 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAn enantioselective synthesis of the C24–C40 fragment of (−)-pulvomycin†
Sandra
Börding
and
Thorsten
Bach
*
Lehrstuhl für Organische Chemie I, Technische Universität München, 85747 Garching, Germany. E-mail: thorsten.bach@ch.tum.de; Fax: +49 89 28913315; Tel: +49 89 28913330
First published on 2nd April 2014
Abstract
The C24–C40 fragment of (−)-pulvomycin was prepared in enantiomerically pure form using a concise synthesis method (15 linear steps from D-fucose, 6.8% overall yield) featuring a diastereoselective addition to an aldehyde, a β-selective glycosylation and a Stille cross-coupling as the key steps.
The antibiotic pulvomycin was first isolated in 1957 from a Streptomyces species but due to the limited analytical data no structure was assigned to the compound.1 In 1963, Akita et al. isolated a natural product from Streptomyces albosporeus var. labilomyceticus,2 which they called labilomycin and which was later shown to be identical to pulvomycin.3 Extensive analytical work by Smith et al. revealed the constitution of the natural product (Fig. 1) as well as the absolute and relative configuration at most stereogenic centers except for C32 and C33.4 The assignment was confirmed and the complete configuration was eventually proven by a crystal structure (1.4 Å resolution) of pulvomycin with the bacterial elongation factor Tu (EF-Tu).5 It is well established that pulvomycin is a potent inhibitor of EF-Tu and it therefore represents a promising lead compound for the development of new antibiotics.6
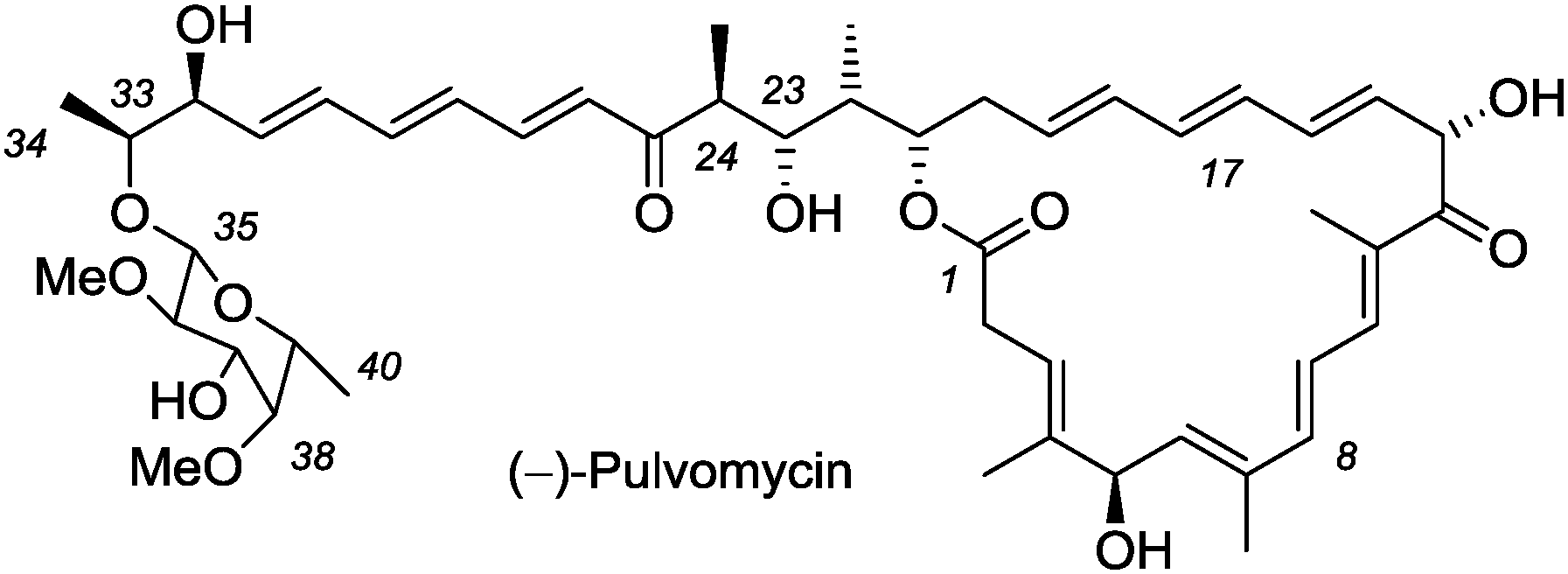 | ||
| Fig. 1 Structure and compound numbering of (−)-pulvomycin. | ||
While synthetic reports on pulvomycin are scarce, the biosynthesis of the pulvomycin aglycone has been elucidated by labeling experiments.7 Our own interest in pulvomycin was triggered by our previous studies on the synthesis8 and antibiotic activity9 of thiazole peptides, such as the GE factors and the amythiamicins. It has been shown that the EF-Tu binding site of pulvomycin is in close proximity to the binding site of thiazole peptides.10 The synthesis of pulvomycin and pulvomycin analogues might consequently help to further investigate the many facets of EF-Tu activity.11 Apart from its biological activity, pulvomycin presents itself as a formidable synthetic challenge due to its complex and labile structure. In this communication we disclose the enantioselective synthesis of a suitably protected C24–C40 fragment 1 (Scheme 1) of pulvomycin.
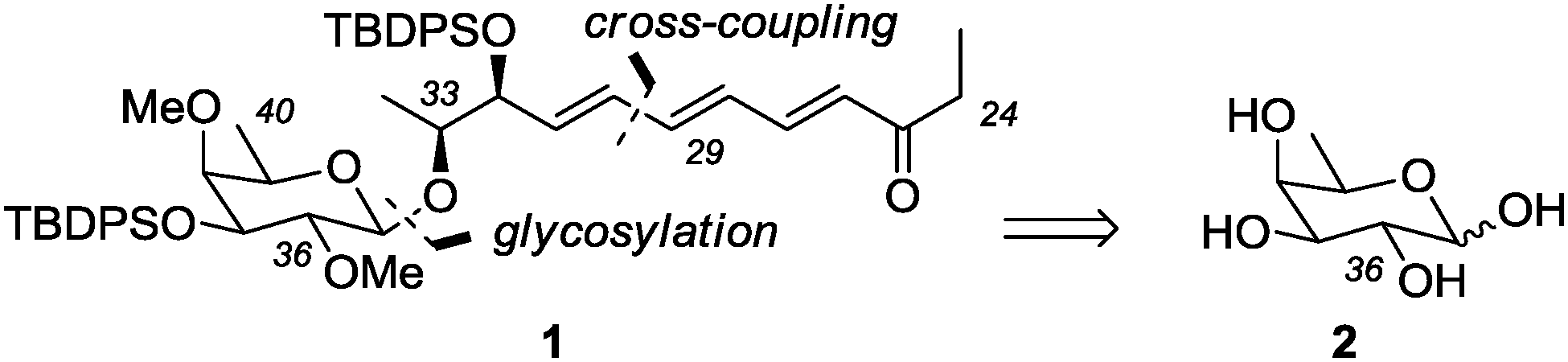 | ||
| Scheme 1 Retrosynthetic disconnection of the title compound 1 leading to D-fucose (2) as an appropriate carbohydrate substrate. | ||
Retrosynthetically, it was envisioned that ketone 1 (TBDPS = tert-butyldiphenylsilyl) could be derived from commercially available D-fucose (2), which shows the correct configuration at the stereogenic centers (C36–C39) of the pyranose ring. In order to establish the desired β-configuration at the glycosidic center an appropriate neighbouring group, e.g. an acetate, was required (at carbon atom C36)12 and the methyl ether linkage was to be introduced after glycosylation. There was precedence for the differentiation of the two equatorial hydroxy groups at C36 and C37 of D-fucose.13
Regarding the C24–C34 fragment, it seemed best to assemble the triene14 after the glycosylation step by an appropriate cross-coupling reaction, e.g. between C29 and C30. The stereogenic center at C33 appeared to be accessible from the chiral pool, e.g. from lactic acid, while the adjacent stereogenic center was to be introduced by a diastereoselective reaction.
The acetylation of D-fucose (2) (Scheme 2) proceeded quantitatively delivering the tetraacetate as an α/β-mixture (α/β = 95/5) of anomers.13 Conversion to the required thioacetal 3 proceeded best in our hands with ethanethiol and BF3·OEt2 in CH2Cl2,15 which delivered depending on the reaction conditions and on the reaction scale variable amounts of separable α/β-isomers (see the ESI† for further details).
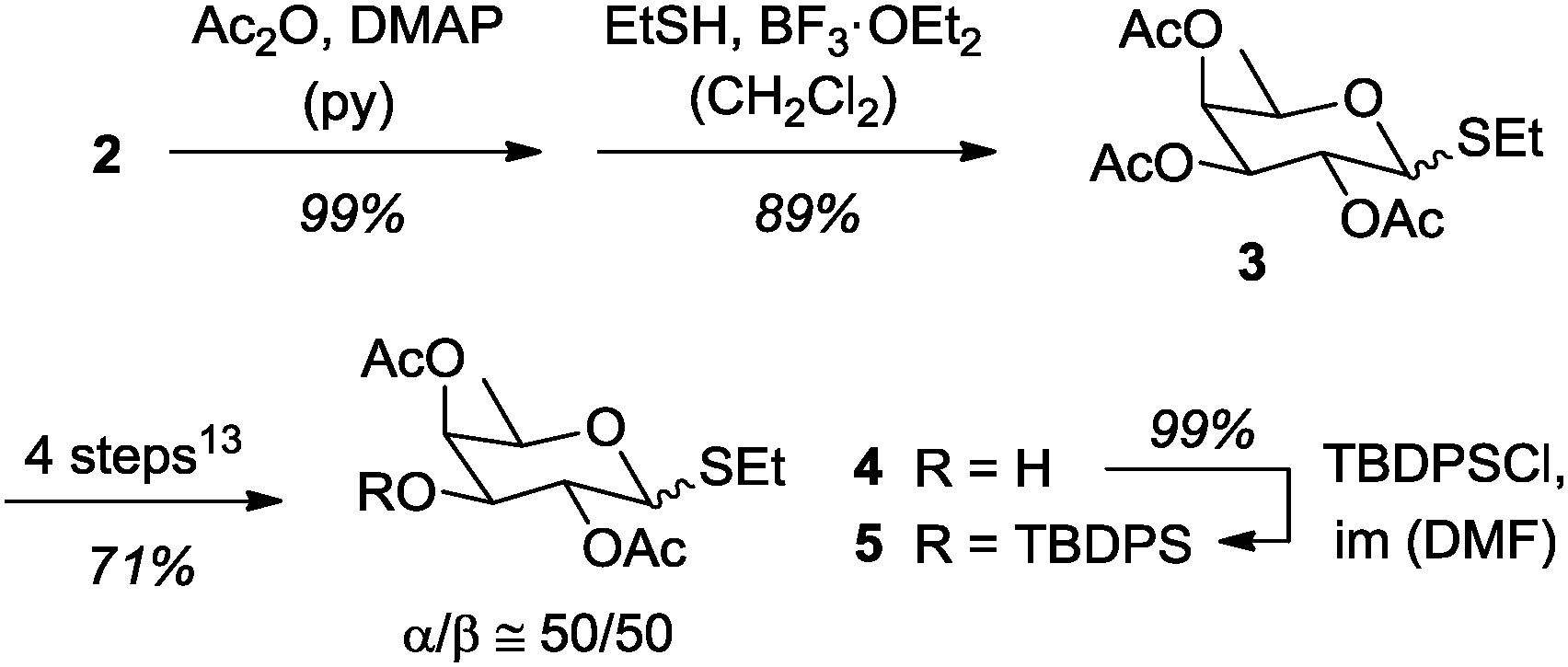 | ||
| Scheme 2 Synthesis of the protected glycosyl donor 5 from D-fucose (2). DMAP = 4-(N,N-dimethylamino)pyridine, py = pyridine, im = imidazole. | ||
Since the relative configuration at the anomeric center was irrelevant for the desired glycosylation reaction, the α/β-mixture of 3 was taken into the four-step procedure previously described for the selective preparation of alcohol β-413 and it furnished the desired product 4 as an α/β-mixture (α/β ≅ 50/50) in a total yield of 60% over six steps from D-fucose (2). Conversion of the equatorial alcohol 4 to silyl ether 5 required elevated temperature (60 °C) and a prolonged reaction time (3 d).
As mentioned above, it was planned to introduce the stereogenic center at C32 by a diastereoselective reaction induced by the adjacent stereogenic center at the carbon atom C33. Surprisingly, the reduction of a (S)-lactate-derived, para-methoxybenzyl (PMB) protected alkynyl ketone16 produced the desired alcohol 7 either in low yields or with insufficient diastereoselectivity (see the ESI† for further details). As an alternative approach, (S)-lactate-derived aldehyde 617 was alkynylated with TMS-acetylene under chelation control18 yielding alcohol 7 and its epimer epi-7 in 81% yield and in a diastereomeric ratio (d.r.) of 87/13 (Scheme 3). The diastereomerically pure product 7 was isolated in 65% yield.
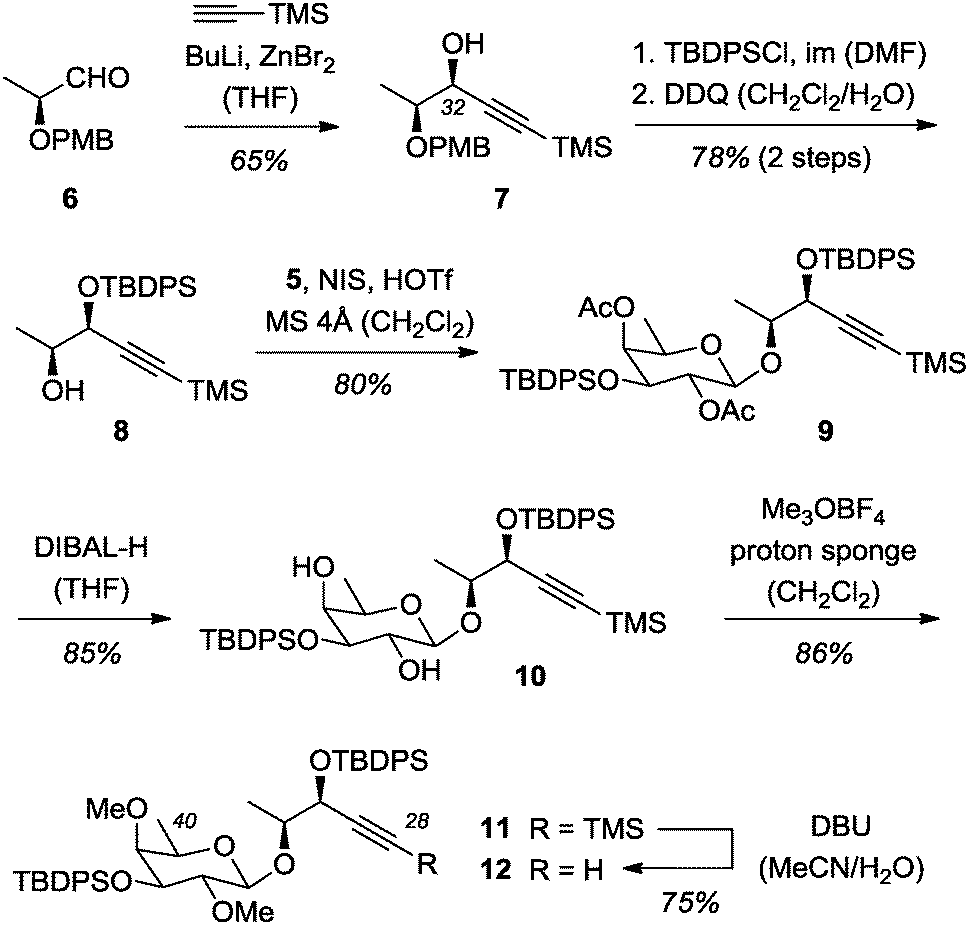 | ||
| Scheme 3 Assembly of the C28–C40 fragment via glycosylation of enantiomerially pure (≥98% ee) alcohol 8 with glycosyl donor 5. | ||
Protection of the secondary alcohol proceeded smoothly at ambient temperature and the PMB group was cleaved oxidatively with 2,3-dicloro-5,6-dicyanobenzoquinone (DDQ)19 to deliver alcohol 8. The enantiomeric excess (ee) of alcohol 8 was established by chiral HPLC analysis and comparison with a racemic sample (see the ESI† for further details). Gratifyingly, the glycosylation reaction, when performed with N-iodosuccinimide (NIS) and trifluoromethanesulfonic acid (HOTf) as activating agents,20 delivered a single diastereomerically pure product 9, which was shown to have the desired β-configuration.21 Reductive removal of the acetyl groups with diisobutylaluminium hydride (DIBAL-H)22 produced 1,3-diol 10, which was converted into the respective dimethylether 11 upon treatment with an excess (10 equiv.) of Meerwein salt and proton sponge [1,8-bis(dimethylamino)naphthalene].23 Less electrophilic methylating reagents (MeOTf, MeI) in combination with appropriate bases failed to react or led to substrate decomposition. Selective desilylation of the alkyne was achieved with 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU).24
With alkyne 12 in hand, various approaches to potential cross-coupling substrates were pursued. It was found that the Pd-catalyzed hydrostannylation with Bu3SnH25 can be successfully performed with alkyne 12 delivering stannane 13 in 44% yield (Scheme 4). Iodide 14 was obtained from stannane 13 upon treatment with iodine in dichloromethane (85% yield).26 The alkyne hept-3-en-1-yne-5-ol27 seemed to be the most suitable precursor for iodide 15 and stannane 16. The compound was available from bis-1,4-(trimethylsilyl)buta-1,3-diyne in four steps and an overall yield of 53% (see the ESI† for further details). Stannylation of hept-3-en-1-yne-5-ol with Bu3SnH was readily achieved employing the Cu-based protocol of Betzer et al.28 to deliver stannane 16 in 79% yield. As for 14, iodide 15 was generated by iodo-de-stannylation employing iodine in dichloromethane (79% yield).
 | ||
| Scheme 4 Stille cross-coupling of building blocks 13 and 15 as key step for the assembly of the title compound. | ||
While attempted Stille cross-coupling reactions29 of stannane 13 and iodide 15 failed, the desired C–C bond formation proceeded smoothly, when performed with the carbohydrate building block as the electrophile. Iodide 14 and stannane 16 underwent a clean cross-coupling employing Pd(MeCN)2Cl2 (10 mol%) as the catalyst.30 Alcohol 17 was obtained in 87% yield and was immediately further oxidized to the desired ketone by treatment with an excess (30 equiv.) of MnO2. Despite a pronounced long wavelength absorption (λmax = 308 nm, ε = 28![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 035 M−1 cm−1 in MeCN), trienone 1 appears to be more stable than alcohol 17 (λmax = 271 nm, ε = 39350 M−1 cm−1 in MeCN; shoulder at λmax = 282 nm, ε = 31180 M−1 cm−1) and could be stored for one week at −25 °C in the dark.
035 M−1 cm−1 in MeCN), trienone 1 appears to be more stable than alcohol 17 (λmax = 271 nm, ε = 39350 M−1 cm−1 in MeCN; shoulder at λmax = 282 nm, ε = 31180 M−1 cm−1) and could be stored for one week at −25 °C in the dark.
In summary, the enantiomerically pure western fragment 1 of (−)-pulvomycin was synthesized in 15 linear steps. The fragment comprises the carbohydrate part (labilose, C35–C40) of the natural product and one of its three triene components (C24–C34). Should an aldol-type reaction of fragment 1 with a suitable Eastern fragment not be successful, stannane 13 and iodide 14 offer suitable options to connect the protected glycoside fragment to the rest of the molecule.
This project was supported by the Deutsche Forschungsgemeinschaft (Ba 1372-18/1), by the TUM Graduate School, and by the Fonds der Chemischen Industrie. Olaf Ackermann and Florian Mayr are acknowledged for help with the HPLC analyses.
Notes and references
- M. Zief, R. Woodside and H. Schmitz, Antibiot. Chemother., 1957, 7, 384–386 CAS.
- (a) E. Akita, K. Maeda and H. Umezawa, J. Antibiot., Ser. A, 1963, 16, 147–151 CAS; (b) E. Akita, K. Maeda and H. Umezawa, J. Antibiot., Ser. A, 1964, 17, 200–217 CAS.
- J. L. Schwartz, M. Tishler, B. H. Arison, H. M. Shafer and S. Omura, J. Antibiot., 1976, 29, 236–241 CrossRef CAS PubMed.
- R. J. Smith, D. H. Williams, J. C. J. Barna, I. R. McDermott, K. Haegele, F. Piriou, J. Wagner and W. Higgins, J. Am. Chem. Soc., 1985, 107, 2849–2857 CrossRef CAS.
- A. Parmeggiani, I. M. Krab, S. Okamura, R. C. Nielsen, J. Nyborg and P. Nissen, Biochemistry, 2006, 45, 6846–6857 CrossRef CAS PubMed.
- For a recent review, see: K. M. G. O'Connell, J. T. Hodgkinson, H. F. Sore, M. Welch, G. P. C. Salmond and D. R. Spring, Angew. Chem., Int. Ed., 2013, 52, 10706–10733 CrossRef PubMed.
- N. D. Priestley and S. Gröger, J. Org. Chem., 1995, 60, 4951–4953 CrossRef CAS.
- (a) O. Delgado, H. M. Müller and T. Bach, Chem. – Eur. J., 2008, 14, 2322–2339 CrossRef CAS PubMed; (b) C. Ammer and T. Bach, Chem. – Eur. J., 2010, 16, 14083–14093 CrossRef CAS PubMed.
- S. Gross, F. Nguyen, M. Bierschenk, D. Sohmen, T. Menzel, I. Antes, D. N. Wilson and T. Bach, ChemMedChem, 2013, 8, 1954–1962 CrossRef CAS PubMed.
- A. Parmeggiani, I. M. Krab, S. Okamura, R. C. Nielsen, J. Nyborg and P. Nissen, Biochemistry, 2006, 45, 6846–6857 CrossRef CAS PubMed.
- Reviews: (a) R. Berisio, A. Ruggiero and L. Vitagliano, Isr. J. Chem., 2010, 50, 71–79 CrossRef CAS; (b) A. Parmeggiani and P. Nissen, FEBS Lett., 2006, 580, 4576–4581 CrossRef CAS PubMed.
- (a) R. U. Lemieux and J.-I. Hayami, Can. J. Chem., 1965, 43, 2162–2173 CrossRef CAS; (b) G.-J. Boons, Contemp. Org. Synth., 1996, 3, 173–200 RSC; (c) T. K. Lindhorst, Essentials of Carbohydrate Chemistry and Biochemistry, Wiley-VCH, Weinheim, 3rd edn, 2007, pp. 157–208 Search PubMed.
- D. Comegna, E. Bedini and M. Parrilli, Tetrahedron, 2008, 64, 3381–3391 CrossRef CAS.
- For selected recent total syntheses of naturally occurring conjugated (E,E,E)-trienes, see: (a) D. J. Del Valle and M. J. Krische, J. Am. Chem. Soc., 2013, 135, 10986–10989 CrossRef CAS PubMed; (b) C. Jahns, T. Hoffmann, S. Müller, K. Gerth, P. Washausen, G. Höfle, H. Reichenbach, M. Kalesse and R. Müller, Angew. Chem., Int. Ed., 2012, 51, 5239–5243 CrossRef CAS PubMed; (c) M. Yoshino, K. Eto, K. Takahashi, J. Ishihara and S. Hatakeyama, Org. Biomol. Chem., 2012, 10, 8164–8174 RSC; (d) P. G. E. Craven and R. J. K. Taylor, Tetrahedron Lett., 2012, 53, 5422–5425 CrossRef CAS; (e) H. J. Jessen, A. Schumacher, F. Schmid, A. Pfaltz and K. Gademann, Org. Lett., 2011, 13, 4368–4370 CrossRef CAS PubMed; (f) D. Amans, V. Bellosta and J. Cossy, Chem. – Eur. J., 2009, 15, 3457–3473 CrossRef CAS PubMed; (g) M. T. Crimmins, H. S. Christie, A. Long and K. Chaudhary, Org. Lett., 2009, 11, 831–834 CrossRef CAS PubMed; (h) I. S. Mitchell, G. Pattenden and J. Stonehouse, Org. Biomol. Chem., 2005, 3, 4412–4431 RSC.
- P. Sjölin, S. K. George, K.-E. Bergquist, S. Roy, A. Svensson and J. Kihlberg, J. Chem. Soc., Perkin Trans. 1, 1999, 1731–1742 RSC.
- The ketone was prepared from literature known Weinreb amide ( V. Convertino, P. Manini, W. B. Schweizer and F. Diederich, Org. Biomol. Chem., 2006, 4, 1206–1208 CAS ) by substitution with the respective magnesium acetylide (see the ESI† for further details).
- W. Yu, Y. Zhang and Z. Jin, Org. Lett., 2001, 3, 1447–1450 CrossRef CAS PubMed.
- K. T. Mead, Tetrahedron Lett., 1987, 28, 1019–1022 CrossRef CAS.
- Y. Oikawa, T. Yoshioka and O. Yonemitsu, Tetrahedron Lett., 1982, 23, 885–888 CrossRef CAS.
- (a) G. H. Veeneman, S. H. van Leeuwen and J. H. van Boom, Tetrahedron Lett., 1990, 31, 1331–1334 CrossRef CAS; (b) P. Konradsson, U. E. Udodong and B. Fraser-Reid, Tetrahedron Lett., 1990, 31, 4313–4316 CrossRef CAS.
- 1H-NMR data of glycoside 9 (500 MHz, CDCl3): δ (ppm) = 0.09 [s, 9H; Si(CH3)3], 0.97 [s, 9H; C(CH3)3], 0.96–0.99 (m, 3H; H3-40), 1.05 [s, 9H; C(CH3)3], 1.30 (d, 3J = 6.3 Hz, 3H; H3-34), 1.46 (s, 3H; C(O)CH3-36), 2.12 [s, 3H; C(O)CH3-38], 3.05 (q, 3J = 6.4 Hz, 1H; H-39), 3.41 (qd, 3J = 6.3, 3.9 Hz, 1 H; H-33), 3.44 (d, 3J = 8.1 Hz, 1 H; Hβ-35), 3.60 (dd, 3J = 9.8, 3.3 Hz, 1H; H-37), 4.19 (d, 3J = 3.9 Hz, 1H; H-32), 4.79 (d, 3J = 3.3 Hz, 1H; H-38), 5.06 (dd, 3J = 9.8, 8.1 Hz, 1H; H-36), 7.23–7.27 (m, 4H; Harom), 7.30–7.48 (m, 10H; Harom), 7.55–7.58 (m, 2H; Harom), 7.60–7.65 (m, 4H; Harom), 7.69–7.73 (m, 2H; Harom).
- F. E. McDonald and M. Wu, Org. Lett., 2002, 4, 3979–3981 CrossRef CAS PubMed.
- B. Wang, T. M. Hansen, T. Wang, D. Wu, L. Weyer, L. Ying, M. M. Engler, M. Sanville, C. Leitheiser, M. Christmann, Y. Lu, J. Chen, N. Zunker, R. D. Cink, F. Ahmed, C.-S. Lee and C. J. Forsyth, J. Am. Chem. Soc., 2010, 133, 1484–1505 CrossRef PubMed.
- C.-E. Yeom, M. J. Kim, W. Choi and B. M. Kim, Synlett, 2008, 565–568 CAS.
- (a) H. X. Zhang, F. Guibe and G. Balavoine, J. Org. Chem., 1990, 55, 1857–1867 CrossRef CAS; (b) J. R. Frost, C. M. Pearson, T. N. Snaddon, R. A. Booth and S. V. Ley, Angew. Chem., Int. Ed., 2012, 51, 9366–9371 CrossRef CAS PubMed.
- R. Alvarez, M. Herrero, S. López and A. R. de Lera, Tetrahedron, 1998, 54, 6793–6810 CrossRef CAS.
- K. Green, J. W. Keeping and V. Thaller, J. Chem. Res., Synop., 1985, 103 CAS; K. Green, J. W. Keeping and V. Thaller, J. Chem. Res., Miniprint, 1985, 1260–1267 Search PubMed.
- J.-F. Betzer, F. Delaloge, B. Muller, A. Pancrazi and J. Prunet, J. Org. Chem., 1997, 62, 7768–7780 CrossRef CAS.
- Reviews: (a) J. K. Stille, Angew. Chem., Int. Ed., 1986, 25, 508–524 CrossRef; (b) T. N. Mitchell, Synthesis, 1992, 803–815 CrossRef CAS; (c) V. Farina, V. Krishnamurthy and W. J. Scott, Org. React., 1997, 50, 1–652 CAS.
- J. K. Stille and B. L. Groh, J. Am. Chem. Soc., 1987, 109, 813–817 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4cc01338g |
| This journal is © The Royal Society of Chemistry 2014 |