 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA two-step O- to C-glycosidic bond rearrangement using complementary glycosyltransferase activities†
Alexander
Gutmann
,
Corinna
Krump
,
Linda
Bungaruang
and
Bernd
Nidetzky
*
Institute of Biotechnology and Biochemical Engineering, Graz University of Technology, Petersgasse 12/1, A-8010 Graz, Austria. E-mail: bernd.nidetzky@tugraz.at
First published on 25th March 2014
Abstract
An efficient 2′-O- to 3′-C-β-D-glucosidic bond rearrangement on the dihydrochalcone phloretin to convert phlorizin into nothofagin was achieved by combining complementary O-glycosyltransferase (OGT) and C-glycosyltransferase (CGT) activities in a one-pot transformation containing catalytic amounts of uridine 5′-diphosphate (UDP). Two separate enzymes or a single engineered dual-specific O/CGT were applied. Overall (quantitative) conversion occurred in two steps via intermediary UDP–glucose and phloretin.
Leloir glycosyltransferases (GTs) are selective catalysts of synthetically useful glycosylation reactions.1,2 Naturally they catalyse glycosyl transfer from an activated donor, typically a nucleoside-diphosphate (NDP) sugar, onto metabolic target acceptor(s) (ESI,† Scheme S1a).3 Recognising the synthetic scope of glycosyltransferase reactions run backwards, researchers introduced two-step exchange reactions to the field (ESI,† Scheme S1b).4–6 Complementary glycosyltransferase activities are combined in a one-pot conversion where NDP-sugar or an acceptor substrate for the actual synthetic transformation is generated in situ from a reactant glycoside and NDP via a reverse glycosyltransferase reaction. The overall catalytic conversion is steered to achieve swapping of glycosyl residues between different acceptor substrates (aglycon exchange),4,6–10 or to result in an alternative glycosylation of a single acceptor compound (sugar exchange).4,7,11 Exchange processes were exploited in different glycosylations of small molecules and enabled glycoengineering of natural products in vitro.12
In this communication, we have discovered that phenolic O- to aromatic C-glycosidic bond rearrangement (Scheme 1; ESI,† Scheme S1c) is achievable by coupled glycosyltransferase reactions. The overall conversion is formally equivalent to a chemical Fries-type rearrangement;13 however, the mechanisms are distinct. Glycosyltransferase conversions involve intermediary release of aglycon and NDP-sugar, followed by regio- and stereoselective C-glycosylation.
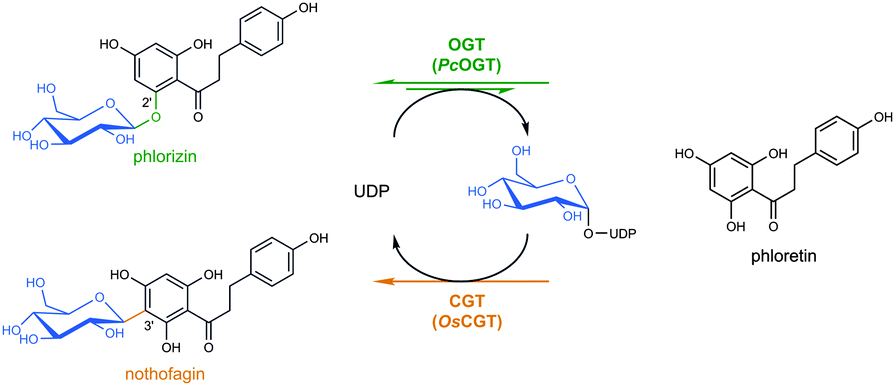 | ||
| Scheme 1 Conversion of phlorizin into nothofagin using a direct O- to C-glucoside rearrangement in two catalytic steps. | ||
A proof of principle was obtained for conversion of phlorizin, the 2′-O-glucoside of the dihydrochalcone phloretin, into the corresponding 3′-C-glucoside nothofagin (Scheme 1). The catalytic process involved O-glucosyltransferase (OGT) and C-glucosyltransferase (CGT) activities, derived from a specificity matched pair of enzymes from pear (Pyrus communis; PcOGT)14 and rice (Oyrza sativa; OsCGT),15 or from an engineered “promiscuous” O/C-glucosyltransferase (O_CGT). Only catalytic amounts of uridine 5′-diphosphate (UDP) were required in the conversion to generate intermediary UDP–glucose and phloretin via reverse reaction of the OGT. The overall two-step rearrangement proceeded under thermodynamic control whereby a large driving force for the second step, aromatic C-glycosylation, enabled nothofagin formation in quantitative yields. The phlorizin–nothofagin pair stands for a number of homologous O- and C-glycosidic natural products, in that the O-glycoside occurs naturally in relatively high abundance over the quite rare C-glycoside.16,17 Fruit trees (Rosaceae) contain large amounts of phlorizin in their bark18 whereas significant quantities of nothofagin were reported only from redbush herbal tea.19 Direct O- to C-glycosidic bond rearrangement makes effective use of both the glycon and the aglycon moiety of the substrate. In principle, therefore, it presents an expedient and particularly atom-efficient transformation of O-glycosides (e.g. flavonoid glycosides) readily available from the natural products pool. Due to their high resistance to hydrolysis, the resulting C-glycosides are of special interest for development of bioactive substances with enhanced in vivo half-lives.16,20
Purified preparations of PcOGT and OsCGT were obtained from E. coli overexpression cultures using His-tag and Strep-tag affinity chromatography, respectively. The Ile121 → Asp variant (I121D) of OsCGT, previously shown to exhibit dual specific O_CGT activity,21 was produced and isolated in the same way as the wild-type enzyme (ESI,† Fig. S1). All reactions were carried out with 20% (by volume) DMSO to enhance phloretin solubility. We confirmed that the yield and the selectivity of the bi-enzymatic rearrangement were not affected by DMSO (ESI,† Fig. S3). Enzymes were fully active and exhibited suitable stability under these conditions. Phloretin, phlorizin, nothofagin, and other phloretin glucosides were identified and quantified by HPLC (ESI,† Fig. S4b).21 UDP–glucose and UDP were measured by capillary zone electrophoresis. OGT and CGT activities were determined using HPLC-based assays.
We noticed that rearrangement of phlorizin to nothofagin could become effective only when main requirements concerning enzyme specificity, reaction kinetics, and reaction thermodynamics in each step were adequately met. PcOGT and OsCGT were known to glucosylate phloretin at position 2′-O14,21 and 3′-C,21,22 respectively. Interference from secondary O-glucoside hydrolase activity in the enzymes used could also be discounted based on earlier evidence.21 Moreover, OsCGT alone was inactive as a “rearrangement enzyme” when phlorizin and UDP were offered as substrates. C-Glucosylation of phloretin from UDP–glucose was known to be largely irreversible (Keq > 400; 30 °C, pH 7.5).23 However, kinetic and thermodynamic characteristics of the PcOGT reaction required clarification. Fig. 1a compares the pH-rate profile for deglucosylation of phlorizin to UDP to the pH-rate profile for glucosylation of phloretin from UDP–glucose. PcOGT was more active in the direction of phlorizin synthesis (18.3 U mg−1) than degradation (5.6 U mg−1) at the respective optimum pH. Interestingly, despite the rather uniform pH effects on enzyme activity in each reaction direction (except for the slight pH range shift), we realised that the reaction equilibrium constant (Keq = [phlorizin][UDP]/[UDP–glucose][phloretin]) increased dramatically in response to pH change from 6.5 to 8.8 (Fig. 1b). Attainment of reaction equilibrium was affirmed rigorously under all conditions, ruling out interference from enzyme activity loss at high or low pH. It was also confirmed that the pH was stable during the conversions. Correlation between log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Keq and pH was linear with a large slope of +1.6 (±0.1), implying that conversion of phlorizin and UDP involves the uptake of proton(s). Potentiometric titration of each compound present in the reaction (ESI,† Fig. S5) revealed the likely importance of protonation of UDP (pKa ∼ 5.6). The immediate ramification of results in Fig. 1b is that exploitation of the PcOGT reverse reaction for supplying substrates in adequate steady-state concentrations to the OsCGT reaction will only be practical at pH 7.5 or lower. Half-saturation constants of OsCGT were determined to be 0.009 mM for phloretin and 0.024 mM for UDP–glucose,23 defining lower limits to the respective substrate concentration for effective utilization of the C-glucosylation activity present.
Keq and pH was linear with a large slope of +1.6 (±0.1), implying that conversion of phlorizin and UDP involves the uptake of proton(s). Potentiometric titration of each compound present in the reaction (ESI,† Fig. S5) revealed the likely importance of protonation of UDP (pKa ∼ 5.6). The immediate ramification of results in Fig. 1b is that exploitation of the PcOGT reverse reaction for supplying substrates in adequate steady-state concentrations to the OsCGT reaction will only be practical at pH 7.5 or lower. Half-saturation constants of OsCGT were determined to be 0.009 mM for phloretin and 0.024 mM for UDP–glucose,23 defining lower limits to the respective substrate concentration for effective utilization of the C-glucosylation activity present.
 | ||
| Fig. 1 pH effects on PcOGT activity for phlorizin synthesis and degradation (a), and on the reaction equilibrium constant (b). (a) Relative activities of PcOGT; black symbols (forward reaction: 0.1 mM phloretin, 0.6 mM UDP–glucose, 100% = 18.3 U mg−1); grey symbols (reverse reaction: 1.0 mM phlorizin, 2.0 mM UDP, 100% = 5.6 U mg−1). The buffers used were citrate (circles), tris (squares), and CAPS (triangles). (b) Keq is for synthesis of phlorizin and UDP from phloretin and UDP–glucose. | ||
Rearrangement of phlorizin (5 mM) into nothofagin was examined in a one-pot reaction that contained UDP (2 mM), PcOGT, and OsCGT. Because the optimum pH for C-glycosylation (pH 8.5)23 did not match pH conditions applicable to phlorizin conversion by PcOGT, we tested the coupled enzyme reaction at different pH values in the range 5.9–7.8. Nothofagin was produced under all conditions used, demonstrating the system's functionality in principle. Fig. 2a compares phlorizin consumption to the corresponding formation of nothofagin and phloretin after 5 h of reaction. Interestingly, whereas the actual phlorizin conversion was not strongly affected by pH change in the applied range, the resulting product distribution, nothofagin compared to phloretin, showed pronounced pH dependence. Accumulation of phloretin at low pH indicated critical limitations due to insufficient OsCGT activity under these conditions. However, C-glucosylation was quite effective in the pH range 6.7–7.8 where only small amounts of phloretin were detected next to the main product nothofagin (2.5 mM; 50% substrate conversion).
 | ||
| Fig. 2 Conversion of phlorizin (5 mM) via O- to C-glycosidic bond rearrangement in the presence of 2 mM UDP using 100 mU mL−1PcOGT and 50 mU mL−1OsCGT. (a) Product distribution after reaction for 5 h at different pH conditions. (b) Reaction time course at pH 7.0. Colours show phlorizin (green), nothofagin (orange), and phloretin (black). | ||
Fig. 2b shows a full time course for enzymatic reaction at pH 7.0, demonstrating that quantitative conversion of phlorizin into nothofagin was made possible under these conditions. Isolation of highly pure nothofagin is typically achieved by reversed phase C-18 HPLC in yields of more than 80%.23 The phloretin concentration attained maximum (∼0.5 mM) early during the reaction, only to drop to a very low level later on. This time dependence was fully consistent with the proposed role of phloretin as an intermediary product in an overall two-step rearrangement process. Evidence that immediately after the reaction started, nothofagin formation was clearly lagging behind the formation of phloretin (see the phloretin “burst” of ∼0.3 mM at t = 0 h in Fig. 2b) gave additional indication of the reaction in two discrete biocatalytic steps.
Kinetic analysis (ESI,† Fig. S6) showed that the phlorizin consumption rate (rP) was hyperbolically dependent on UDP and phlorizin concentration, resulting in apparent half-saturation constants of 1.22 ± 0.16 mM (phlorizin) and 0.074 ± 0.010 mM (UDP).
The overall rearrangement rate (rN; nothofagin formation) paralleled rP under all conditions, except at high phlorizin concentration (≥3 mM) where rN dropped off strongly in relation to rP. The behaviour of rN is explained by substrate inhibition of OsCGT at high phloretin concentration.23 However, disparity between rP and rN did not diminish the nothofagin yield when 10 mM phlorizin was applied (ESI,† Fig. S4a). High rearrangement rates could be maintained with as little as 0.25 mM UDP (ESI,† Fig. S6b) confirming the requirement for only catalytic amounts of UDP in the overall conversion.
We recognised the interesting possibility of performing the two-step rearrangement using only a single enzyme that exhibits OGT as well as CGT activity. The I121D mutant of OsCGT which catalyses glucosyl transfer to O2 and O4, next to C3 of the phloretin acceptor, was considered useful.21 Kinetic properties of the mutant required adaptation of reaction conditions for synthesis of nothofagin from phlorizin (ESI,† Methods, Fig. S7–S9). In particular, the UDP concentration was increased to 5 mM to promote an otherwise inefficient reverse reaction from phlorizin substrate, which was applied at a lowered concentration of 0.5 mM. Additionally, UDP–glucose was supplied at 1.0 mM to drive the C-glucosylation.
Fig. 3 displays the time course of reaction catalysed by I121D mutant under these conditions. Turnover frequency of the mutant (∼0.001 s−1)21 restricted the amount of enzyme activity usable in the reaction, resulting in relatively slow conversion. However, all of the initial phlorizin was converted, and nothofagin was obtained in 70% yield. Unlike in the reaction catalysed by coupled OGT and CGT (Fig. 2b), the phloretin concentration increased steadily during the mutant-catalysed reaction, approaching a maximum value of ∼0.1 mM at the time when phlorizin was depleted (Fig. 3). Therefore, this indicates that C-glucosylation was rate limiting for the overall rearrangement catalysed by the mutant. Interestingly, small amounts of phloretin 4′-O-glucoside were also produced and used up later in the conversion (Fig. 3). Therefore, this implied O2 to O4 positional rearrangement in phloretin O-glucosides catalysed by the mutant.
 | ||
| Fig. 3 Conversion of 0.5 mM phlorizin (green) by I121D mutant of OsCGT (2.5 mU mL−1) in the presence of 5 mM UDP and 1 mM UDP–glucose. Further symbols: phloretin (black), nothofagin (orange), 4′-O-glucoside of phloretin (blue). | ||
Summarising, the biocatalytic rearrangement discovered in this study is a remarkable chemical transformation that might open up new opportunities for synthesis of aromatic C-glycosidic natural products or natural product-like structures from (readily available) phenolic O-glycosidic substrates. Unlike glycosyltransferase exchange reactions in which overcoming thermodynamic restrictions presents the main issue,4,8,24 the example of phlorizin conversion into nothofagin shows that (quantitative) O- to C-glycosidic bond rearrangement is promoted by a large driving force on the C-glycosylation. While the study provides a clear proof of principle, expansion of the synthetic scope of the biocatalytic rearrangement will be important. Development of new pairs of complementary enzymes through discovery work and protein engineering is required whereby identification of new CGT enzymes will be the key.
Financial support from the Austrian Science Funds (FWF DK Molecular Enzymology W901-B05) and from Ramkhamhaeng University (Bangkok, Thailand) is gratefully acknowledged.
Notes and references
- M. M. Palcic, Curr. Opin. Chem. Biol., 2011, 15, 226–233 CrossRef CAS PubMed.
- C. J. Thibodeaux, C. E. Melançon III and H. W. Liu, Angew. Chem., Int. Ed., 2008, 47, 9814–9859 CrossRef CAS PubMed.
- L. L. Lairson, B. Henrissat, G. J. Davies and S. G. Withers, Annu. Rev. Biochem., 2008, 77, 521–555 CrossRef CAS PubMed.
- C. Zhang, B. R. Griffith, Q. Fu, C. Albermann, X. Fu, I. K. Lee, L. Li and J. S. Thorson, Science, 2006, 313, 1291–1294 CrossRef CAS PubMed.
- H. B. Bode and R. Müller, Angew. Chem., Int. Ed., 2007, 46, 2147–2150 CrossRef CAS PubMed.
- A. Minami, K. Kakinuma and T. Eguchi, Tetrahedron Lett., 2005, 46, 6187–6190 CrossRef CAS PubMed.
- R. Chen, H. Zhang, G. Zhang, S. Li, G. Zhang, Y. Zhu, J. Liu and C. Zhang, J. Am. Chem. Soc., 2013, 135, 12152–12155 CrossRef CAS PubMed.
- R. W. Gantt, P. Peltier-Pain, W. J. Cournoyer and J. S. Thorson, Nat. Chem. Biol., 2011, 7, 685–691 CrossRef CAS PubMed.
- C. Zhang, C. Albermann, X. Fu and J. S. Thorson, J. Am. Chem. Soc., 2006, 128, 16420–16421 CrossRef CAS PubMed.
- C. Zhang, Q. Fu, C. Albermann, L. Li and J. S. Thorson, ChemBioChem, 2007, 8, 385–390 CrossRef CAS PubMed.
- C. Zhang, E. Bitto, R. D. Goff, S. Singh, C. A. Bingman, B. R. Griffith, C. Albermann, G. N. Phillips and J. S. Thorson, Chem. Biol., 2008, 15, 842–853 CrossRef CAS PubMed.
- R. W. Gantt, P. Peltier-Pain and J. S. Thorson, Nat. Prod. Rep., 2011, 28, 1811–1853 RSC.
- R. G. dos Santos, A. R. Jesus, J. M. Caio and A. P. Rauter, Curr. Org. Chem., 2011, 15, 128–148 CrossRef CAS.
- C. Gosch, H. Halbwirth, B. Schneider, D. Hölscher and K. Stich, Plant Sci., 2010, 178, 299–306 CrossRef CAS PubMed.
- M. Brazier-Hicks, K. M. Evans, M. C. Gershater, H. Puschmann, P. G. Steel and R. Edwards, J. Biol. Chem., 2009, 284, 17926–17934 CrossRef CAS PubMed.
- T. Bililign, B. R. Griffith and J. S. Thorson, Nat. Prod. Rep., 2005, 22, 742–760 RSC.
- J. Härle, S. Günther, B. Lauinger, M. Weber, B. Kammerer, D. L. Zechel, A. Luzhetskyy and A. Bechthold, Chem. Biol., 2011, 18, 520–530 CrossRef PubMed.
- J. R. L. Ehrenkranz, N. G. Lewis, C. R. Kahn and J. Roth, Diabetes/Metab. Res. Rev., 2005, 21, 31–38 CrossRef CAS PubMed.
- E. Joubert, Food Chem., 1996, 55, 403–411 CrossRef CAS.
- C. Dürr, D. Hoffmeister, S. E. Wohlert, K. Ichinose, M. Weber, U. von Mulert, J. S. Thorson and A. Bechthold, Angew. Chem., Int. Ed., 2004, 43, 2962–2965 CrossRef PubMed.
- A. Gutmann and B. Nidetzky, Angew. Chem., Int. Ed., 2012, 51, 12879–12883 CrossRef CAS PubMed.
- M. Brazier-Hicks and R. Edwards, Metab. Eng., 2013, 16, 11–20 CrossRef CAS PubMed.
- L. Bungaruang, A. Gutmann and B. Nidetzky, Adv. Synth. Catal., 2013, 355, 2757–2763 CrossRef CAS PubMed.
- P. Peltier-Pain, K. Marchillo, M. Zhou, D. R. Andes and J. S. Thorson, Org. Lett., 2012, 14, 5086–5089 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures used; the scheme of reverse glycosyltransferase reactions; enzyme purity; deglycosylation of phlorizin by PcOGT; the effect of cosolvents and substrate concentrations on rearrangement; HPLC trace of phloretin (glycosides); potentiometric titrations; optimisation of conversion by OsCGT_I121D. See DOI: 10.1039/c4cc00536h |
| This journal is © The Royal Society of Chemistry 2014 |