 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Peptide ligation from alkoxyamine based radical addition†
Thomas
Trimaille
*a,
Laurent
Autissier
a,
Mamy Daniel
Rakotonirina
a,
Yohann
Guillaneuf
a,
Claude
Villard
b,
Denis
Bertin
a,
Didier
Gigmes
a and
Kamel
Mabrouk
*a
aAix-Marseille Université, CNRS, UMR 7273, 13397 Marseille Cedex 20, France. E-mail: thomas.trimaille@univ-amu.fr; kamel.mabrouk@univ-amu.fr; Fax: +33 4 9128 8758; Tel: +33 4 9128 8083
bAix-Marseille Université, INSERM, UMR 911 CRO2, 13385 Marseille, France
First published on 17th January 2014
Abstract
Intermolecular radical 1,2-addition (IRA) of N-tert-butyl-N-(1-diethylphosphono-2,2-dimethylpropyl)aminoxyl (SG1) based alkoxyamines onto activated olefins is used as a tool for peptide ligation. This strategy relies on simple peptide pre-derivatization to obtain (i) a SG1 nitroxide functionalized resin peptide at its N-terminus (SG1-peptide alkoxyamine), (ii) a vinyl functionalized peptide (either at its C-terminus or N-terminus), and does not require any coupling agents.
The search for increasingly versatile and efficient ligation–conjugation reactions is a major concern in peptide science, as they can provide valuable edifices in crucial applications such as vaccine/drug design,1–4 antigen capture,5 or biological mechanism elucidation.6 Many chemoselective ligation methods have emerged over the two past decades for coupling of peptide fragments or for peptide modification with biomolecules of interest.7 These methods include native chemical ligation,8,9 imine10 or oxime11 ligation, Staudinger ligation,12–14 triazole formation through alkyne–azide addition,15 decarboxylative condensations of N-alkylhydroxylamines and α-ketoacids16 or, more recently, bis(2-sulfanylethyl)amino native peptide ligation.17 However, to our knowledge, free radical chemistry based reactions have been poorly exploited as a peptide conjugation/ligation strategy, with few studies limited to thiol–ene click,18 radical-based carbonyl addition,19 or carboaminoxylation explored by Studer et al. to immobilize peptides/sugars on polymers.20 In previous studies we reported a novel and efficient free radical reaction, namely intermolecular radical 1,2-addition (IRA) of N-tert-butyl-N-(1-diethylphosphono-2,2-dimethylpropyl)aminoxyl (SG1) based alkoxyamines onto activated vinyl functionalized compounds (Scheme 1),21,22 that enabled the preparation of a range of novel alkoxyamines of interest for use as initiator-controller in the field of controlled radical polymerization. The success of this reaction relies on the higher homolytic dissociation rate constant (kd) value of the starting tertiary stabilized alkoxyamine 1 compared to the one of the alkoxyamine 3 resulting from the addition on the double bond of vinyl compound 2, which bears a less stabilized and bulky secondary radical as the alkyl part. If the reaction temperature is carefully chosen, only the cleavage of the alkoxyamine 1 occurs, and a modest to good yield of the 1,2 adduct 3 is obtained and no oligomerization occurs. The tuning of the experimental conditions thus requires the determination of the dissociation rate constant of the starting alkoxyamines and the use of a kinetic model that we developed previously.22
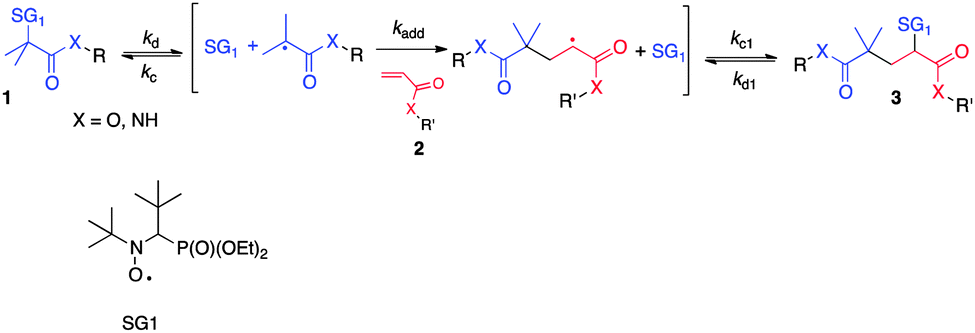 | ||
| Scheme 1 General intermolecular radical 1,2-addition (IRA) of SG1-based alkoxyamines onto activated olefins. | ||
We focus here on the potential of this radical reaction as a novel method for peptide conjugation, termed Alkoxyamine Peptide Ligation (APL). Due to the radical mechanism involved, unprotected lateral functional groups of the peptides are not expected to be involved in side reactions, making the APL approach potentially attractive and versatile. Our strategy simply implies the functionalization of one peptide on its N- or C-terminus with a vinyl group (R′ as the peptidyl moiety, Scheme 1) and that of the other one on its N-terminus with a SG1 moiety (R as the peptidyl moiety, Scheme 1). In our approach, the latter is advantageously kept on the resin to react with the vinyl peptide via the IRA reaction. This strategy produces the desired peptide conjugate after simple cleavage from the resin. As a proof of concept, and as classically reported for any new ligation strategy,13,14,16,17 we worked on model peptide fragments to demonstrate our conjugation approach, namely GGGWIKVAV and GGG/RGDK based peptidyl moieties.
The resin SG1-modified peptide 5 (SG1-MAMA-GGGWIKVAV, containing the laminin-derived IKVAV sequence, Scheme 2) was obtained as previously described.23 Typically, the SG1 moiety was introduced at the N-terminus of the GGGWIKVAV peptide (4), as the last step of Fmoc/t-Bu solid-phase peptide synthesis (SPPS), through a reaction between the carboxylic acid function of the MAMA-SG1 (BlocBuilder MA) alkoxyamine and the peptide terminal amine (PyBOP as a coupling agent), as shown in Scheme 2. Peptide 5 was kept on the resin for further IRA conjugation. The degree of alkoxyamine functionalization and the dissociation rate constant (kd) were assessed by Electron Spin Resonance (ESR), by monitoring the concentration of released SG1 nitroxide upon heating the macroalkoxyamine bound resin. The degree of functionalization was obtained by comparing the final concentration of released nitroxide to the theoretical amount of SG1 bound to the resin, and kd was determined from fitting of the profile of released SG1 vs. time by a first-order kinetic law. The degree of functionalization was found to be 92%, which is a nearly quantitative functionalization of the peptide-bound resin. Experiments were first performed in tert-butyl benzene as the solvent to compare the dissociation rate constant as well as the activation energy with the data of the literature (see ESI† section for details). The activation energy (Ea) was measured to be 119.3 kJ mol−1 (Fig. S1, ESI†), higher than that of the BlocBuilder alkoxyamine (112.3 kJ mol−1), as already observed for other amide-functionalized alkoxyamines derived from BlocBuilder.24 This increase has been ascribed to an intramolecular H-bonding that occurs between the N–H and both the P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and N–O–C groups. The kd measurement was further performed in DMF, as the latter was envisioned for performing the on-resin ligation reaction. Indeed, this solvent has been widely described as the solvent of choice for performing on-resin coupling reactions,25 including on-resin ligations,26 due to its suitable properties of resin-peptide solvation and presentation. In this solvent, a strong increase of the kd value was observed that leads to an activation energy of 109 kJ mol−1 (Fig. S2, ESI†). Some authors27 already observed an increase of the kd value in polar solvents and especially in DMF, but such a difference was barely obtained. This phenomenon could be due to both the disappearance of the H-bonding and an increase of the kd value with the polarity. Such a high dissociation rate is a very interesting feature as it allows to envision to perform the 1,2 radical addition ligation reaction at mild temperatures (70–90 °C) in the convenient DMF solvent.
O and N–O–C groups. The kd measurement was further performed in DMF, as the latter was envisioned for performing the on-resin ligation reaction. Indeed, this solvent has been widely described as the solvent of choice for performing on-resin coupling reactions,25 including on-resin ligations,26 due to its suitable properties of resin-peptide solvation and presentation. In this solvent, a strong increase of the kd value was observed that leads to an activation energy of 109 kJ mol−1 (Fig. S2, ESI†). Some authors27 already observed an increase of the kd value in polar solvents and especially in DMF, but such a difference was barely obtained. This phenomenon could be due to both the disappearance of the H-bonding and an increase of the kd value with the polarity. Such a high dissociation rate is a very interesting feature as it allows to envision to perform the 1,2 radical addition ligation reaction at mild temperatures (70–90 °C) in the convenient DMF solvent.
 | ||
| Scheme 2 The peptide conjugation strategy based on the intermolecular radical 1,2-addition (IRA), via vinyl functionalization of the peptide at the N-terminus (left pathway) or at the C-terminus lysine (right pathway). | ||
In a first approach to prove the IRA-based conjugation concept and to optimize the reaction conditions, the vinyl moiety on the GGG peptide was similarly introduced at the N-terminus through a straightforward reaction of acrylic acid with the peptide α-amine as the last step of the SPPS (Scheme 2, left pathway). After cleavage from the resin, the vinyl peptide 6 (=GGG, M = 242.1) was involved in the IRA reaction with the SG1 resin peptide in DMF (Scheme 2), at various temperatures and reaction times. It should be noted here that, as ligation is performed on the resin, the HPLC characterization of the reaction was systematically performed after TFA cleavage. Although maintained on-resin for the ligation process, the SG1 peptide was of good purity as assessed by HPLC after TFA cleavage (Fig. 1, top, starting time reference chromatogram). It should also be noted that whereas typical solid phase based reactions are usually performed with 3- or 4-fold molar excess of the reactant in solution compared to that on the support, the IRA was here carried out with only a slight excess of vinyl peptide (∼1.2 eq./SG1-peptide), to avoid problems of oligomerization.21 The resulting compound 8 was cleaved from the resin (TFA treatment), and further precipitated in diethyl ether, to afford the peptide 10. HPLC-UV/MS analysis of the crude product showed the presence of the expected product (10, M = 1433.78), whatever the conditions used (Fig. 1). The ligation product is, as for the starting SG1 peptide, devoid of the t-Bu group adjacent to the nitrogen of the SG1 moiety, which is known to leave upon TFA cleavage, as previously described.23,28 At 90 °C, conversion of the SG1 resin-peptide into the conjugation product occurred quite efficiently in the course of time. However, the presence of a side product was observed in significant amounts. This product corresponds to the starting GGGWIKVAV peptide having lost the labile SG1 upon heating (substitution with hydrogen (M = 954.57) or hydroxyl (M = 970.56), Fig. S3, ESI†).29 This was confirmed by performing a reference reaction (DMF, 3 h, 90 °C) in the absence of vinyl peptide, which indeed led to the formation of the hydrogen/hydroxyl substituted product. Interestingly, when decreasing the temperature to 70 °C and increasing the reaction time (72 h), the formation of the side product could be strongly minimized to the benefit of the conjugation product (Fig. 1, bottom). Based on the chromatogram peak areas, it was determined a 58% conjugation yield, 4% remaining starting SG1 resin peptide and 38% side product formation. The ligation product was purified by HPLC (Fig. 2; see Fig. S4 (ESI†) for MS analysis and complete structure of the peptide) and obtained in 35% yield. Its structure was further confirmed by MS/MS analysis (Fig. S5, ESI†).
 | ||
| Fig. 1 HPLC-UV/MS monitoring of the intermolecular radical 1,2-addition (IRA) of the =GGG peptide 6 on the SG1 resin peptide 5, leading to the ligation product 10 (M = 1433.78), at 90 °C (0 to 3 hours) and at 70 °C after 72 h. | ||
 | ||
| Fig. 2 HPLC analysis of the purified peptide adduct 10 (M = 1433.78) obtained by intermolecular radical 1,2-addition (IRA) from the =GGG peptide 6 at 70 °C after 72 h. | ||
The RGDK peptide model (fibronectin-derived peptide), acrylated at the N-terminus (7, M = 527.3), was then similarly involved in the IRA process, with a view to assess the chemoselectivity of the reaction in the presence of various functional groups (carboxylic acid, amine, guanidine). Again the ligation was successful, with a conversion yield of about 50% (HPLC/MS in Fig. S6 (ESI†) for the crude mixture, Fig. S7 (ESI†) for the purified product, 30% yield). The ligation product (11) appeared in the form of two peaks (17.0 and 17.4 min) presenting the same expected mass (1719.0). No side product was detected (except again the initial resin peptide devoid of SG1, M = 954.57 and 970.56), showing the good chemoselectivity of the process.
To finally show the versatility of the IRA based conjugation, we used a peptide functionalized by a vinyl moiety at its C-terminal lysine (12, GGGK=, M = 370.2, Scheme 2, right pathway), which allows to envision the non-native ligation reaction with the N-terminal SG1 functionalized peptide SG1-MAMA-GGGWIKVAV (5). To introduce the vinyl moiety on the peptide, the monomethoxytrityl (Mmt) protecting group of the lysine ε-amine was selectively removed for an ε-amine reaction with acryloyl chloride, after incorporation of the last amino-acid (Fmoc-Gly-OH) along the SPPS process. Again, the ligation product (14, M = 1561.87) was successfully obtained (Fig. S8, ESI†). The conversion yield was lower than that previously observed in the N-terminus approach under the same reaction conditions (either at 90 °C or 70 °C), but could be increased to 20% by increasing the amounts of acrylated peptide (2 eq./SG1-peptide). The purified peptide (Fig. S9, ESI†) was isolated at a yield of 15%. The lower reactivity could be ascribed to the “axial” position of the double bond (lysine end-functionalization) in the peptidic chain, as compared to that of the N-terminal approach.
In conclusion, in this work we have demonstrated the efficiency of Alkoxyamine Peptide Ligation (APL), a novel straightforward radical chemistry based strategy for peptide conjugation through Intermolecular Radical 1,2-Addition (IRA) between a peptide alkene and a peptide alkoxyamine. The reaction proceeds without any coupling agents and in the solid phase (use of the SG1 functionalized peptide bound to the resin), making further purification of the peptide adduct easier. This conjugation approach can thus represent a breakthrough in the field of peptide ligation, and is currently under further investigation in our group with respect to medium-sized peptide coupling and possible requirements for orthogonal protection of methionine and cysteine residues during the radical process.
The authors wish to acknowledge Aix-Marseille Université and Arkema for financial support.
Notes and references
- J. Chen, S. Jaracz, X. Zhao, S. Chen and I. Ojima, Expert Opin. Drug Delivery, 2005, 5, 873 CrossRef PubMed.
- F. Hudecz, Z. Bánóczi and G. Csík, Med. Res. Rev., 2005, 6, 679 CrossRef PubMed.
- K. S. Rosenthal, H. Mao, W. I. Horne, C. Wright and D. Zimmerman, Vaccine, 1999, 17, 535 CrossRef CAS.
- G. A. Cremer, N. Bureaud, D. Lelievre, V. Piller, F. Piller and A. Delmas, Chem.–Eur. J., 2004, 10, 6353 CrossRef CAS PubMed.
- Y. Jung, H. J. Kang, J. M. Lee, S. O. Jung, W. S. Yun, S. J. Chung and B. H. Chung, Anal. Biochem., 2008, 374, 99 CrossRef CAS PubMed.
- S. Futaki, Adv. Drug. Delivery Rev., 2005, 57, 547 CrossRef CAS PubMed.
- (a) C. P. R. Hackenberger and D. Schwarzer, Angew. Chem., Int. Ed., 2008, 47, 10030 CrossRef CAS PubMed; (b) G. A. Eggimann, S. Buschor, T. Darbre and J. L. Reymond, Org. Biomol. Chem., 2013, 11, 6717 RSC; (c) M. Galibert, O. Renaudet, P. Dumy and D. Boturyn, Angew. Chem., 2011, 123, 1941 CrossRef.
- P. E. Dawson, T. W. Muir, I. Clark-Lewis and S. B. H. Kent, Science, 1994, 266, 776 CAS.
- P. E. Dawson and S. B. H. Kent, Annu. Rev. Biochem., 2000, 69, 923 CrossRef CAS PubMed.
- C. F. Liu and J. P. Tam, Proc. Natl. Acad. Sci. U. S. A., 1994, 91, 6584 CrossRef CAS.
- J. Shao and J. P. Tam, J. Am. Chem. Soc., 1995, 117, 3893 CrossRef CAS.
- E. Saxon and C. R. Bertozzi, Science, 2000, 287, 2007 CrossRef CAS.
- B. L. Nilsson, L. L. Kiessling and R. T. Raines, Org. Lett., 2000, 2, 1939 CrossRef CAS PubMed.
- R. Merkx, D. T. S. Rijkers, J. Kemmink and R. M. J. Liskamp, Tetrahedron Lett., 2003, 44, 4515 CrossRef CAS.
- (a) V. Aucagne, I. E. Valverde, P. Marceau, M. Galibert, N. Dendane and A. F. Delmas, Angew. Chem., 2012, 124, 11482 CrossRef; (b) Y. L. Angell and K. Burgess, Chem. Soc. Rev., 2007, 36, 1674 RSC.
- J. W. Bode, R. M. Fox and K. D. Baucom, Angew. Chem., Int. Ed., 2006, 45, 1248 CrossRef CAS PubMed.
- N. Ollivier, J. Dheur, R. Mhidia, A. Blanpain and O. Melnyk, Org. Lett., 2010, 12, 5238 CrossRef CAS PubMed.
- S. Wittrock, T. Becker and H. Kunz, Angew. Chem., Int. Ed., 2007, 46, 5226 CrossRef CAS PubMed.
- T. Mittag, K. L. Christensen, N. C. Nielsen and T. Skrydstrup, J. Org. Chem., 2008, 73, 1088 CrossRef CAS PubMed.
- J. P. Lindner and A. Studer, Chem.–Eur. J., 2011, 17, 4090 CrossRef CAS PubMed.
- P. E. Dufils, N. Chagneux, D. Gigmes, T. Trimaille, S. R. A. Marque, D. Bertin and P. Tordo, Polymer, 2007, 48, 5219 CrossRef CAS PubMed.
- D. Gigmes, P. E. Dufils, D. Glé, D. Bertin, C. Lefay and Y. Guillaneuf, Polym. Chem., 2011, 2, 1624 RSC.
- T. Trimaille, K. Mabrouk, V. Monnier, L. Charles, D. Bertin and D. Gigmes, Macromolecules, 2010, 43, 4864 CrossRef CAS.
- N. Chagneux, T. Trimaille, M. Rollet, E. Beaudoin, P. Gérard, D. Bertin and D. Gigmes, Macromolecules, 2009, 42, 9435 CrossRef CAS.
- (a) C. K. Taylor, P. W. Abel, M. Hulce and D. D. Smith, J. Pept. Res., 2005, 65, 84 CrossRef CAS PubMed; (b) E. M. Cilli, E. Oliveira, R. Marchetto and C. R. Nakaie, J. Org. Chem., 1996, 61, 8992 CrossRef CAS PubMed.
- J. Tulla-Puche and G. Barany, J. Org. Chem., 2004, 69, 4101 CrossRef CAS PubMed.
- (a) G. Audran, P. Brémond, S. R. A. Marque and G. Obame, Polym. Chem., 2012, 3, 2901 RSC; (b) S. Harrisson, P. Couvreur and J. Nicolas, Macromol. Rapid Commun., 2012, 33, 805 CrossRef CAS PubMed; (c) K. H. Kuo, W. Y. Chiu and K. C. Cheng, Polym. Int., 2008, 57, 730 CrossRef CAS.
- C. Barrère, C. Chendo, T. N. T. Phan, V. Monnier, T. Trimaille, S. Humbel, S. Viel, D. Gigmes and L. Charles, Chem.–Eur. J., 2012, 18, 7916 CrossRef PubMed.
- The first hydrogen derived compound arises from H abstraction on the peptidyl radical formed SG1-peptide dissociation. As for the hydroxylated it most probably arises from oxygen addition on the peptidyl radical, followed by formation of a tetroxide and further homolytic dissociation, leading to the alkoxyl radical and subsequent H abstraction ( P. Brémond, et al. , Tetrahedron Lett., 2012, 53, 4543 CrossRef PubMed ). Indeed, despite degassing conditions used for 1,2-addition, residual oxygen is reported to induce side products, unless using drastic vacuum conditions to enable complete absence of oxygen ( M. Edeleva, et al. , J. Polym. Sci., Part A: Polym. Chem., 2012, 51, 1323 CrossRef ).
Footnote |
| † Electronic supplementary information (ESI) available: Details of synthesis, experimental procedures and peptide characterization. See DOI: 10.1039/c3cc49389j |
| This journal is © The Royal Society of Chemistry 2014 |