 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCatalytic metal-free Si–N cross-dehydrocoupling†
Lutz
Greb‡
,
Sergej
Tamke‡
and
Jan
Paradies
*
Institute for Organic Chemistry, Karlsruhe Institute of Technology (KIT), Fritz-Haber-Weg 6, D-76131 Karlsruhe, Germany. E-mail: jan.paradies@kit.edu
First published on 10th January 2014
Abstract
The metal-free B(C6F5)3 catalyzed dehydrocoupling of hydrosilanes with anilines, carbazoles and indoles is reported. For anilines and carbazoles the reaction proceeds by the liberation of H2 as the sole Si–N coupling byproduct. Indoles react with diphenyl(methyl) hydrosilane to give N-silyl indolines with high diastereoselectivity (d.r. 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) in excellent yields. A mechanism for this Si–N coupling/hydrogenation sequence is proposed.
:1) in excellent yields. A mechanism for this Si–N coupling/hydrogenation sequence is proposed.
The cross-dehydrocoupling is an efficient methodology for the connection of two molecular entities.1 Especially the dehydrocoupling of Si–H and N–H fragments provides an environmentally benign access to silyl-protected amines.2 These ubiquitous structural motifs are usually obtained by the reaction of halosilanes with deprotonated amines, the generation of which often requires strong bases.3 This is not only of concern for atom efficiency but also for functional group tolerance. In light of this, the Si–N dehydrocoupling proved very useful, e.g. for the protection of indoles using Zn(OTf)2 (10 mol%) in the presence of 0.5–1.0 equiv. of pyridine.4 Oestreich's sulfur-bridged Ru–arene complex5 is particularly effective in the base-free dehydrocoupling of silanes with other nitrogen-containing heterocycles, e.g. indole, carbazole and pyrrole derivatives using only 1 mol% of catalyst loading.6 However, a metal-free variant has not yet been disclosed.7
We have shown earlier that the H2-activation product 1 of the frustrated Lewis pair (FLP) consisting of 2/3 is a transient species which readily releases H2 at room temperature (Scheme 1, top).8 Accordingly, the isostructural intermediate iso-1, generated through the silyl-transfer from the silane 4 to the aniline 5a, should readily liberate H2 with concomitant release of the Si–N coupling product 6 (Scheme 1, bottom). As a potential silyl-transfer catalyst, borane 2 has attracted significant attention in hydrosilylation of aldehydes, ketones, imines and olefins.9 An analogous mechanism was only recently proposed by Oestreich as a competing pathway in the borane-promoted imine reduction with hydrosilanes.9a
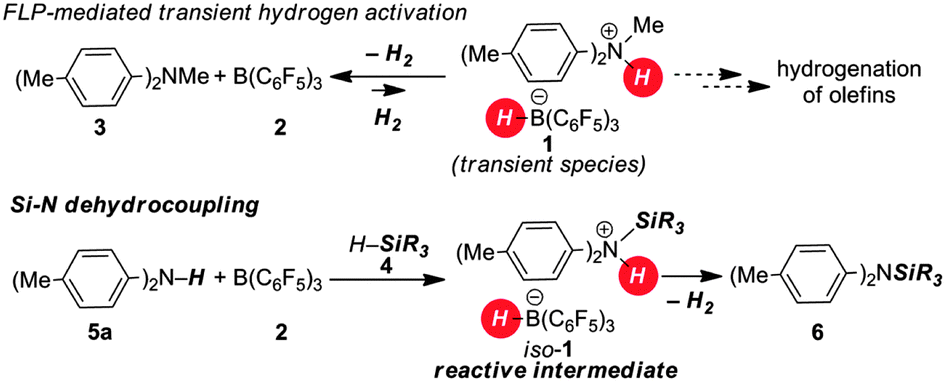 | ||
| Scheme 1 Conceptional outline for the Si–N dehydrocoupling. | ||
Indeed, when bis(4-toloyl)amine (5a) was reacted with diphenyl(methyl) silane (4a) in the presence of 5 mol% B(C6F5)3 (2) at room temperature, the silylamine 6a was obtained in 95% yield accompanied with the evolution of H2 (Table 1, entry 1). In the absence of the catalyst, the formation of 6a was not observed even when a mixture of 5a and 4a was heated to 90 °C for 12 h (Table 1, entry 2). The catalyst loading was reduced to 1 mol% with slight erosion in yield (73%, entry 3). Lower catalyst loadings of 0.1 mol% led to significantly reduced yields (entry 4). Further experiments were carried out with 1 mol% of 2 as catalyst.
|
|
|||||
|---|---|---|---|---|---|
| Entry | t [h] | T [°C] | Product | Yield [%] | |
| a Reactions were performed on a 1.0 mmol scale, 3 M in CH2Cl2. b 5 mol% 2. c Absence of 2. d 0.1 mol% B(C6F5)3. e 10 mol% 2, 0.1 mmol scale, 3 M in CD2Cl2, yield determined by 1H NMR. f 2 mol% 2. | |||||
| Diarylamines | 1 | 1 | 25 |
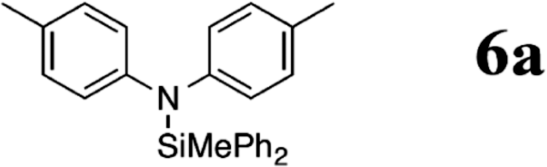
|
95b |
| 2 | 12 | 90 | 0c | ||
| 3 | 1 | 25 | 73 | ||
| 4 | 10 | 25 | 32d | ||
| 5 | 1 | 25 |
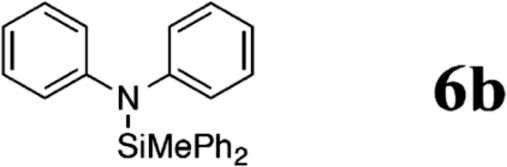
|
91 | |
| 6 | 1 | 25 |
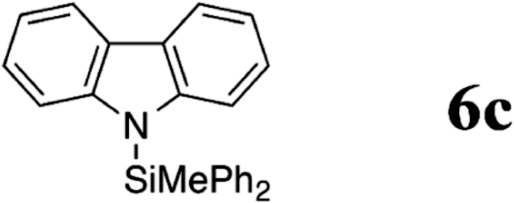
|
97 | |
| 7 | 1 | 25 |

|
83 | |
| 8 | 24 | 25 |
|
95e | |
| 9 | 1 | 25 |
|
95e | |
| 10 | 1 | 25 |

|
97 | |
| Anilines | 11 | 72 | 70 |
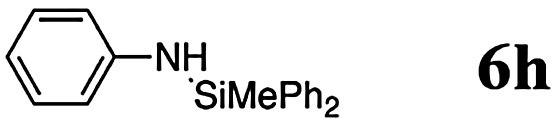
|
90b |
| 12 | 48 | 70 |
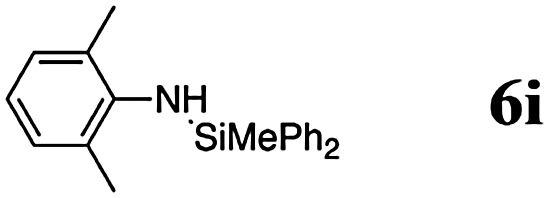
|
90 | |
| 13 | 48 | 70 |

|
93 | |
| 14 | 36 | 60 |

|
97 | |
| 15 | 24 | 60 |
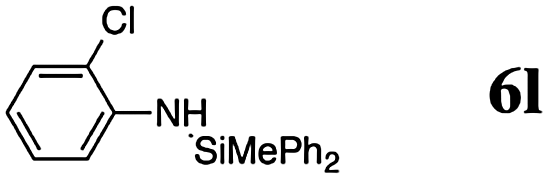
|
91 | |
| 16 | 36 | 25 |
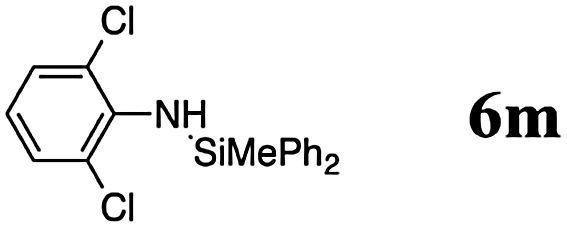
|
88 | |
| 17 | 24 | 25 |
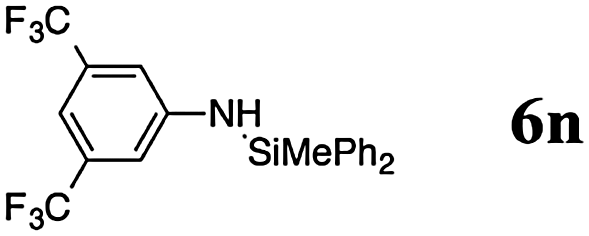
|
97 | |
| Diamines | 18 | 24 | 25 |
|
26f |
| 19 | 24 | 70 |

|
92b | |
| 20 | 24 | 60 |
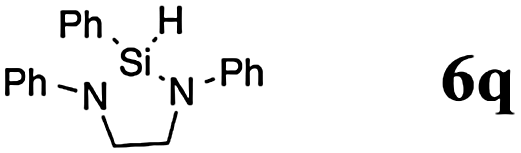
|
83 | |
| Indoles | 21 | 144 | 70 |

|
50e |
| 22 | 24 | 70 |
|
81 | |
| 23 | 24 | 70 |

|
96 | |
| 24 | 24 | 70 |
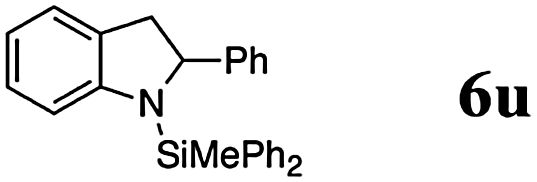
|
97 | |
| 25 | 24 | 70 |
|
92 | |
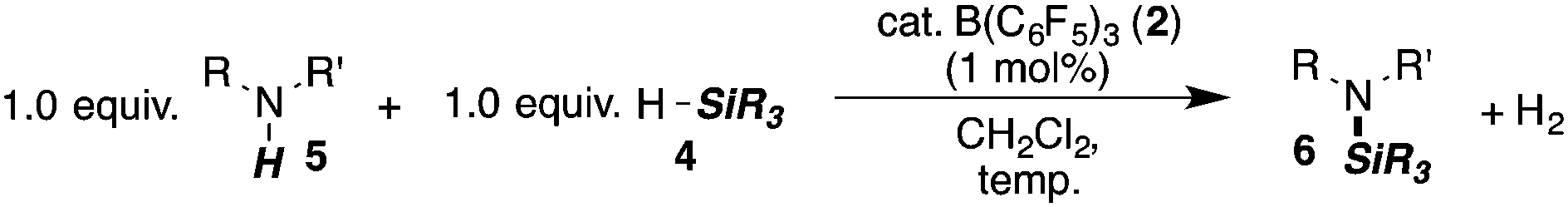
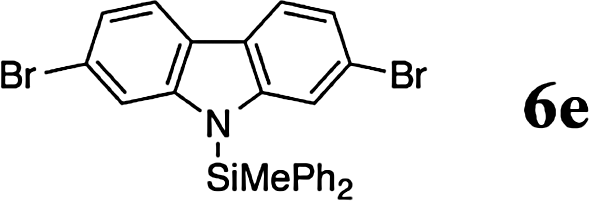
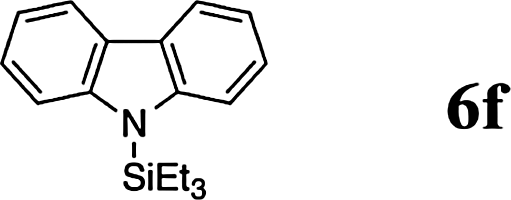
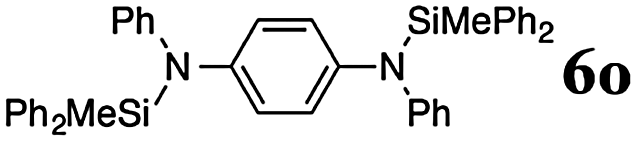
The reaction displays a remarkable substrate scope. Besides diphenylamine derivatives (5a and 5b, entries 3 and 5), carbazole derivatives 5c-f also proved to be viable substrates and the products 6c-f were obtained in 83–97% yields (entries 6–9). The bibromo derivative required 70 °C to undergo Si–N cross-dehydrocoupling in 51% yield without the observation of dehalogenation (entry 8). The reduced yield was attributed to the very low solubility of 5e in toluene. Other silanes were also useful in the Si–N coupling reaction. Triethylsilane (4b) or 1,1,3,3-tetramethyldisiloxane (4c) readily reacted with carbazole (5c) or bis(4-tolyl)amine (5a) in high yields (entries 9 and 10). The silylation of primary aniline derivatives proceeded at 60–70 °C in excellent yields (88–97%, entries 11–15).10 The electron-deficient anilines 5m and 5n were reactive even at room temperature and 6m and 6n were obtained in 88% and 97% yields (entries 16 and 17). Also the two diamines N,N′-(diphenyl)-1,4-phenylene diamine (5o) and N,N′-(diphenyl)-ethylene diamine (5p) underwent silylation with diphenylmethyl silane (4a) in high yields (entries 18 and 19).
Accordingly, the reaction of 5p with phenylsilane (4d) provided the cyclic product 6q in 83% yield (entry 20).
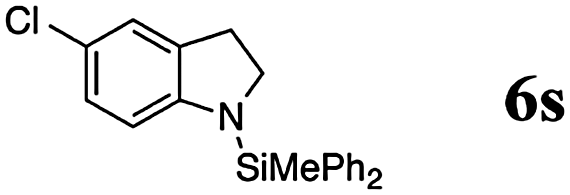
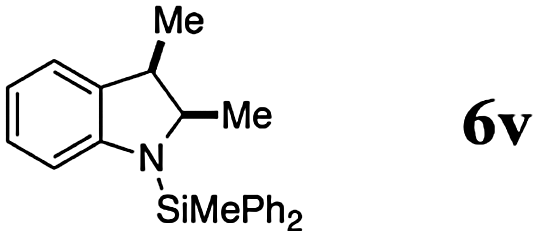
Finally, we investigated the potential of the Si–N dehydrocoupling for pyrrole and indole derivatives. While pyrrole-derivatives were unreactive under our reaction conditions,11 the indole-derivatives 5r–v displayed high reactivity. The indoles 5r–v were chemospecifically converted into the 1-silylated indoline derivatives 6r–v (entries 21–25) without the formation of unsaturated side products arising from N or C3-silylation.12 Indole (5r) required prolonged reaction time (144 h, entry 21) for the domino silylation/reduction sequence and indoline (6r) was obtained in 50% yield. The less electron-rich 6-chloroindole (5s) was transformed into 6s in excellent yield in only 24 h (95%, entry 22). Substituents in position 2 were well tolerated and the 2-methyl and 2-phenyl indolines 5t and 5u were obtained in quantitative yields (96% and 97%, entries 3–5). 2,3-dimethylindole (5v) was diastereoselectively reduced to cis-2,3-dimethyl indoline (6v) in quantitative yield (98%, d.r. 10:1).13
The high chemospecificity and diastereoselectivity prompted us to investigate the Si–N cross coupling/hydrogenation reaction of 5v with 4a in detail (Scheme 2). Only resonances of the starting materials and the product 6v were observed when the reaction was monitored by 1H NMR (1 mol% 3, [D8]-toluene). Neither the resonance of FLP-activated H2 nor the resonance of dissolved H2 was observed by 1H NMR. Deuterium labeling experiments were conducted to investigate the fate of the hydridic and protic hydrogen atoms in silane 4a and indole 5v. The reaction of 1-D-2,3-dimethyl indole (1-D-5v, 95% D) with H–SiMePh2 (4a) gave exclusively cis-3-D-2,3-dimethyl indoline (3-D-6v) in high yields (97%, 92% D-incorporation, Scheme 2a). The reaction of D–SiMePh2 (D-2a, 95% D) with 5v provided exclusively cis-2-D-2,3-dimethyl indoline (2-D-6v) in 96% yield with 92% D-incorporation at position 2. Together the chemoselective deuteration and the absence of dissolved or FLP-activated H2 or HD14 strongly support a N-silylation/rearrangement/reduction mechanism (Scheme 3). The product of the B(C6F5)3-catalyzed silyl-transfer to 5v is 1-silyl-1-H-indol-1-ium 6, which rearranges to the more stable 1-silyl-3-H-indol-1-ium 7. Alternatively, an intermolecular proton-transfer might be conceivable. However, according to our cross experiment using 5t–u and 1-silyl-indole 8, the sigmatropic rearrangement mechanism is more likely (Scheme 2c). The indole derivatives 5t and 5u were equally reactive as 5v (96–98%, 24 h, see Table 1, entries 23–25) and should be readily protonated by transiently formed 6 (formed by the reaction of 5 and 4a, compare Scheme 3). However, the reaction of an equimolar mixture of 8, 5t–u, and 4a in the presence of 10 mol% 2 produced 6t or 6u as the product (6u/6v >95:5; 6t:6v >90:10). This is a strong indication that intermolecular proton-transfer is not operative in the silylation/hydrogenation reaction sequence. The final step in the catalytic cycle is the hydride transfer from [H–B(C6F5)3] to the highly electrophilic iminium species 7 from the least hindered side liberating cis-6v and the catalyst 2.
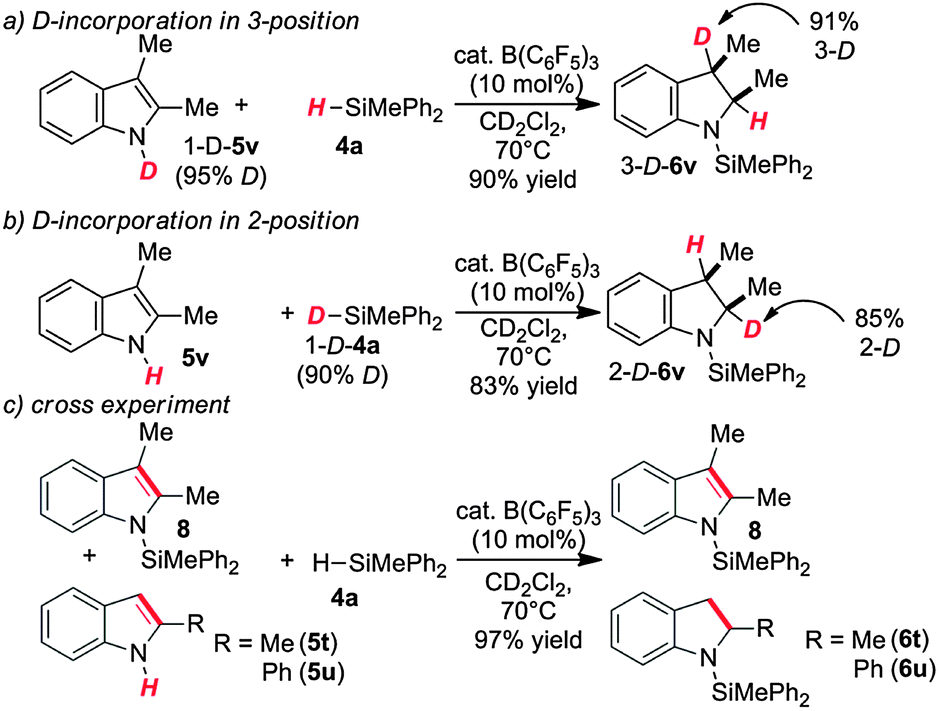 | ||
| Scheme 2 Isotope labelling experiments with (a) 1-D-2,3-dimethylindole (1-(D)-4v), with (b) D-SiMePh2 (D-4a) and (c) cross experiment. | ||
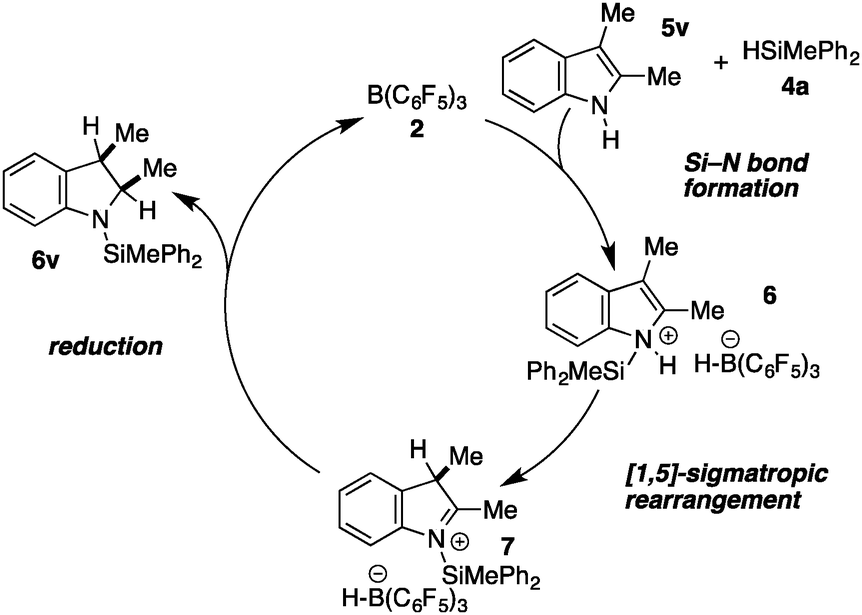 | ||
| Scheme 3 Proposed catalytic cycle for the Si–N coupling/hydrogenation domino reaction. | ||
In summary, we have developed the metal-free Si–N cross-dehydrocoupling for primary and secondary aryl amines having solely molecular hydrogen as byproduct. Indole derivatives undergo N-silylation followed by a rearrangement/reduction sequence to furnish indolines in high yields and high diastereoselectivity (d.r. 10:1).
Notes and references
- For reviews, see: (a) C. S. Yeung and V. M. Dong, Chem. Rev., 2011, 111, 1215 CrossRef CAS PubMed; (b) C. J. Scheuermann, Chem.–Asian J., 2010, 5, 436 CrossRef CAS PubMed; (c) G. E. Dobereiner and R. H. Crabtree, Chem. Rev., 2010, 681 CrossRef CAS PubMed; (d) C. J. Li, Acc. Chem. Res., 2009, 42, 335 CrossRef CAS PubMed ; for recent examples, see: ; (e) X. M. Jie, Y. P. Shang, P. Hu and W. P. Su, Angew. Chem., Int. Ed., 2013, 52, 3630 CrossRef CAS PubMed; (f) N. Kuhl, M. N. Hopkinson and F. Glorius, Angew. Chem., Int. Ed., 2012, 51, 8230 CrossRef CAS PubMed.
- For reviews, see: (a) R. Waterman, Chem. Soc. Rev., 2013, 42, 5629 RSC; (b) E. M. Leitao, T. Jurca and I. Manners, Nat. Chem., 2013, 5, 817 CrossRef CAS PubMed; (c) M. S. Hill, D. J. Liptrot, D. J. MacDougall, M. F. Mahon and T. P. Robinson, Chem. Sci., 2013, 4, 4212 RSC; (d) J. Y. Corey, Chem. Rev., 2011, 111, 863 CrossRef CAS PubMed; (e) J. F. Harrod, Coord. Chem. Rev., 2000, 206, 493 CrossRef; (f) J. A. Reichl and D. H. Berry, Adv. Organomet. Chem., 1998, 43, 197 CrossRef ; for pioneering work, see: ; (g) E. Matarasso-Tchiroukhine, Chem. Commun., 1990, 681 RSC ; for recent examples, see: ; (h) J. F. Dunne, S. R. Neal, J. Engelkemier, A. Ellern and A. D. Sadow, J. Am. Chem. Soc., 2011, 133, 16782 CrossRef CAS PubMed; (i) C. K. Toh, H. T. Poh, C. S. Lim and W. Y. Fan, J. Organomet. Chem., 2012, 717, 9 CrossRef CAS PubMed; (j) F. Buch and S. Harder, Organometallics, 2007, 26, 5132 CrossRef CAS; (k) A. Iida, A. Horii, T. Misaki and Y. Tanabe, Synthesis, 2005, 2677 CAS; (l) F. Lunzer, C. Marschner and S. Landgraf, J. Organomet. Chem., 1998, 568, 253 CrossRef CAS; (m) H. Q. Liu and J. F. Harrod, Can. J. Chem., 1992, 70, 107 CrossRef CAS; (n) H. Q. Liu and J. F. Harrod, Organometallics, 1992, 11, 822 CrossRef CAS; (o) W. D. Wang and R. Eisenberg, Organometallics, 1991, 10, 2222 CrossRef CAS; (p) Y. D. Blum, K. B. Schwartz and R. M. Laine, J. Mater. Sci., 1989, 24, 1707 CrossRef CAS; (q) Y. Blum and R. M. Laine, Organometallics, 1986, 5, 2081 CrossRef CAS.
- (a) P. G. M. Wuts and T. W. Greene, Protective Group in Organic Chemistry, John Wiley & Sons, Inc., Hoboken, New Jersey, 4th edn, 2007 Search PubMed; (b) S. Djuric, J. Venit and P. Magnus, Tetrahedron Lett., 1981, 22, 1787 CrossRef CAS.
- T. Tsuchimoto, Y. Iketani and M. Sekine, Chem.–Eur. J., 2012, 18, 9500 CrossRef CAS PubMed.
- (a) T. Stahl, H. F. T. Klare and M. Oestreich, J. Am. Chem. Soc., 2013, 135, 1248 CrossRef CAS PubMed; (b) T. Stahl, K. Muether, Y. Ohki, K. Tatsumi and M. Oestreich, J. Am. Chem. Soc., 2013, 135, 10978 CrossRef CAS PubMed; (c) C. D. F. Konigs, H. F. T. Klare, Y. Ohki, K. Tatsumi and M. Oestreich, Org. Lett., 2012, 14, 2842 CrossRef CAS PubMed; (d) H. F. T. Klare, M. Oestreich, J.-i. Ito, H. Nishiyama, Y. Ohki and K. Tatsumi, J. Am. Chem. Soc., 2011, 133, 3312 CrossRef CAS PubMed; (e) Y. Ohki, Y. Takikawa, H. Sadohara, C. Kesenheimer, B. Engendahl, E. Kapatina and K. Tatsumi, Chem.–Asian J., 2008, 3, 1625 CrossRef CAS PubMed.
- C. D. F. Koenigs, M. F. Mueller, N. Aiguabella, H. F. T. Klare and M. Oestreich, Chem. Commun., 2013, 49, 1506 RSC.
- For metal-free silylation of OH-groups, see: (a) D. J. Gao and C. M. Cui, Chem.–Eur. J., 2013, 19, 11143 CrossRef CAS PubMed; (b) for metal-free silylation of P–P bonds, see: S. J. Geier and D. W. Stephan, Chem. Commun., 2010, 46, 1026 RSC.
- (a) L. Greb, S. Tussing, B. Schirmer, P. Oña-Burgos, K. Kaupmees, M. Lokov, I. Leito, S. Grimme and J. Paradies, Chem. Sci., 2013, 4, 2788 RSC; (b) L. Greb, P. Oña-Burgos, B. Schirmer, S. Grimme, D. W. Stephan and J. Paradies, Angew. Chem., Int. Ed., 2012, 51, 10164 CrossRef CAS PubMed.
- (a) J. Hermeke, M. Mewald and M. Oestreich, J. Am. Chem. Soc., 2013, 46, 17537 CrossRef PubMed; (b) L. Greb, P. Oña-Burgos, A. Kubas, F. C. Falk, F. Breher, K. Fink and J. Paradies, Dalton Trans., 2012, 40, 9056 RSC; (c) W. E. Piers, A. J. V. Marwitz and L. G. Mercier, Inorg. Chem., 2011, 50, 12252 CrossRef CAS PubMed; (d) A. Berkefeld, W. E. Piers and M. Parvez, J. Am. Chem. Soc., 2010, 132, 10660 CrossRef CAS PubMed; (e) J. M. Blackwell, D. J. Morrison and W. E. Piers, Tetrahedron, 2002, 58, 8247 CrossRef CAS; (f) M. Rubin, T. Schwier and V. Gevorgyan, J. Org. Chem., 2002, 67, 1936 CrossRef CAS PubMed; (g) D. J. Parks, J. M. Blackwell and W. E. Piers, J. Org. Chem., 2000, 65, 3090 CrossRef CAS PubMed; (h) J. M. Blackwell, E. R. Sonmor, T. Scoccitti and W. E. Piers, Org. Lett., 2000, 2, 3921 CrossRef CAS PubMed; (i) W. E. Piers and T. Chivers, Chem. Soc. Rev., 1997, 26, 345 RSC; (j) D. J. Parks and W. E. Piers, J. Am. Chem. Soc., 1996, 118, 9440 CrossRef CAS.
- The elevated temperature of 60–70 °C was required to thermally cleave the aniline/B(C6F5)3 adduct as evidenced by 11B NMR.
- Neither N- nor C3-silylation products were observed, see: L. D. Curless, E. R. Clark, J. J. Dunsford and M. J. Ingleson, Chem. Commun., 2014 10.1039/c3cc47372d.
- For B(C6F5)3-catalyzed 1,4-hydrosilylation of 2-phenyl chinoline, see: S. J. Geier, P. A. Chase and D. W. Stephan, Chem. Commun., 2010, 46, 4884 RSC.
- Determined by deprotection of 6v and comparison of the 1H NMR spectra of the resulting 1-H indoline with literature reported NMR data: F. O. Arp and G. C. Fu, J. Am. Chem. Soc., 2006, 128, 14264 CrossRef CAS PubMed.
- Although the hydrogenation of N-methyl indoles is reported (10 mol% 2, 103 bar H2, 80 °C, see: D. W. Stephan, S. Greenberg, T. W. Graham, P. Chase, J. J. Hastie, S. J. Geier, J. M. Farrell, C. C. Brown, Z. M. Heiden, G. C. Welch and M. Ullrich, Inorg. Chem., 2011, 50, 12338 CrossRef CAS PubMed ) the hydrogenation of 1-silyl-2,3-dimethyl indole (4 bar H2) in the presence of 5 mol% 2 in toluene at 70 °C did not furnish indoline 6v.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental procedures, analytical data. See DOI: 10.1039/c3cc49558b |
| ‡ Both authors contributed evenly. |
| This journal is © The Royal Society of Chemistry 2014 |