lithiation and subsequent Pd-catalysed Suzuki cross-coupling reactions. The choice of central aryl synthon 1 was made on the basis that: (i) the hexyl group would improve the solubility of the intermediates and final products, since one of the drawbacks of rigid ligands is often poor solubility of products; (ii) fluorine atoms are ortho-directing in the lithiation with n-BuLi, which will ensure the necessary regiochemistry for synthesis of 1,3-diboronic acids; and (iii) the F atoms will block undesired, competitive metallation of the C4 and C6 positions of the aryl ring upon reaction with iridium(III) chloride, which is known to be the predominant mode of binding for unsubstituted dpyb.10 The key intermediate is the boronic acid 4, from which a variety of bridging bis-terdentate N∧C∧N–N∧C∧N ligands can be prepared in one step simply by reaction with an appropriate dihalogenated heterocycle. For example, we prepared three such ligands L1H2, L2H2 and L3H2 by the cross-coupling of 4 with 4,6-dichloropyrimidine, 2,5-dibromopyrazine or 3,6-dichloropyridazine respectively, under standard Suzuki conditions. Other linking heterocycles, such as naphthyridines or phenanthrolines, can equally be introduced at this stage.
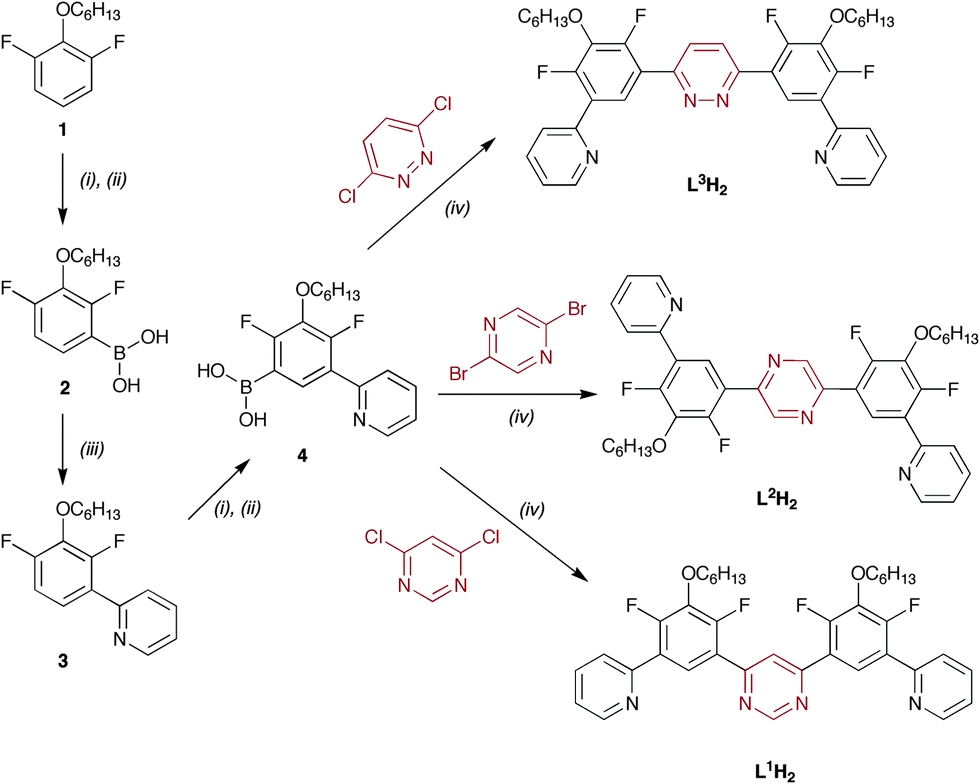 | ||
| Scheme 1 Synthesis of cyclometallating bis-terdentate ligands. (i) BuLi; (ii) B(OiPr)3 then H3O+; (iii) 2-bromopyridine, Pd cat.; (iv) Pd cat. | ||
In the first instance, we have explored the complexation chemistry of the pyrimidine-based proligand L1H2 with iridium(III). 1,3-Di(2-pyridyl)-2,6-difluorobenzene (F2dpybH) is known to react with IrCl3·3H2O in ethoxyethanol/water to give a dichloro-bridged dimer [Ir(F2dpyb)Cl(μ-Cl)]2.14 Applying the same conditions to L1H2 with 2 equivalents of IrCl3·3H2O led to a similarly bridged tetranuclear, dimeric complex [Ir2L1Cl2(μ-Cl)2]2 in which, according to NMR, L1 is doubly metallated, bridging two metal centres (Scheme 2). Treatment of this intermediate with 2-(p-tolyl)pyridine (ptpyH) in toluene in the presence of silver triflate led to the cleavage of the chloro bridges and introduction of a C∧N-bound tolylpyridine into the coordination sphere of each metal ion, generating Ir2L1(ptpy)2Cl2. The coordination sphere is completed by one monodentate chloride ligand remaining on each metal. When the reaction was carried out with only one equivalent of IrCl3·3H2O relative to L1H2, the corresponding mononuclear complex IrL1H(ptpy)Cl was obtained, in which L1H is singly metallated only.
 | ||
| Scheme 2 Synthesis of Ir2L1(ptpy)2Cl2; schematic illustration of the relative disposition of the Cl ligands (centre), and corresponding structures obtained by X-ray diffraction (right; for clarity, the structures are shown without H atoms, solvent molecules and minor disorder components). | ||
Owing to the strong trans effect of metallated carbon atoms, the ptpy ligand is introduced in such a way that its pyridyl ring, not the aryl ring, is disposed trans to the central aryl ring of the N∧C∧N unit.10,15 A single isomer of IrL1H(ptpy)Cl is therefore isolated (as a racemic mixture of enantiomers). In the case of the dinuclear Ir2L1(ptpy)2Cl2, three stereoisomers are formed: one meso form with an internal mirror plane, and a racemic pair of enantiomers in which the two chloride ligands are disposed on opposite sides of the plane of L1 (Scheme 2). The meso and rac isomers have different physical properties and were easily separated by conventional column chromatography. The isolated yields were in the region of 25% for each form. The structures of the meso form and the rac pair were determined by X-ray diffraction (Scheme 2).‡ The coordination of two Ir(III) cations favours the adoption of a fully planar conformation by the bis-terdentate ligand L1, and the Ir–Cl bonds are almost perpendicular to the plane of L1. The L1 ligands in the two structures are essentially identical apart from the hexyl substituents, and the main differences are in the orientations of the ptpy ligands and the conformations of the flexible hexyl chains, most of which display disorder.
The absorption spectrum of the mononuclear complex IrL1H(ptpy)Cl displays very intense bands in the UV region, attributable to π–π* transitions within the ligand, together with a series of moderately intense bands in the visible region (Fig. 1, Table 1). By analogy with typical Ir(III) complexes with cyclometallating aryl-heterocycle ligands,4d,16 the latter can be attributed to singlet and triplet charge-transfer transitions, involving filled orbitals localised predominantly on the metal and cyclometallating aryl rings, and vacant orbitals on the heterocycle. It is notable, however, that these bands extend to longer wavelength than the corresponding bands in, for example, Ir(F2dpyb)(ppy)Cl.14 This is readily rationalised on the basis that pyrimidine has a lower-energy π* orbital than pyridine and hence the energy of the charge-transfer transition will be lowered.
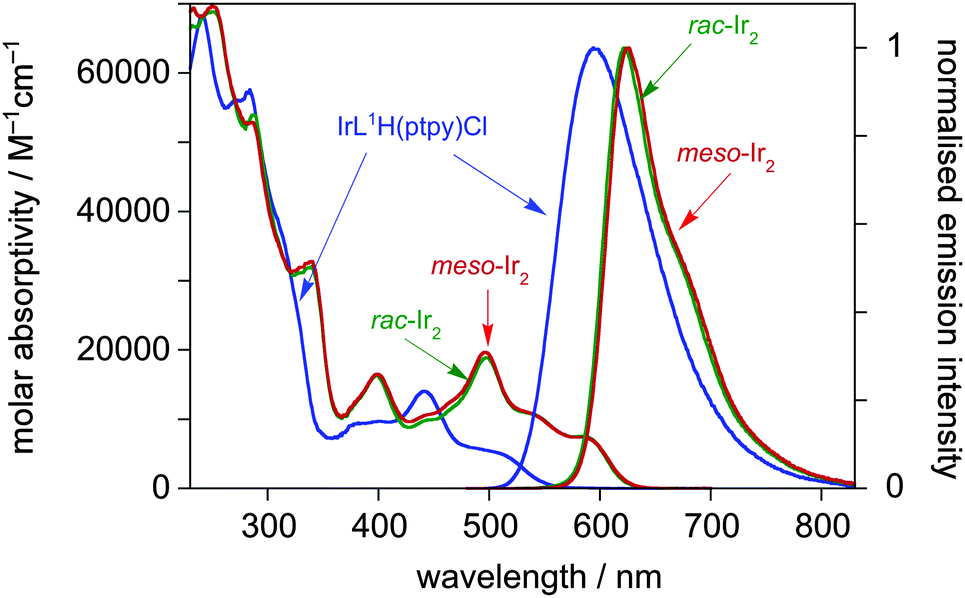 | ||
| Fig. 1 Absorption and photoluminescence spectra of IrL1H(ptpy)Cl (blue lines) and the rac and meso forms of Ir2L1(ptpy)2Cl2 (green and red lines respectively) in solution in CH2Cl2 at 298 K. | ||
| Complex | Isomer | λ absmax/nm (ε/M−1 cm−1) | λ emmax/nm | Φ | τ/nsc |
k
r/105 s−1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) d d |
k
nr/105 s−1d |
Emission at 77 Ke | |
|---|---|---|---|---|---|---|---|---|---|
| λ max/nm | τ/ns | ||||||||
|
a In deoxygenated CH2Cl2 at 298 K except where stated otherwise.
b Luminescence quantum yield determined using Ru(bpy)3Cl2 in water as the standard; estimated uncertainty in absolute values is ±20% or better, while the error on relative values amongst the three complexes is <5%.
c Values in parentheses refer to air-equilibrated solutions; estimated uncertainty in lifetime values is ±10% or better.
d
k
r and knr estimated as described in the text, assuming that the emissive state is formed with unitary efficiency upon light absorption.
e In diethyl ether/isopentane/ethanol (2:2:1 v/v).
|
|||||||||
| IrL1H(ptpy)Cl | — | 242 (68100), 283 (57100), 385 sh (9410), 402 (9700), 441 (14000), 501 (5400) |
595 | 0.59 | 2800 [450] | 2.1 | 1.5 | 562 | 13000 |
| Ir2L1(ptpy)2Cl2 | rac | 250 (68800), 287 (54000), 339 (31900), 398 (16300), 448 (9930), 498 (18900), 537 (10800), 583 (7550) |
622 | 0.65 | 760 [520] | 8.6 | 4.6 | 617, 666 | 3700 |
| Ir2L1(ptpy)2Cl2 | meso | 250 (69700), 286 (52900), 340 (32800), 399 (16500), 447 (10700), 497 (19700), 537 (10900), 582 (7420) |
625 | 0.65 | 730 [460] | 8.9 | 4.8 | 620, 672 | 3500 |
The photophysical properties of the meso and rac forms of Ir2L1(ptpy)2Cl2 are very similar to one another: their absorption spectra are almost identical, with differences in λmax values being within the uncertainty of the measurement (Fig. 1). The introduction of a second metal ion into the system – i.e. going from IrL1H(ptpy)Cl to meso- or rac-Ir2L1(ptpy)2Cl2 – is accompanied by a large red shift of the bands in the visible region (e.g. the lowest-energy band shifts by around 80 nm). Evidently, coordination of a second Ir(III) cation to the other N atom of the pyrimidine ring will stabilise further the pyrimidine-based π* orbital, leading to the observed red shift, irrespective of which isomer is formed. The behaviour in this respect is similar to that observed recently for multinuclear complexes with bis-N∧C-coordinating diphenylpyrimidine ligands,5 and is reminiscent of earlier studies on Ru(II) complexes of dipyridylpyrimidine.2a
All three complexes are intensely photoluminescent in solution at room temperature. The emission of the mononuclear complex is in the orange-red region of the spectrum, peaking at λmax = 595 nm, with a quantum yield of 0.59 and a luminescence lifetime of 2.8 μs under deoxygenated conditions (Fig. 1, Table 1). As in absorption, the introduction of the second metal ion to generate meso- or rac-Ir2L1(ptpy)2Cl2 is accompanied by a red shift (albeit a smaller one than in absorption), and the meso and rac emission spectra are essentially identical to one another. Remarkably, the quantum yield is enhanced for the dinuclear complexes, despite the red shift. Typically, quantum yields tend to drop off with decreasing excited state energy, as non-radiative decay is facilitated. Some insight into the possible reasons for the increase in Φ can be obtained by considering the rate constants of radiative and non-radiative decay, kr and knr, respectively. Assuming that the emissive excited state is formed with unitary efficiency, the rate constants can be estimated from the lifetime and quantum yield as follows: kr = Φ/τ and knr = τ−1 − kr. Such an analysis reveals that, although the non-radiative decay constants are indeed somewhat higher in the binuclear complexes, this is more than offset by a substantial 4-fold increase in kr (Table 1).
The increase in kr upon introduction of the second metal ion is intriguing. Other things being equal, a decrease in the radiative rate would be expected as the energy decreases, according to the Einstein coefficient which depends on ν3. At least two effects may be at work here. Firstly, the fact that T1 → S0 phosphorescence from such metal complexes is observed at room temperature is due to the high spin–orbit coupling constant of the heavy metal ion (ζ = 3909 cm−1 for Ir), which breaks down the ΔS = 0 spin selection rule for radiative decay. The presence of a second metal ion may be augmenting the spin–orbit coupling effect.5 Secondly, it may be noted that the red-shift in emission on going from mono- to dinuclear complexes is smaller than the red-shift of the lowest-energy absorption band, which indicates that the energy gap between S1 and T1 is lower in the dinuclear systems. Using the λmax values as a guide to the energies, we can estimate that ΔE(S1 − T1) is 3200 cm−1 for IrL1H(ptpy)Cl but only around 1100 cm−1 for the dinuclear complexes. Spin–orbit coupling pathways are complicated and not fully understood, but it is well-established that the T1 state must mix with higher-lying 1MLCT states for phosphorescence to be promoted.17 The efficiency with which this occurs is inversely proportional to the energy gap between the T1 state and the higher singlet state. The observation of a lower ΔE(S1 − T1) in the dinuclear complexes may thus be indicative of more efficient SOC pathways.
In summary, we have presented a new family of cyclometallating bis(N∧C∧N)-coordinating ligands and their application in the synthesis of dinuclear Ir(III) complexes. The synthetic methodology, centred around cross-couplings of synthon 4, allows facile variation of the bridging unit within the ligands, and provides access to a variety of possible structures. In comparison to classical coordination complexes of polypyridines, cyclometallating ligands offer both structural and photophysical advantages. The strong trans effect of cyclometallating carbon atoms dictates the orientation of the N∧C ligand in the coordination sphere, such that only those isomers in which the heterocycle of the N∧C ligand is trans to the carbon of the N∧C∧N unit are formed. The di-iridium complexes are amongst the very best phosphorescent red emitters known to date.18 Phosphorescent emission at 625 nm with 65% quantum yields and emission lifetimes <1 μs render these materials strong candidates for consideration as OLED dopants. Moreover, the strong absorption in the orange-to-red region is a highly desirable attribute for sensitisers in both dye-sensitised solar cells and in photocatalysed water splitting, where iridium(III) complexes are attracting more and more attention.19 Thus, the structural and emissive properties of this new class of complexes are of interest for exploitation in metallo-supramolecular chemistry and the design of new functional materials.
We thank EPSRC (grant ref. EP/I014942/1) for support of this work, and Diamond Light Source for access to synchrotron single-crystal diffraction beamline I19. Mass spectra were acquired at the EPSRC UK National Mass Spectrometry Facility at Swansea University.
Notes and references
- G. S. Hanan, C. R. Arana, J.-M. Lehn and D. Fenske, Angew. Chem., Int. Ed. Engl., 1995, 34, 1122 CrossRef CAS PubMed; M. W. Cooke, D. Chartrand and G. S. Hanan, Coord. Chem. Rev., 2008, 252, 903 CrossRef PubMed.
- For example: (a) S. Serroni, A. Juris, S. Campagna, M. Venturi, G. Denti and V. Balzani, J. Am. Chem. Soc., 1994, 116, 9086 CrossRef CAS; (b) P. Ceroni, A. Credi, V. Balzani, S. Campagna, G. S. Hanan, C. R. Arana and J.-M. Lehn, Eur. J. Inorg. Chem., 1999, 1409 CrossRef CAS; (c) A. Credi, V. Balzani, S. Campagna, G. S. Hanan, C. R. Arana and J.-M. Lehn, Chem. Phys. Lett., 1995, 243, 102 CrossRef CAS; (d) G. S. Hanan, C. R. Arana, J.-M. Lehn, G. Baum and D. Fenske, Chem. – Eur. J., 1996, 2, 1292–1302 CrossRef CAS PubMed.
- A. Hagfeldt, G. Boschloo, L. Sun, L. Kloo and H. Pettersson, Chem. Rev., 2010, 110, 6595 CrossRef CAS PubMed; N. Kaveevivitchai, R. Chitta, R. Zong, M. El Ojaimi and R. P. Thummel, J. Am. Chem. Soc., 2012, 134, 10721 CrossRef PubMed; Z. Deng, H.-W. Tseng, R. Zong, D. Wang and R. Thummel, Inorg. Chem., 2008, 47, 1835 CrossRef PubMed; V. Balzani, S. Campagna, G. Denti, A. Juris, S. Serroni and M. Venturi, Acc. Chem. Res., 1998, 31, 26 CrossRef.
- (a) M. A. Baldo, D. F. O'Brien, Y. You, A. Shoustikov, S. Sibley, M. E. Thompson and S. R. Forrest, Nature, 1998, 395, 151 CrossRef CAS PubMed; (b) Highly Efficient OLEDs with Phosphorescent Materials, ed. H. Yersin, Wiley-VCH, Weinheim, Germany, 2007 Search PubMed; (c) Y. Chi and P. T. Chou, Chem. Soc. Rev., 2010, 39, 638 RSC; (d) L. F. Gildea and J. A. G. Williams, Iridium and platinum complexes for OLEDs, in Organic Light-Emitting Diodes: Materials, Devices and Applications, ed. A. Buckley, Woodhead, Cambridge, 2013 Search PubMed.
- V. N. Kozhevnikov, M. C. Durrant and J. A. G. Williams, Inorg. Chem., 2011, 50, 6304 CrossRef CAS PubMed; S. Culham, P.-H. Lanoë, V. L. Whittle, M. C. Durrant, J. A. G. Williams and V. N. Kozhevnikov, Inorg. Chem., 2013, 52, 10992 CrossRef PubMed.
- J.-P. Sauvage, J.-P. Collin, J.-C. Chambron, S. Guillerez, C. Coudret, V. Balzani, F. Barigelletti, L. De Cola and L. Flamigni, Chem. Rev., 1994, 94, 993 CrossRef CAS.
- L. Hammarström, F. Barigelletti, L. Flamigni, M. T. Indelli, N. Armaroli, G. Calogero, M. Guardigli, A. Sour, J. P. Collin and J. P. Sauvage, J. Phys. Chem. A, 1997, 101, 9061 CrossRef CAS; M. Maestri, N. Armaroli, V. Balzani, E. C. Constable and A. M. W. Cargill Thompson, Inorg. Chem., 1995, 34, 2759 CrossRef.
- M. Beley, J. P. Collin, R. Louis, B. Metz and J. P. Sauvage, J. Am. Chem. Soc., 1991, 113, 8521 CrossRef CAS; M. Beley, J. P. Collin and J. P. Sauvage, Inorg. Chem., 1993, 32, 4549 CrossRef.
- J. A. G. Williams, Chem. Soc. Rev., 2009, 38, 1783 RSC.
- A. J. Wilkinson, A. E. Goeta, C. E. Foster and J. A. G. Williams, Inorg. Chem., 2004, 43, 6513 CrossRef CAS PubMed; A. J. Wilkinson, H. Puschmann, J. A. K. Howard, C. E. Foster and J. A. G. Williams, Inorg. Chem., 2006, 45, 8685 CrossRef PubMed; L. F. Gildea, A. S. Batsanov and J. A. G. Williams, Dalton Trans., 2013, 42, 10388 RSC.
- S. J. Farley, D. L. Rochester, A. L. Thompson, J. A. K. Howard and J. A. G. Williams, Inorg. Chem., 2005, 44, 9690 CrossRef CAS PubMed; V. N. Kozhevnikov, B. Donnio and D. W. Bruce, Angew. Chem., Int. Ed., 2008, 47, 6286 CrossRef PubMed; S. Develay, O. Blackburn, A. L. Thompson and J. A. G. Williams, Inorg. Chem., 2008, 47, 11129 CrossRef PubMed.
- A. Rausch, L. Murphy, J. A. G. Williams and H. Yersin, Inorg. Chem., 2012, 51, 312 CrossRef CAS PubMed; A. Rausch, L. Murphy, J. A. G. Williams and H. Yersin, Inorg. Chem., 2009, 48, 11407 CrossRef PubMed.
- V. L. Whittle and J. A. G. Williams, Inorg. Chem., 2008, 47, 6596 CrossRef CAS PubMed.
- P. Brulatti, R. J. Gildea, J. A. K. Howard, V. Fattori, M. Cocchi and J. A. G. Williams, Inorg. Chem., 2012, 51, 3813 CrossRef CAS PubMed.
- J. Kuwabara, T. Namekawa, M. Haga and T. Kanbara, Dalton Trans., 2012, 41 Search PubMed.
- A. B. Tamayo, B. D. Alleyne, P. I. Djuorovich, S. Lamansky, I. Tsyba, N. N. Ho, R. Bau and M. E. Thompson, J. Am. Chem. Soc., 2003, 125, 7377 CrossRef CAS PubMed.
- A. F. Rausch, H. H. H. Homeier, P. I. Djurovich, M. E. Thompson and H. Yersin, Proc. SPIE-Int. Soc. Opt. Eng., 2007, 6655, F6550 CrossRef CAS PubMed; H. Yersin, A. F. Rausch, R. Czerwieniec, T. Hofbeck and T. Fischer, Coord. Chem. Rev., 2011, 255, 2622 CrossRef PubMed; P. T. Chou, Y. Chi, M. W. Chung and C. C. Lin, Coord. Chem. Rev., 2011, 255, 2653 CrossRef PubMed.
- S. Lamansky, P. Djurovich, D. Murphy, F. Abdel-Razzaq, H. E. Lee, C. Adachi, P. E. Burrows, S. R. Forrest and M. E. Thompson, J. Am. Chem. Soc., 2001, 123, 4304 CrossRef CAS PubMed; A. Tsuboyama, H. Iwawaki, M. Furugori, T. Mukaide, J. Kamatani, S. Igawa, T. Moriyama, S. Miura, T. Takiguchi, S. Okada, M. Hoshini and K. Ueno, J. Am. Chem. Soc., 2003, 125, 12971 CrossRef PubMed; Y. J. Su, H. L. Huang, C. L. Li, C. H. Chien, Y. T. Tao, P. T. Chou, S. Datta and R. S. Liu, Adv. Mater., 2003, 15, 224 CrossRef PubMed; F. M. Hwang, H. Y. Chen, P. S. Chen, C. S. Liu, Y. Chi, C. F. Shu, F. I. Wu, P. T. Chou, S. M. Peng and G. H. Lee, Inorg. Chem., 2005, 44, 1344 CrossRef PubMed; H. Bronstein, C. E. Finlayson, K. R. Kirov, R. H. Friend and C. K. Williams, Organometallics, 2008, 27, 2980 CrossRef.
- For a very recent example featuring structurally related mononuclear Ir complexes, see: D. N. Chirdon, W. J. Transue, H. N. Kagalwala, A. Kaur, A. B. Maurer, T. Pintauer and S. Bernhard, Inorg. Chem., 2014, 53, 1487 CrossRef CAS PubMed.
- SHELXTL, Bruker AXS Inc., Madison, WI, USA, 2013 Search PubMed; G. M. Sheldrick, SHELXL-2013, University of Göttingen, 2013 Search PubMed.
- A. L. Spek, Acta Crystallogr., Sect. D, 2009, 65, 148 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Tables of crystallographic results, emission spectra of the Ir complexes at 77 K. CCDC 990003 and 990004. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4cc01808g |
| ‡ Data were collected at beamline I19 of Diamond Light Source (λ = 0.6889 Å). Structure solution and refinement20 included modelling of disorder for 3 of the 4 hexyl chains, inclusion of ordered solvent molecules in both structures with the aid of restraints, and SQUEEZE treatment21 of additional disordered solvent in the meso structure. |
| This journal is © The Royal Society of Chemistry 2014 |