 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A gold-catalysed fully intermolecular oxidation and sulfur-ylide formation sequence on ynamides†
Mickaël Dos
Santos
and
Paul W.
Davies
*
School of Chemistry, University of Birmingham, Edgbaston, B15 2TT, Birmingham, UK. E-mail: p.w.davies@bham.ac.uk; Tel: +44 (0)121 4144408
First published on 16th April 2014
Abstract
An efficient C–O, C–S and C–C bond-forming sequence leads to functionalised compounds bearing sulfur-substituted quaternary carbons. Ynamides are employed as diazo-equivalents to access the [2,3]-sigmatropic rearrangements of allyl sulfonium ylides by a three-component chemoselective oxidation and intermolecular ylide formation.
The [2,3]-sigmatropic rearrangement of allyl sulfonium ylides is a potent method for the formation of Csp3–Csp3 bonds.1 A significant hydrocarbon functionalisation process is achieved when the rearrangement is coupled with in situ ylide formation through reaction of a sulfide with a metal carbene, formed in situ from a sacrificial functionality such as the diazo-group (Doyle–Kirmse reaction, Scheme 1).2,3 We4 and others5 have been engaged in efforts to access the synthetic potential of these ylides using methods to generate carbenoids that avoid the pre-installation and use of potentially hazardous high-energy diazo groups. Our initial studies established that sulfur ylides can be prepared from the intermolecular reaction of sulfides with gold-carbenes (Scheme 2).4a,d,6 However, the use of propargylic carboxylates as carbenoid precursors impacted on the subsequent ylidic rearrangements to generally afford products isomeric to those from [2,3]-sigmatropic pathways. More-congested centres and synthetically valuable sulfur-substituted quaternary carbons were inaccessible as terminal alkynes and unsubstituted allyl groups on the sulfide7 were required.4a,d We subsequently established that sigmatropic-rearrangements of sulfur ylides were accessible through an intramolecular gold or platinum catalysed cycloisomerisation of alkynyl allyl sulfoxides.4b,8 Here we report a diazo-free oxidation-ylide formation sequence to access sulfur-ylide rearrangements by a selective and efficient fully intermolecular transformation of ynamides.
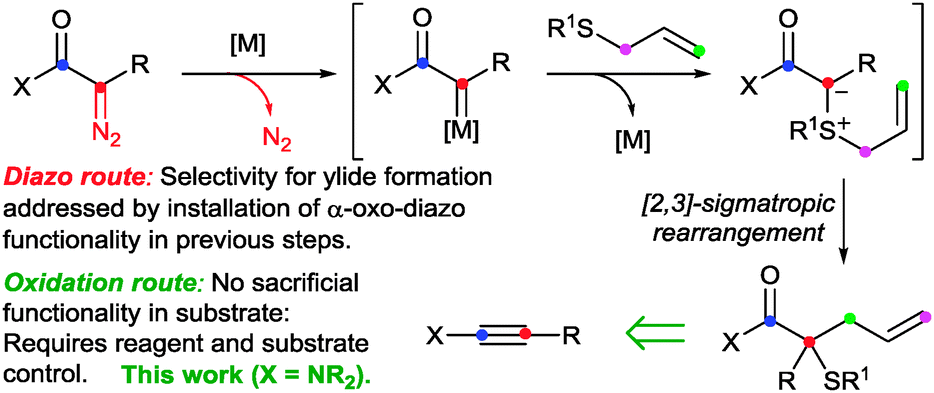 | ||
| Scheme 1 The Doyle–Kirmse reaction and an alternate oxidation based route. | ||
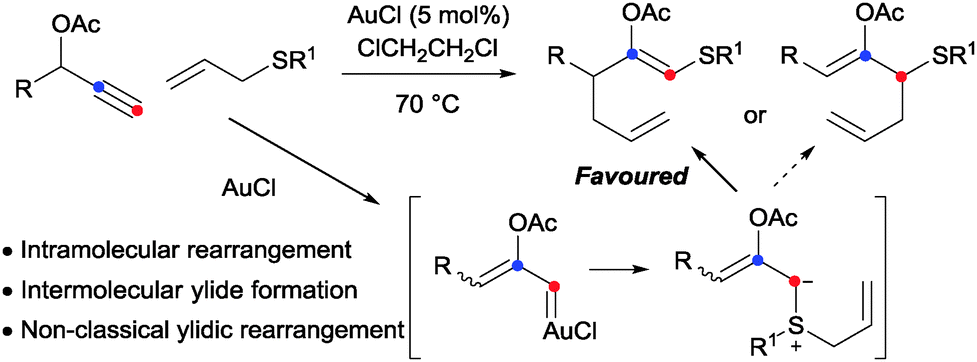 | ||
| Scheme 2 Intermolecular trapping of gold carbenes with allyl sulfides: ref. 4a,d | ||
In light of our,9 and others10 studies into gold-catalysed intermolecular atom/group-transfer onto ynamides A,11 we questioned whether ynamides could be used to replace diazo compounds in intermolecular sulfonium ylide formation to form quaternary carbons (Schemes 1 and 3).
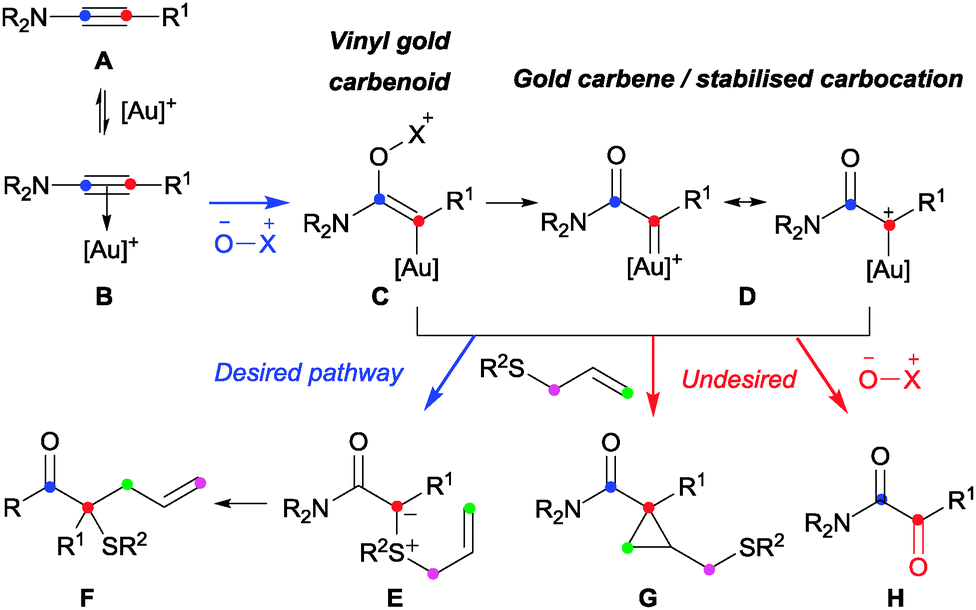 | ||
| Scheme 3 A gold-catalysed intermolecular oxidation – intermolecular trapping manifold for ynamides. | ||
Significant transformations have resulted from α-oxo-gold carbene formation by intermolecular oxidation of a C–C triple bond.9,10,12 While these predominantly feature subsequent intramolecular cyclisations, there are striking exceptions in the terminal alkyne series where the mono-substituted gold carbene is trapped with intermolecular oxygen-, nitrogen- and halide nucleophiles other than the oxidant.13 Similar three-component couplings have not however been reported in the ynamide series and it is notable that the resulting 1,1-disubstituted organogold species C/D appears prone to oxidation even when intramolecular pathways are available.9,10,14 A successful outcome therefore requires a high level of reagent-compatibility and selectivity: the sulfide must not affect activation or oxidation of the ynamide (A → B → C).15 Ylide formation must then compete successfully with cyclopropanation (G) and oxidation (H) of organogold species C and/or D.
After exploring a variety of parameters including catalyst, solvent and oxidant with various ynamides (see ESI†), reaction conditions were found to effect the complexity-increasing cascade from ynamides 1 into the functionalised tertiary thioethers 4 (Scheme 4). Competing double-oxidation to form imido-ketones (H) was observed throughout this study alongside incomplete consumption of 1, and this pathway was exclusively observed using Au(III) or NHC–Au(I) catalysts. However, good conversion and selectivity for the formation of 4 was achieved using a combination of cationic gold phosphite complex Au-I alongside methylpicolinate N-oxide 3. The reactions were run in dichloromethane at room temperature and reagents 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2:3 were added at the start of the reaction in a near-stoichiometric 1:1.2:1.3 ratio respectively (Scheme 4). Recourse to continual or portion-wise slow-addition strategies that maintain a low concentration of oxidant to favour reaction with the desired nucleophile was avoided. This preference was maintained throughout the study in order to explore the extent and influences on the observed chemoselective preference for sulfide attack at the organogold intermediate.13,16
:2:3 were added at the start of the reaction in a near-stoichiometric 1:1.2:1.3 ratio respectively (Scheme 4). Recourse to continual or portion-wise slow-addition strategies that maintain a low concentration of oxidant to favour reaction with the desired nucleophile was avoided. This preference was maintained throughout the study in order to explore the extent and influences on the observed chemoselective preference for sulfide attack at the organogold intermediate.13,16
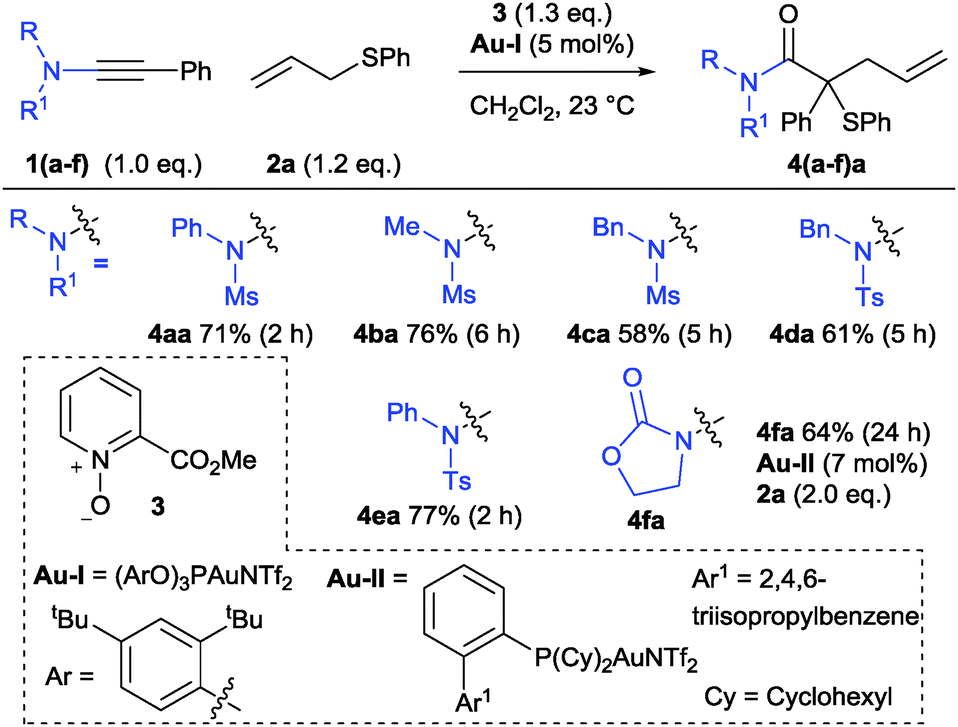 | ||
| Scheme 4 The ynamide-based Doyle–Kirmse type reaction. | ||
Ynamides bearing N-phenyl and N-methyl groups reacted similarly (4bavs.4aa) while an N-benzyl equivalent was less suited affording a lower yield of 4ca with competing over-oxidation observed.17 The trend between N-phenyl and N-methyl was maintained in the p-tolylsulfonamide series, albeit with slightly higher yields (Scheme 4, 4da and 4ea). In both cases, products from cyclisation of C/D onto the N-substituent were not observed. An ynamide prepared from imidazole was unreactive. Oxazolidinone-derived ynamide 1f could be used to form 4fa but greater conversion and selectivity for ylide formation was achieved using a bulky phosphine-derived catalyst Au-II in place of the phosphite–gold complex, identifying potentially significant influencing factors between different types of ynamides (see ESI†).
The sulfonamide-derived ynamides were used throughout the remainder of this study into structural effects on the relative efficacy of the standard conditions. The reaction remained effective in the presence of S-aryl, S-benzyl- and S-alkyl allyl sulfides 2b–e as well as bisallylsulfide 2f (Scheme 5). Unsurprisingly, a more electron-deficient aryl bromide substituent renders the ylide pathway less competitive against oxidation with ∼30% yield of the over-oxidation product determined from the crude reaction mixture.
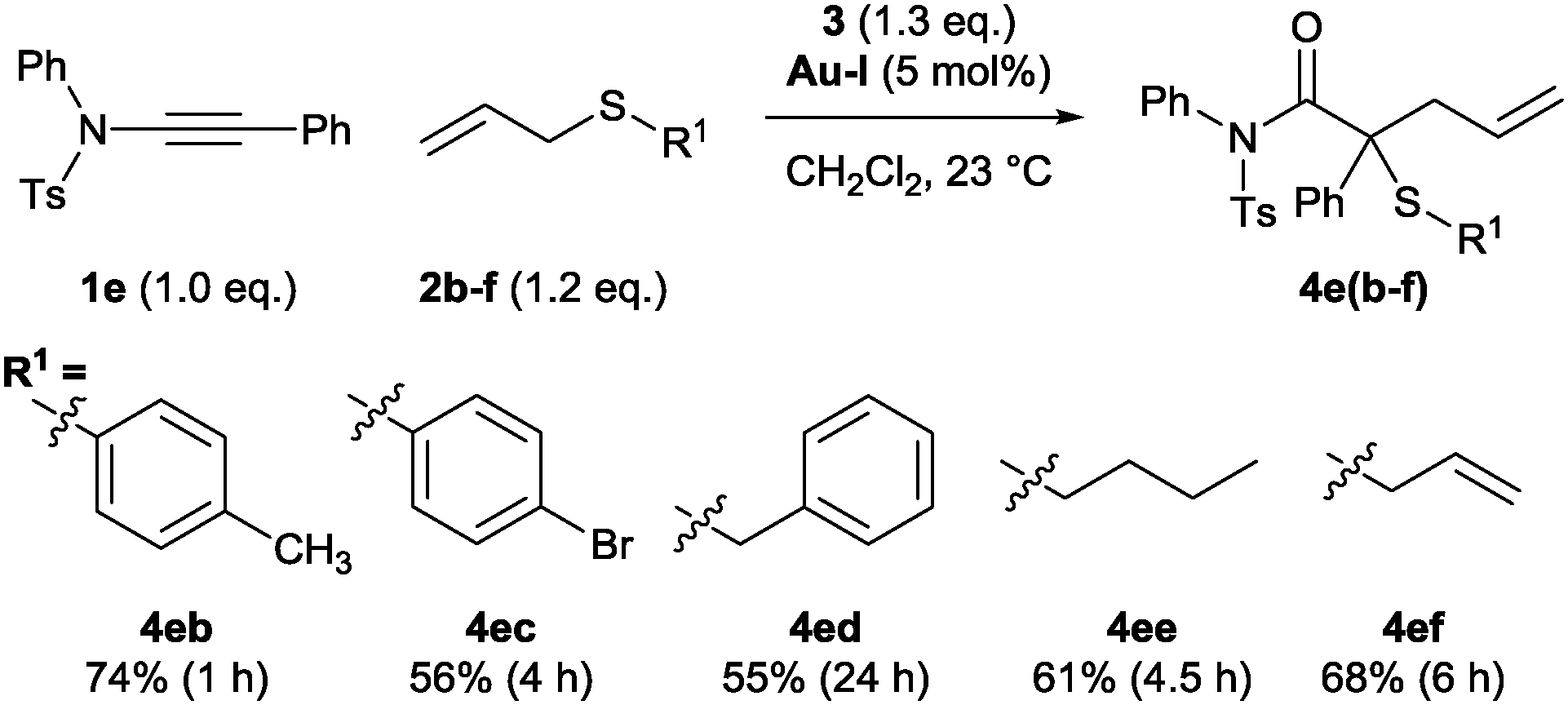 | ||
| Scheme 5 Effect of the non-migrating group on the allyl sulfide. | ||
The role of the ynamide C-substituent was next tested under the standard conditions (Scheme 6). None of the desired product from ylide formation was observed when anisole-derived ynamide 1g was reacted with sulfides 2a or 2b with the over-oxidation product 5 instead predominating. A preference for over-oxidation against an intramolecular reaction has recently been reported when using p-anisole derived ynamide.10f In contrast, both inductively and mesomerically electron-withdrawing groups were well tolerated (4hb and 4ib). Alkyl-substituted gold carbenoids are prone to 1,2-insertion9a and this intramolecular pathway was indeed preferred, affording 6 with low conversion of the hexyne-derived ynamide 1j. A conjugated ene-ynamide 1k did not follow a recently reported 4-π-electrocyclisation pathway10c instead reacting productively to afford functionalised tertiary allylic thioether 4ka.
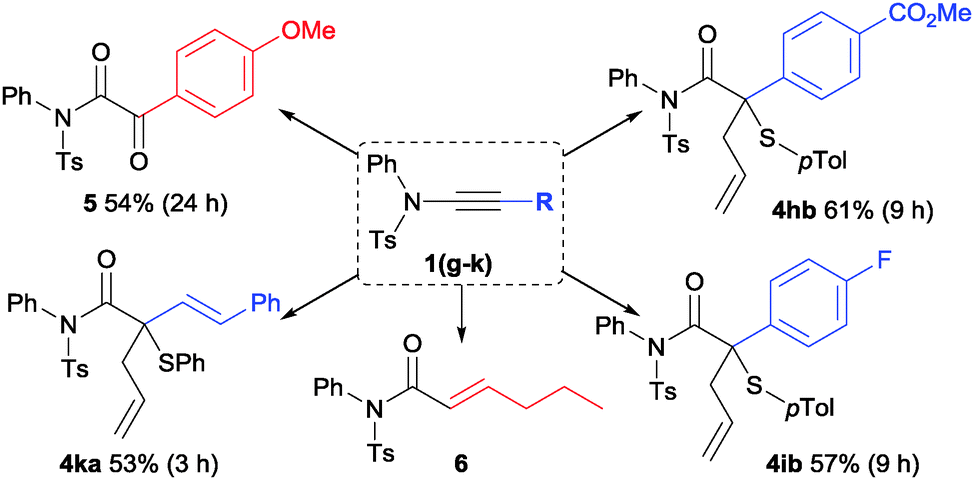 | ||
| Scheme 6 Effect of the ynamide C-substituent on the reaction outcome. | ||
By analogy to the effects encountered in diazo-derived metal carbene chemistry, changing the ynamide C-substituent was expected to influence the reactivity of the resulting carbenoid.2 However, the impact of the electronic influence on the chemoselectivity of these ynamide-based reactions is informative: the formation of the desired compound 4 requires the active organogold species to react preferentially with the polarisable sulfur nucleophile 2 in preference to the dipolar oxygen-nucleophile 3. Nucleophilic attack can occur either alongside- or after elimination of methylpicolinate (Scheme 3, C → Evs.C → D → E).8–10,12,13 Considering the contrasting natures of the nucleophiles, a favourable attack of the sulfide on the vinyl gold carbenoid C, bearing a relatively-uncharged carbon centre, appears more likely than on the cationic carbon of gold carbene D. The strongly π-acidic phosphite ligand affords significant cationic character to D by decreasing the ability of gold to donate electron density.18 However, these same electronic characteristics should aid the desired chemoselectivity by disfavouring the elimination of methylpicolinate required to form D. On this basis, the reactivity of electron-rich ynamide 1g (R1 = p-MeOC6H4) can be explained by the mesomeric contribution from the anisole MeO-group aiding elimination of the nucleofuge to form D and disfavouring ylide formation.
Sulfides with substituted allyl units were then explored to ascertain if a [2,3]-sigmatropic rearrangement was operative. Pleasingly, and in contrast with other systems,7 both 2-vinyltetrahydrothiophene 2g and cinnamyl derivative 2h underwent clean reactions. The functionalised 8-membered sulfur heterocycle 4eg and the tertiary thioether bearing a vicinal tertiary stereogenic centre 4eh are the products expected from sulfur-ylide formation and [2,3]-sigmatropic rearrangement with allylic inversion (Scheme 7).‡2,6,19 Interestingly, formation of 4eh proceeded with higher diastereoselectivity than is normally observed from the reaction of substituted allyl sulfides 2h with metal carbenes derived from donor–acceptor diazocompounds.6,19 The relative stereochemistry of the major diastereoisomer was confirmed by X-ray analysis (Scheme 7) and is consistent with a preference for the cinnamyl phenyl group to be positioned anti to the amide group across an envelope transition state. Rapid access to these N,N-disubstituted amide derivatives by the ynamide strategy may therefore provide additional synthetic advantages.
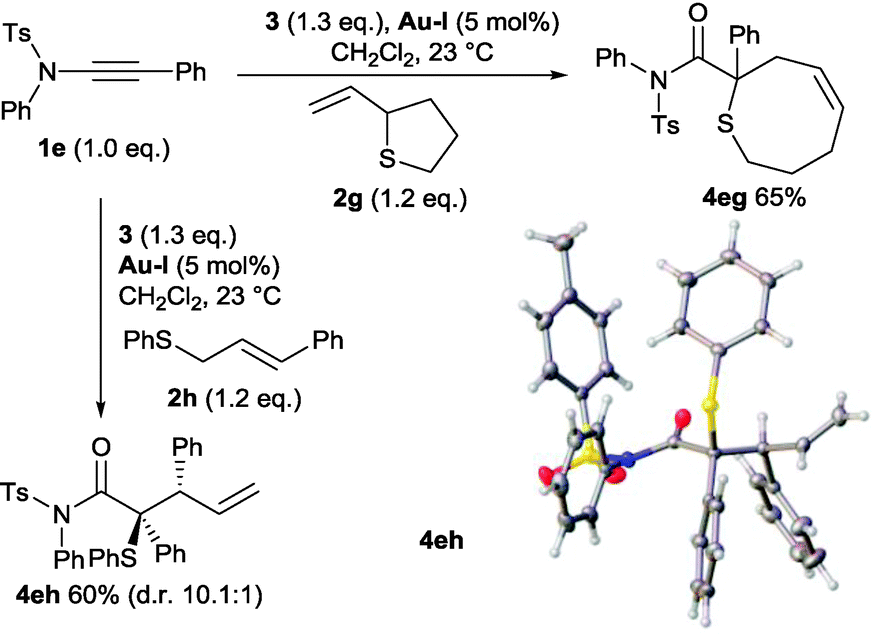 | ||
| Scheme 7 Reactions of ynamide-derived sulfur ylides displaying allylic inversion. Crystal structure of 4eh with ellipsoids drawn at the 50% probability level. | ||
In conclusion, a gold-catalysed cascade reaction is reported for the practical complexity-increasing synthesis of functionalised tertiary thioethers directly from ynamides. Selectivity for sulfur ylide formation over competing processes is rationalised and can be achieved when all reagents are added at the start of the process with a near-stoichiometric reagent loading. The products are consistent with those expected from [2,3]-sigmatropic rearrangement of allyl sulfonium ylides. This report demonstrates that ynamides can be used as synthetically attractive replacements to 1,1-disubstituted diazocompounds in intermolecular processes.
The authors thank the European Union FP7 Marie-Curie IEF for funding (Fellowship to MDS: ESAMY). We thank the EPSRC UK National Crystallography Service at the University of Southampton for the collection of the crystallographic data20 and Dr Louise Male (University of Birmingham) for its analysis. The facilities used in this research were part supported through Birmingham Science City AM2 by Advantage West Midlands and the European Regional Development Fund.
Notes and references
- Reviews: (a) J. S. Clark, Nitrogen, Oxygen and Sulfur Ylide Chemistry: A Practical Approach in Chemistry, Oxford University Press, Oxford, 2002 Search PubMed; (b) Sulfur Mediated Rearrangements: Topics in Current Chemistry, ed. E. Schaumann, vol. 275, Springer, Heidelberg, 2007 Search PubMed.
- (a) W. Kirmse and M. Kapps, Chem. Ber., 1968, 101, 994 CrossRef CAS . Reviews: ; (b) M. P. Doyle, W. H. Tamblyn and V. Bagheri, J. Org. Chem., 1981, 46, 5094 CrossRef CAS; (c) M. P. Doyle, M. A. McKervey and T. Ye, Modern Catalytic Methods for Organic Synthesis with Diazo Compounds, Wiley-Interscience, New York, 1998 Search PubMed; (d) A.-H. Li, L.-X. Dai and V. K. Aggarwal, Chem. Rev., 1997, 97, 2341 CrossRef CAS PubMed; (e) D. M. Hodgson, F. Y. T. M. Pierard and P. A. Stupple, Chem. Soc. Rev., 2001, 30, 50 RSC ; for a recent example; (f) M. S. Holzwarth, I. Alt and B. Plietker, Angew. Chem., Int. Ed., 2012, 51, 5351 CrossRef CAS PubMed.
- Use of tosyl hydrazone diazo-precursors: (a) V. K. Aggarwal and C. L. Winn, Acc. Chem. Res., 2004, 37, 611 CrossRef CAS PubMed; (b) Y. Li, Z. Huang, X. Wu, P.-F. Xu, J. Jin, Y. Zhang and J. Wang, Tetrahedron, 2012, 68, 5234 CrossRef CAS.
- (a) P. W. Davies and S. J.-C. Albrecht, Chem. Commun., 2008, 238 RSC; (b) P. W. Davies and S. J.-C. Albrecht, Angew. Chem., Int. Ed., 2009, 48, 8372 CrossRef CAS PubMed; (c) P. W. Davies, Pure Appl. Chem., 2010, 82, 1537 CrossRef CAS; (d) P. W. Davies and S. J.-C. Albrecht, Synlett, 2012, 70 CrossRef CAS.
- (a) Y. Kato, K. Miki, K. F. Nishino, K. Ohe and S. Uemura, Org. Lett., 2003, 5, 2619 CrossRef CAS PubMed; (b) D. Yadagiri and P. Anbarasan, Chem. – Eur. J., 2013, 19, 15115 CrossRef CAS PubMed.
- P. W. Davies, S. J.-C. Albrecht and G. Assanelli, Org. Biomol. Chem., 2009, 7, 1276 CAS.
- Cyclopropanation not ylide formation was observed when allyl sulfides bearing more substituted alkenes were employed, see ref. 4d.
- Alkynyl sulfoxide reactions: (a) N. D. Shapiro and F. D. Toste, J. Am. Chem. Soc., 2007, 129, 4160 CrossRef CAS PubMed; (b) G. Li and L. Zhang, Angew. Chem., Int. Ed., 2007, 46, 5156 CrossRef CAS PubMed; (c) B. Lu, Y. Li, Y. Wang, D. H. Aue, Y. Luo and L. Zhang, J. Am. Chem. Soc., 2013, 135, 8512 CrossRef CAS PubMed.
- (a) P. W. Davies, A. Cremonesi and N. Martin, Chem. Commun., 2011, 47, 379 RSC; (b) P. W. Davies, A. Cremonesi and L. Dumitrescu, Angew. Chem., Int. Ed., 2011, 50, 8931 CrossRef CAS PubMed.
- (a) D. Vasu, H.-H. Hung, S. Bhunia, S. A. Gawade, A. Das and R.-S. Liu, Angew. Chem., Int. Ed., 2011, 50, 6911 CrossRef CAS PubMed; (b) A. Mukherjee, R. B. Dater, R. Chaudhuri, S. Bhunia, S. N. Karad and R.-S. Liu, J. Am. Chem. Soc., 2011, 133, 15372 CrossRef CAS PubMed; (c) S. Bhunia, C.-J. Chang and R.-S. Liu, Org. Lett., 2012, 14, 5522 CrossRef CAS PubMed; (d) R. B. Dateer, K. Pati and R.-S. Liu, Chem. Commun., 2012, 48, 7200 RSC; (e) L.-Q. Yang, K.-B. Wang and C.-Y. Li, Eur. J. Org. Chem., 2013, 2775 CrossRef CAS; (f) K.-B. Wang, R.-Q. Ran, S.-D. Xiu and C.-Y. Li, Org. Lett., 2013, 15, 2374 CrossRef CAS PubMed; (g) R. Liu, G. N. Winston-McPherson, Z.-Y. Yang, X. Zhou, W. Song, I. A. Guzei, X. Xu and W. Tang, J. Am. Chem. Soc., 2013, 135, 8201 CrossRef CAS PubMed; Sulfoxides: (h) C.-W. Li, K. Pati, G.-Y. Lin, S. A. Sohel, H.-H. Hung and R.-S. Liu, Angew. Chem., Int. Ed., 2010, 49, 9891 CrossRef CAS PubMed; (i) C.-F. Xu, M. Xu, Y.-X. Jia and C.-Y. Li, Org. Lett., 2011, 13, 1556 CrossRef CAS PubMed ; Nitrene addition; (j) C. Li and L. Zhang, Org. Lett., 2011, 13, 1738 CrossRef CAS PubMed.
- Ynamide preparations: (a) Y. Zhang, R. P. Hsung, M. R. Tracey, K. C. M. Kurtz and E. L. Vera, Org. Lett., 2004, 6, 1151 CrossRef CAS PubMed; (b) A. Coste, G. Karthikeyan, F. Couty and G. Evano, Angew. Chem., Int. Ed., 2009, 48, 4381 CrossRef CAS PubMed; (c) T. Hamada, X. Ye and S. S. Stahl, J. Am. Chem. Soc., 2008, 130, 833 CrossRef CAS PubMed ; Reviews; (d) K. A. DeKorver, H. Li, A. G. Lohse, R. Hayashi, Z. Lu, Y. Zhang and R. P. Hsung, Chem. Rev., 2010, 110, 5064 CrossRef CAS PubMed; (e) G. Evano, A. Coste and K. Jouvin, Angew. Chem., Int. Ed., 2010, 49, 2840 CrossRef CAS PubMed.
- For the first report and then representative examples from other groups: (a) L. Ye, W. He and L. Zhang, J. Am. Chem. Soc., 2010, 132, 8550 CrossRef CAS PubMed; (b) L. Zhang, Acc. Chem. Res., 2014, 47, 877 CrossRef CAS PubMed; (c) M. Xu, T.-T. Ren and C.-Y. Li, Org. Lett., 2012, 14, 4902 CrossRef CAS PubMed; (d) A. S. K. Hashmi, T. Wang, S. Shi and M. Rudolph, J. Org. Chem., 2012, 77, 7761 CrossRef CAS PubMed; (e) D. Qian and J. Zhang, Chem. Commun., 2012, 48, 7082 RSC; (f) J. Fu, H. Shang, Z. Wang, L. Chang, W. Shao, Z. Yang and Y. Tang, Angew. Chem., Int. Ed., 2013, 52, 4198 CrossRef CAS PubMed; (g) S. Bhunia, S. Ghorpade, D. B. Huple and R.-S. Liu, Angew. Chem., Int. Ed., 2013, 52, 4229 CrossRef PubMed; (h) G. Henrion, T. E. J. Chavas, X. Le Goff and F. Gagosz, Angew. Chem., Int. Ed., 2013, 52, 6277 CrossRef CAS PubMed.
- (a) W. He, L. Xie, Y. Xu, J. Xiang and L. Zhang, Org. Biomol. Chem., 2012, 10, 3168 RSC; (b) Y. Luo, K. Ji, Y. Li and L. Zhang, J. Am. Chem. Soc., 2012, 134, 17412 CrossRef CAS PubMed; (c) K. Ji, Y. Zhao and L. Zhang, Angew. Chem., Int. Ed., 2013, 52, 6508 CrossRef CAS PubMed; (d) L. Xie, Z. Liang, D. Yan, W. He and J. Xiang, Synlett, 2013, 1809 CAS; (e) C. Wu, Z. Liang, D. Yan, W. He and J. Xiang, Synthesis, 2013, 2605 Search PubMed.
- For two-component intermolecular metathesis, see ref. 10b.
- Gold carbenes with thiols: R. J. Mudd, P. C. Young, J. A. Jordan-Hore, G. M. Rosair and A.-L. Lee, J. Org. Chem., 2012, 77, 7633 CrossRef CAS PubMed.
- Regular portionwise addition of 3 affords 81% (by 1H NMR) by maintaining a low concentration of oxidant relative to the sulfide.
- Co-elution prevented accurate yields of over-oxidised product.
- D. Benitez, N. D. Shapiro, E. Tkatchouk, Y. Wang, W. A. Goddard III and F. D. Toste, Nat. Chem., 2009, 1, 482 CrossRef CAS PubMed.
- (a) T. Fukudat, R. Irie and T. Katsuki, Tetrahedron, 1999, 55, 649 CrossRef; (b) S. Kitagaki, Y. Yanamoto, H. Okubo, M. Nakajima and S. Hashimoto, Heterocycles, 2001, 54, 623 CrossRef CAS; (c) Y. Nishibayashi, K. Ohe and S. Uemura, J. Chem. Soc., Chem. Commun., 1995, 1245 RSC; (d) X. Zhang, Z. Qu, Z. Ma, W. Shi, X. Jin and J. Wang, J. Org. Chem., 2002, 67, 5621 CrossRef CAS PubMed.
- S. J. Coles and P. A. Gale, Chem. Sci., 2012, 3, 683 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Synthetic procedures and characterisation for all compounds. CCDC 984457. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4cc01059k |
| ‡ Crystal data for 4eh: C36H31NO3S2, M = 589.74, monoclinic, a = 16.0175(3) Å, b = 9.6290(2) Å, c = 19.5279(5) Å, β = 98.570(1)°, U = 2978.2(1) Å3, T = 120(2) K, space group P21/c, Z = 4, 34108 reflections measured, 6813 unique (Rint = 0.0607) which were used in all calculations. The final R1 was 0.0458 (I > 2σ(I)) and wR(F2) was 0.1150 (all data). |
| This journal is © The Royal Society of Chemistry 2014 |