 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Poly(3-alkylthiophene)s show unexpected second-order nonlinear optical response†
S.
Deckers
a,
S.
Vandendriessche
a,
D.
Cornelis
b,
F.
Monnaie
b,
G.
Koeckelberghs
b,
I.
Asselberghs
c,
T.
Verbiest
a and
M. A.
van der Veen
*d
aLaboratory for Molecular Electronics and Photonics, University of Leuven, Celestijnenlaan 200D, 3001 Leuven, Belgium
bLaboratory for Polymer Synthesis, University of Leuven, Celestijnen 200F, 3001 Leuven, Belgium
cImec, Kapeldreef 75, 3001 Leuven, Belgium
dCatalysis Engineering, Chemical Engineering Department, Delft University of Technology, Julianalaan 136, BL 2826 Delft, The Netherlands. E-mail: m.a.vanderveen@tudelft.nl
First published on 16th January 2014
Abstract
Regioregular poly(3-hexylthiophene)s with chain lengths varying from 5 to 100 monomers are synthesized. Poly(3-hexylthiophene)s show in solution an unexpectedly significant second-order nonlinear optical response. The increase in transition dipole moment upon oligomerisation causes the significant second-order nonlinear optical response.
Second-order nonlinear optical materials are applied as electro-optical modulators, for frequency doubling, terahertz generation,1 and are more intensively investigated as materials for optical computing.2 So far, nearly all commercially used materials are inorganic materials. Organic materials however show faster response times,3 and have more process flexibility, providing more potential for downscaling into functional devices. In this context, active polymers are of particular interest due to their increased stability over functionalised blends.
The versatile use of poly(3-alkylthiophene)s and conjugated polymers in general is widely explored in research fields such as organic field effect transistors (OFET), solar cells, organic light emitting diodes (OLED) and nonlinear optical devices.4 The broad applicability of polythiophenes resulted in a large product diversity as well as in a wide range of synthetic routes to produce them in reasonable yields and obtain meso and nano scale architectures.5 Although already investigated for their third order nonlinear optical response,6 their second order nonlinear optical response has not been investigated thoroughly. This is largely due to the fact that the structure of polythiophenes does not follow the typical donor – conjugated structure – acceptor paradigm, and therefore no significant second-order nonlinear effect is expected. Typically, high second-order nonlinear optical responses of materials are expected in extended conjugated systems that are non-centrosymmetrically organized.7 The dominant strategy is to utilize dipolar chromophores, either in the polymer backbone covalently bonded to the side chains or via doping polymers with chromophores.8
Previously, a scaling study of the second-order nonlinear optical response has been conducted on non-conjugated oligonucleotides up to 50 monomers.9 However, to the best of our knowledge, scaling studies of conjugated polymers are limited to 4–5 monomers.10 Herein, we report for the first time the second-order nonlinear optical response of conjugated polymers upon increasing the chain length to 100 units. More specifically, regioregular poly(3-hexylthiophene)s (P3HT) are studied. Despite the limited donor strength of the alkyl substituents, an appreciably large second-order nonlinear optical response is observed. Upon increasing the polymerization degree the materials show a dramatic increase in the second-order response due to an increase in the transition dipole moment.
A series of varying chain lengths of regioregular head-to-tail coupled P3HT polymers (see Scheme 1) is synthesized. The small polydispersities (∼1.2) were confirmed by gel permeation chromatography. The degree of polymerization was determined by 1H NMR by end-group quantification.11 We obtained a series ranging from 5 to 100 repeating monomer units (see ESI†).
 | ||
| Scheme 1 Head-to-tail coupled regioregular poly(3-alkylthiophene). | ||
The second-order nonlinear optical response of the polymers was determined in solution via a Hyper-Rayleigh Scattering (HRS) experiment at a fundamental input wavelength of 800 nm. The incoherent scattered light at the second harmonic wavelength of 400 nm is detected in a perpendicular geometry. To separate the frequency doubled light from two photon absorption fluorescence at the same frequency, we take advantage of the time-delay of fluorescence versus the quasi instant generation of frequency doubled light. This is done in the frequency domain according to the method described by Olbrechts et al.12 Further details of the experimental set-up and conditions can be found in the ESI.†
We report the directly measured hyperpolarizability β, as well as the static hyperpolarizability β0 in order to exclude contributions from resonance enhancement. The static hyperpolarizability β0 is a frequency independent quantity that allows comparison of molecules that have different electronic resonances. It thus allows us to compare the performance of different organic molecules which is of relevance to optoelectronic applications. We thus adjust the red-shift in electronic resonance frequency upon increasing the chain length of P3HT by applying the homogeneously damped two-state model to derive the static hyperpolarizability β0 from the measured hyperpolarizability β (see ESI†).13
The P3HT oligomers/polymers are dissolved in a good solvent, chloroform, at a typical concentration of 0.1 mM of monomer units to ensure complete solubility. In Fig. 1, we show the observed hyper-Rayleigh response per polymer chain βpol as a function of the number of monomer units per polymer chain n. An apparent linear increase of the HRS-response upon chain lengthening is observed (the fitted line is guidance to the eye). However, when rescaling the data towards the number of monomer units in the polymer backbone (βpol/n), a totally different trend is observed (Fig. 2). The depiction of βpol/n is a means of comparing the amount of second-order nonlinear optical response generated by molecules of different molecular weight as it depicts the hyperpolarizability for the same density of material. Initially the HRS response βpol/n increases upon chain lengthening, but saturation occurs for chain lengths longer than 15 repeating monomer units. The same trend is observed for the directly measured βpol/n as well as for the static βpol,0/n (see Fig. 2). At this point, it is worthwhile mentioning that the monomer unit by itself has a very small hyperpolarizability that cannot be measured using our HRS set-up. This implies that the 2nd order NLO response does not originate from the 3-alkylthiophene monomer itself, but from a cooperative effect of the monomers within one oligomer–polymer chain such as the delocalized π-conjugated backbone.
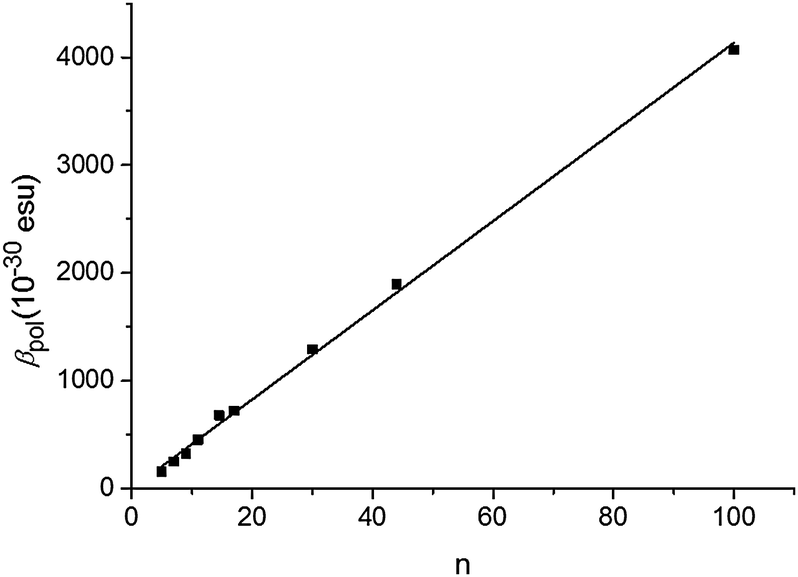 | ||
| Fig. 1 Hyperpolarizability βpolversus the number of monomers n of P3HT. | ||
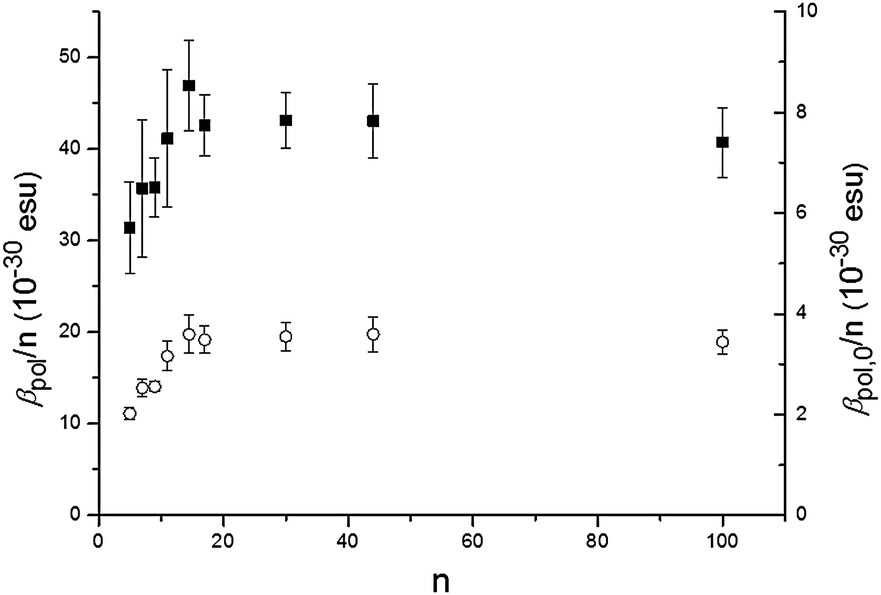 | ||
| Fig. 2 Hyperpolarizability per monomer unit βpol/n (filled squares) and the static hyperpolarizability per monomer unit βpol,0/n (open circles) as a function of the number of monomer units n of P3HT. | ||
To understand which properties of molecules contribute to the second-order nonlinear response, we provide the description of the static hyperpolarizability β0 within the two-state model where two states provide the dominant contribution to hyper-Rayleigh scattering14
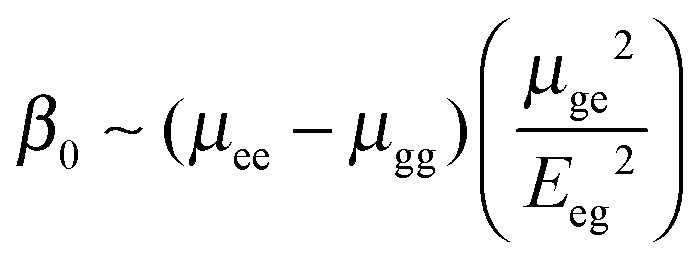
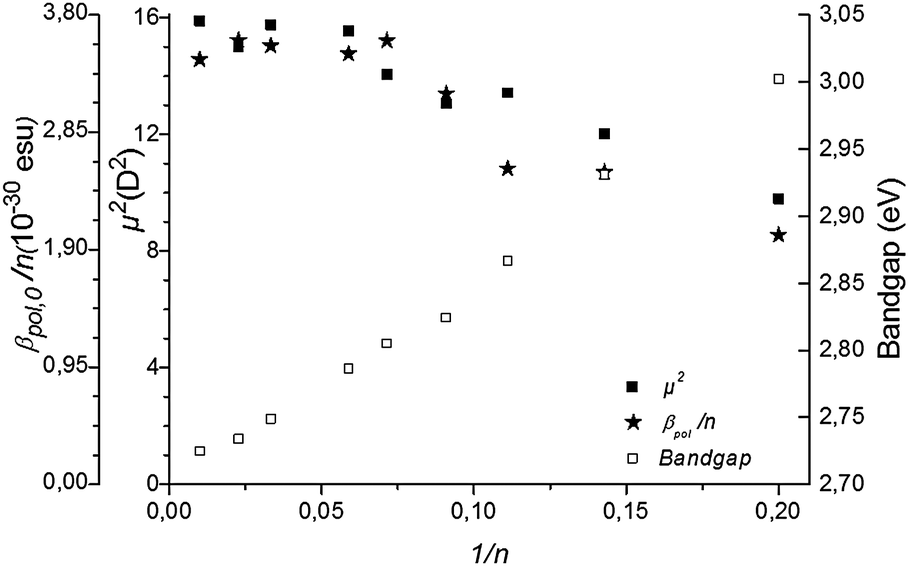 | ||
| Fig. 3 The transition dipole moment μge2 and the static hyperpolarizability per monomer unit βpol,0/n and the bandgap are given as a function of the inverse of the number of monomer units n of P3HT. | ||
Several effects can cause the non-centrosymmetry required for the second order NLO effect. The alkyl substituents, all in the 3-position in the regioregular P3HT, cause breaking of non-centrosymmetry (see Scheme 1). The effect on the NLO response is expected to be relatively small due to the limited donor strength of alkyl substituents. A non-planar conformation of the backbone15 with helical structure can also cause asymmetry. As non-centrosymmetry is a requirement for the second-order NLO effect, the conformation will have an effect on the second order NLO response. Experimental and theoretical studies have shown that the persistence length – a measure of the length of rodlike or straight conformation within a polymer – of P3HT in a good solvent is about 10 units.16 Yet, the effect of conformation is not clear from the data. Indeed, as can be seen from Fig. 2, the trend in the hyperpolarizability is largely explained by electronic effects.
As mentioned, the second-order nonlinear optical response of the monomer could not be measured. This means that the extension of conjugation leads to a dramatic enhancement of the second-order nonlinear optical properties of the material. This is also in line with the theoretical results that predict a generally larger than 3-fold increase of the hyperpolarizability upon extending from the thiophene monomer to the trimer.17 If we compare the static hyperpolarizability βpol,0 for 5 units thus having 32‡ conjugated electrons, 17.7 ± 1.2 × 10−30 esu, with p-nitroaniline, a benchmark NLO molecule, extended with two additional benzene rings to 4-nitro-4′′-amino-p-terphenyl, thus having 22 conjugated electrons, we find a very similar hyperpolarizability β, namely 16 × 10−30 esu.18 The second-order nonlinear optical response of P3HT is thus very significant, while it was expected to be negligible. The best performing organic molecules typically have a hyperpolarizability one order of magnitude larger,19 but polythiophenes outperform these complex organic molecules with respect to easy synthesis and processability into devices.5,20 Moreover, in the solid-state polythiophenes display much more planar conformations with higher electron delocalization and less bending can be achieved.21 Based on the results reported here, we conclude that such solid-state polythiophenes could have an even larger second-order nonlinear optical response.
In conclusion, we found for oligomers of P3HT in solution that they, despite the limited donor strength of the substituents, show an unexpectedly significant second-order nonlinear optical response. This response is in the order of a benchmark nonlinear optical molecule. The dramatic increase in hyperpolarizability with chain length is largely attributed to the increased conjugation length. This is the first systematic study of second-order nonlinear scattering response of a conjugated polymer as a function of chain length.
MAvdV and SV acknowledge the Scientific Research Fund Flanders (FWO) for their personal fellowships. We are also grateful to the Onderzoeksfonds KU Leuven/Research Fund KU Leuven for a GOA and CREA grant.
Notes and references
- (a) L. R. Dalton, P. A. Sullivan and D. H. Bale, Chem. Rev., 2010, 110, 25–55 CrossRef CAS PubMed; (b) M. Lee, H. E. Katz, C. Erben, D. M. Gill, P. Gopalan, J. D. Heber and D. J. McGee, Science, 2002, 298, 1401–1403 CrossRef CAS PubMed; (c) X. Zheng, C. V. McLaughlin, P. Cunningham and L. M. Hayden, J. Nanoelectron. Optoelectron., 2007, 2, 58–76 CrossRef PubMed; (d) L. R. Dalton, S. J. Benight, L. E. Johnson, D. B. Knorr, I. Kosilkin, B. E. Eichinger, B. H. Robinson, A. K.-Y. Jen and R. M. Overney, Chem. Mater., 2011, 23, 430–445 CrossRef CAS.
- J. L. O`Brien, Science, 2007, 318, 1567–1570 CrossRef PubMed.
- J. M. Cole, Philos. Trans. R. Soc., A, 2003, 361, 2751–2770 CrossRef CAS PubMed.
- (a) G. Wang, J. Swensen, D. Moses and A. J. Heeger, J. Appl. Phys., 2003, 93, 6137 CrossRef CAS PubMed; (b) G. Li, V. Shrotriya, J. Huang, Y. Yao, T. Moriarty, K. Emery and Y. Yang, Nat. Mater., 2005, 4, 864 CrossRef CAS; (c) N. R. Armstrong, W. Wang, D. M. Alloway, D. Placencia, E. Ratcliff and M. Brumbach, Macromol. Rapid Commun., 2009, 30, 717–731 CrossRef CAS PubMed.
- (a) B. H. Cumpston, S. P. Ananthavel, S. Barlow, D. L. Dyer, J. E. Ehrlich, L. L. Erskine, A. A. Heikal, S. M. Kuebler, M. Rumi, X. Wu, S. R. Marder and J. W. Perry, Nature, 1999, 398, 51–54 CrossRef CAS PubMed; (b) G. Zotti, B. Vercelli and A. Berlin, Acc. Chem. Res., 2008, 41, 1098–1109 CrossRef CAS PubMed; (c) I. Osaka and R. D. McCullough, Acc. Chem. Res., 2008, 41, 1202–1214 CrossRef CAS PubMed.
- (a) S. Kishino, Y. Ueno, K. Ochiai, M. Rikukawa, K. Sanui, T. Kobayashi, H. Kunugita and K. Ema, Phys. Rev. B, 1998, 58, 430–433 CrossRef; (b) M.-T. Zhao, B. P. Singh and P. N. Prasad, J. Chem. Phys., 1988, 89, 5535 CrossRef CAS PubMed; (c) Y. Verbandt, H. Thienpont, I. Veretennicoff, P. Geerlings and G. L. J. a. Rikken, Chem. Phys. Lett., 1997, 270, 471–475 CrossRef CAS; (d) T. Bjornholm, D. R. Greve, T. Geisler, J. C. Petersen, M. Jayaraman and R. D. McCullough, Adv. Mater., 1996, 8, 920 CrossRef; (e) T. Bjornholm, D. R. Greve, T. Geisler, J. C. Petersen, M. Jayaraman and R. D. McCullough, Synth. Met., 1997, 84, 531–532 CrossRef CAS.
- T. Verbiest, K. Clays and V. Rodriguez, Second-Order Nonlinear Optical Characterization Techniques, CRC Press, Boca Raton, 2009 Search PubMed.
- (a) P. N. Prasad and D. J. Williams, Introduction to nonlinear optical effects in molecules & polymers, Wiley-Interscience, New York, 1991 Search PubMed; (b) M. J. Cho, D. H. Choi, P. A. Sullivan, A. J. P. Akelaitis and L. R. Dalton, Prog. Polym. Sci., 2008, 33, 1013–1058 CrossRef CAS PubMed.
- Thèse EPFL nr 2414 (2001). Rinuy, Juliette: Nonlinear optics of proteins at liquid interfaces and of DNA oligonucleotides in liquid phase.
- M. G. Kuzyk, J. Pérez-Moreno and S. Shafei, Phys. Rep., 2013, 529, 297–398 CrossRef PubMed.
- M. Verswyvel, F. Monnaie and G. Koeckelberghs, Macromolecules, 2011, 44, 8489–8498 CrossRef.
- G. Olbrechts, R. Strobbe, K. Clays and A. Persoons, Rev. Sci. Instrum., 1998, 69, 2233–2241 CrossRef CAS PubMed.
- B. J. Orr and J. F. Ward, Mol. Phys., 1971, 20, 513 CrossRef CAS.
- (a) C. B. Gorman and S. R. Marder, Proc. Natl. Acad. Sci. U. S. A., 1993, 90, 11297–11301 CrossRef CAS; (b) J. L. Oudar and D. S. Chemla, J. Chem. Phys., 1977, 66, 2664 CrossRef CAS PubMed; (c) J. Zyss and J. L. Oudar, Phys. Rev. A, 1982, 26, 2016 CrossRef; (d) J. L. Brdas, F. Meyers, B. M. Pierce and J. Zyss, J. Am. Chem. Soc., 1992, 114, 4928 CrossRef.
- S. R. Bhatta, Y. Y. Yimer, M. Tsige and D. S. Perry, Comput. Theor. Chem., 2012, 995, 36–42 CrossRef PubMed.
- (a) G. M. Heffner and D. S. Person, Macromolecules, 1991, 24, 6295 CrossRef CAS; (b) P. V. Shibaev, K. Schaumburg, T. Bjornholm and K. Norgaard, Synth. Met., 1998, 97, 97–104 CrossRef CAS; (c) B. McCulloch, V. Ho, M. Hoarfrost, C. Stanley, C. Do, W. T. Heller and R. A. Segalman, Macromolecules, 2013, 46, 1899–1907 CrossRef CAS.
- J. Waite and M. G. Papadopoulos, J. Phys. Chem., 1990, 94, 6244–6249 CrossRef CAS.
- L.-T. Cheng, W. Tam, S. R. Marder, A. E. Stiegman, G. Rikken and C. W. Spangler, J. Phys. Chem., 1991, 95, 10643 CrossRef CAS.
- (a) B. Coe, J. A. Harris, I. Asselberghs, K. Clays, G. Olbrechts, A. Persoons, J. T. Hupp, R. C. Johnson, S. J. Coles, M. B. Hursthouse and K. Nakatani, Adv. Funct. Mater., 2002, 12, 110–116 CrossRef CAS; (b) J. Pérez-Moreno, Y. Zhao, K. Clays and M. Kuzyk, Opt. Lett., 2007, 32, 59–61 CrossRef.
- (a) C. L. Gettinger, A. J. Heeger, J. M. Drake and D. J. Pine, J. Chem. Phys., 1994, 101, 1673–1678 CrossRef CAS PubMed; (b) R. R. Tykwinski, U. Gubler, R. E. Martin, C. Bosshard and P. Gu, J. Phys. Chem. B, 1998, 102, 4451–4465 CrossRef CAS; (c) J. Hou, M.-H. Park, S. Zhang, Y. Yao, L.-M. Chen, J.-H. Li and Y. Yang, Macromolecules, 2008, 41, 6012–6018 CrossRef CAS.
- (a) M. Chang, D. Choi, B. Fu and E. Reichmanis, ACS Nano, 2013, 7, 5402–5413 CrossRef CAS PubMed; (b) A. R. Aiyar, J.-I. Hong, R. Nambiar, D. M. Collard and E. Reichmanis, Adv. Funct. Mater., 2011, 21, 2652–2659 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Synthesis of P3HT, details of UV-VIS and HRS set-up and measurements and homogeneously damped two-level model. See DOI: 10.1039/c3cc48099b |
| ‡ Each of the five thiophene units has 6 conjugated electrons. In addition, due to the synthesis procedure a bromo substituent is present at one end of the oligomer. This brings the total of conjugated electrons to 32. |
| This journal is © The Royal Society of Chemistry 2014 |