 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
‘Click cyclic ADP-ribose’: a neutral second messenger mimic†
Joanna M.
Swarbrick
a,
Richard
Graeff
b,
Clive
Garnham
c,
Mark P.
Thomas
a,
Antony
Galione
c and
Barry V. L.
Potter
*b
aWolfson Laboratory of Medicinal Chemistry, Dept. of Pharmacy and Pharmacology, University of Bath, Bath, BA2 7AY, UK. E-mail: B.V.L.Potter@bath.ac.uk; Fax: +44-(0)1225-386114; Tel: +44-(0)1225-386639
bDepartment of Physiology, University of Hong Kong, Hong Kong, China
cDepartment of Pharmacology, University of Oxford, Mansfield Road, Oxford OX1 3QT, UK
First published on 15th January 2014
Abstract
Analogues of the potent Ca2+ releasing second messenger cyclic ADP-ribose (cADPR) with a 1,2,3-triazole pyrophosphate bioisostere were synthesised by click-mediated macrocyclisation. The ability to activate Ca2+ release was surprisingly retained, and hydrolysis of cADPR by CD38 could also be inhibited, illustrating the potential of this approach to design drug-like signalling pathway modulators.
Intracellular Ca2+ plays a key role in many processes including cell division, muscle contraction, cell death and fertilisation.1 The spatial and temporal release of Ca2+ within the cell is controlled by second messengers. In contrast to the now well characterized IP3,2 the three Ca2+ mobilizing nucleotides adenosine 5′-diphosphate ribose (ADPR), cyclic-ADPR (cADPR), and nicotinic acid adenine dinucleotide phosphate (NAADP) are relatively new second messengers whose specialised roles are only just emerging. Currently, the only examples of neutral, active, drug-like molecules able to modulate one of these critical pathways are the NAADP signalling probe ‘Ned-19’, that was identified by virtual screening,3 and very recently SAN2589 and SAN4825, identified by high-throughput screening, and specific to the cardiac cADPR signalling pathway.4
In humans cADPR (1, Fig. 1),5 like ADPR and NAADP, is formed from nicotinamide adenine dinucleotide by CD38, a multifunctional ADP-ribosyl cyclase6 involved in disease states including AIDs, leukaemia, diabetes and inflammation.7 Two opposing orientations of CD38 have been reported, allowing the catalytic domain to be both extra- or intracellular and resolving a long-standing topological paradox.8 Ca2+ release by cADPR occurs at the ryanodine receptor and may require accessory proteins; the mechanism remains controversial.9 The cADPR signalling pathway is thus in pressing need of new modulators for chemical biological intervention and as prototype therapeutic candidates. However, its charged pyrophosphate motif is unattractive in molecules for medicinal and clinical development, due to obvious difficulties with membrane permeability and stability. Furthermore, cADPR is readily hydrolysed in both neutral aqueous solution and under physiological conditions.10
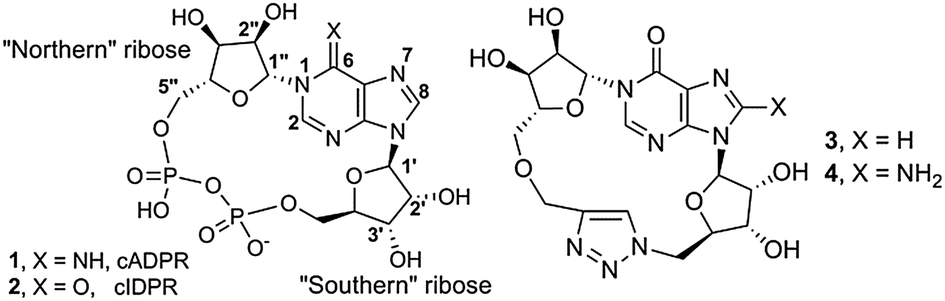 | ||
| Fig. 1 Structure of cADPR, cIDPR and 1,4-triazole analogues 3 and 4. | ||
Despite the preparation of numerous cADPR analogues,11 previous work has failed to address the critical issue of the pyrophosphate. Furthermore, analogue synthesis has been limited in scope, yield and number by either the substrate specificity of Aplysia californica cyclase12 or because of arduous multi-step synthetic routes.13 While introduction of a third phosphate maintains potent agonist activity,14 the pyrophosphate region of cADPR is demonstrably sensitive to conservative modifications: a methylenebisphosphonate substitution is less active15 and sulphur or selenium-substituted pyrophosphates demonstrate contrasting activities between diastereoisomers in ether-substituted analogues.16 Alkylating the pyrophosphate has been used to generate “caged cADPR” and derivatives as tools with improved membrane permeability, but such analogues themselves are biologically inactive.17 Such approaches do not address hydrolysis of the pyrophosphate bond18 and the dramatic impact on activity observed with such minor alterations seems to suggest that complete removal of the charged motif would likely be fruitless for the generation of active compounds.
Pyrophosphates are one of the most challenging functionalities to replace in the generation of biologically active, drug-like compounds.19 They are negatively charged at physiological pH20 and this usually corresponds to positively charged or electron poor residues in a binding counterpart, a consideration for any neutral replacement.21 1,2,3-Triazoles are neutral, but possess nitrogen lone pairs that may supply electron density during binding. While present in some linear inhibitors22 and carbohydrate analogues,23 no such substitution has been explored in any cyclic nucleotide.
To avoid instability and remove the partial positive charge at N1, we chose an N1-cyclic inosine 5′-diphosphate ribose (cIDPR, 2)24 scaffold. This 6–NH2 → 6![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O substitution in cADPR led to an analogue that inhibits hydrolysis of cADPR by CD3825 and is an agonist for Ca2+ release in permeabilized T-cells with essentially equivalent potency to cADPR.24 Therefore, we designed a suitable N1-ribosylated inosine26 precursor to explore Huisgen 1,3-dipolar cycloaddition.27
O substitution in cADPR led to an analogue that inhibits hydrolysis of cADPR by CD3825 and is an agonist for Ca2+ release in permeabilized T-cells with essentially equivalent potency to cADPR.24 Therefore, we designed a suitable N1-ribosylated inosine26 precursor to explore Huisgen 1,3-dipolar cycloaddition.27
A 5′-azido group was installed on 2′,3′-O-isopropylidene adenosine (5, Scheme 1), under Mitsunobu-like conditions.28 The azide was installed prior to the 8-bromo substituent that is required to promote N1-ribosylation,26 to circumvent concomitant nucleophilic substitution of the 8-Br generating an unwanted 8-N3 product with an undesirable second site for click cyclisation. Subsequently, the 8-Br substituent was installed by treatment of 6 with Br2 in dioxane–NaOAc buffer at pH 4 to generate protected 5′-azido-5′-deoxy-8-bromoadenosine 7. This was followed by conversion to the corresponding 8-bromoinosine 8 by treatment with a large excess of sodium nitrite in aqueous acetic acid.
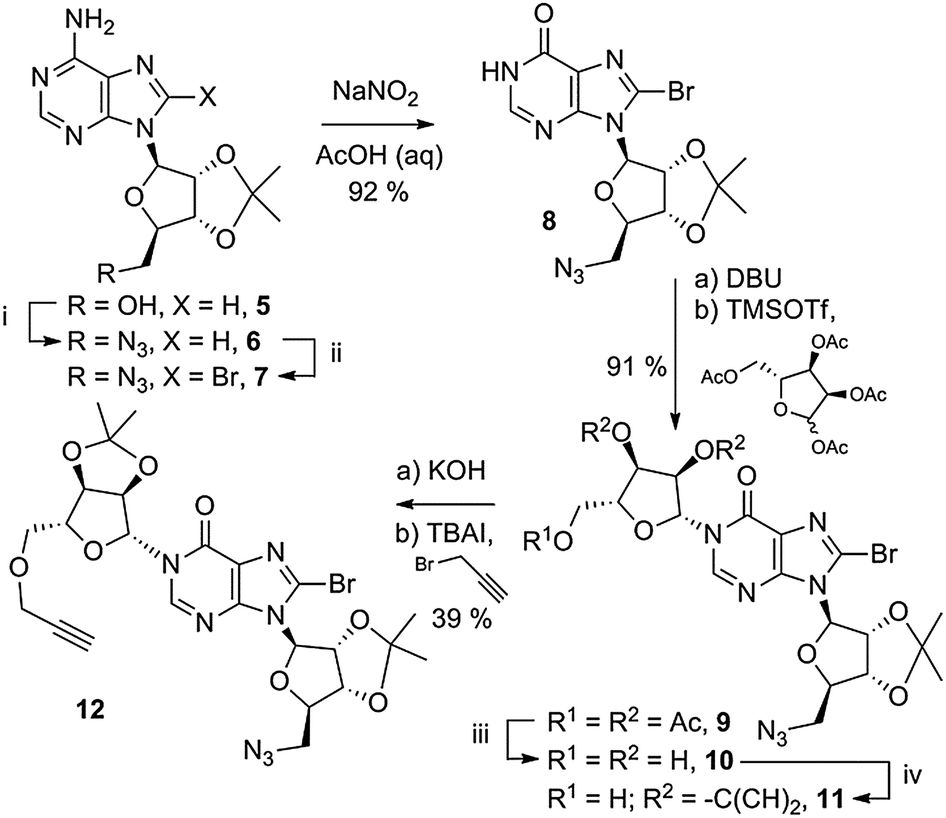 | ||
| Scheme 1 Synthesis of linear precursor suitable for click cyclisation. Reagents and conditions (i) (a) DPPA, DBU; (b) NaN3, TBAI, 15-crown-5, 100%; (ii) Br2, pH 4, 99%; (iii) NH3, MeOH, 81%; (iv) Me2C(OMe)2, acetone, pTsOH, 93%. | ||
5′-Azido-5′-deoxy-2′,3′-O-isopropylidene-8-bromoinosine 8 was coupled to the tri-O-acetyl protected “northern” ribose unit in 91% yield. No evidence of competing O-6 alkylation was observed. The three acetyl esters were removed using methanolic ammonia to generate triol 10 and the 2′′,3′′-diol was selectively reprotected as an isopropylidene ketal in intermediate 11. Initial attempts to introduce a 5′′-O-propargyl ether to 11 were not successful. Deprotonation with NaH led to rapid decomposition of the starting material and deprotonation with DBU followed by addition of propargyl-chloride recovered only unreacted starting material. Upon treatment of 11 with KOH in MeCN, a white precipitate was observed and no further reaction occurred. Optimising the solvent conditions to dioxane–toluene (2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 v/v) in which the anion of 11 remained in solution, followed by addition of the more reactive electrophile propargyl-bromide generated the desired ether 12 in moderate yield.
:1 v/v) in which the anion of 11 remained in solution, followed by addition of the more reactive electrophile propargyl-bromide generated the desired ether 12 in moderate yield.
In initial attempts to promote click cyclisation, using in situ reduction of Cu(II), a dilute solution of 12 was added to a solution of copper sulfate and sodium ascorbate by syringe pump, but after 72 hours no reaction was observed. In contrast, direct addition of Cu(I)I to a 1 mg mL−1 degassed solution of 12 and diisopropylethylamine in THF27b followed by 16 h reflux generated a single product. 1H-NMR spectroscopy of the crude material revealed that an exceedingly clean conversion had taken place, characterised by a new singlet at 7.15 ppm indicative of a 1,4-disubstituted-1,2,3-triazole and disappearance of the alkynyl proton (δ = 2.46 ppm). No evidence of any intermolecular reaction was observed. Treatment of 1,4-disubstituted triazole 13 with aq. TFA then removed both isopropylidene ketal protecting groups to generate 14 (Scheme 2).
 | ||
| Scheme 2 Synthesis of 8-H and 8-NH2 click cADPR analogues 3 and 4. | ||
The resulting 8-bromotriazole derivative 14 is insoluble in D2O. However, reduction of the 8-bromo substituent with Pd/C under an atmosphere of H2 generated the water-soluble cyclic triazole 3. Treatment of 14 with sodium azide in DMF generated the 8-azidotriazole 15, also insoluble in aqueous media. Reduction of the 8-azido to the 8-amino group by hydrogenation generated a second water soluble analogue 4. No decomposition of the cyclic 1,4-disubstituted triazole analogues was observed during deprotection with TFA or at elevated reaction temperatures (70 °C), suggesting that they are chemically stable.
The ability of 3 and 4 to inhibit the enzyme activity of CD38 was assessed. Both novel compounds inhibit hydrolysis of cADPR by CD38, suggesting that they block entry of cADPR into the binding pocket. Non-substituted 3 is a slightly poorer inhibitor (IC50 396 μM, Fig. 2) than the parent template cIDPR (IC50 260 μM, ref. 25). However, introduction of the 8-amino substituent in 4 improves inhibition (IC50 185 μM). The similarity in inhibition shown by cIDPR, 3 and 4 suggests that the ability of these analogues to enter the binding pocket is surprisingly not reduced by the pyrophosphate → 1,2,3-triazole substitution. In fact, the reduction in macrocycle flexibility caused by the planar triazole may even reduce the overall size of the ligand, making it easier for 3 and 4 to enter the binding pocket.
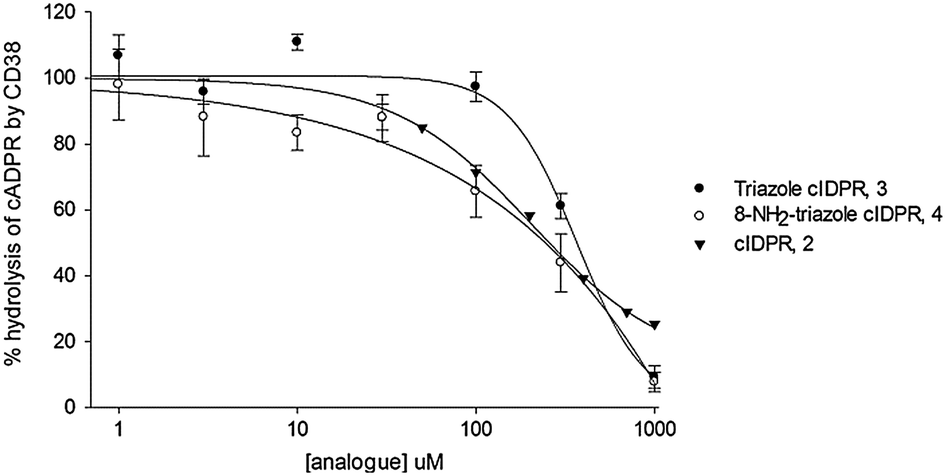 | ||
| Fig. 2 Inhibition of CD38 mediated cADPR hydrolysis by 3 and 4. | ||
To rationalise the observed activity, 3 and 4 were each docked 25 times into the co-crystal structure of CD38 and cIDPR25 with the cIDPR ligand removed. The majority of the docked ligands had the same pose (Fig. 3a and b, Fig. S1, ESI†) in which the “northern” ribose of the ligand overlays that of the crystal structure ligand almost perfectly and the 1,4-disubstituted triazole lies between the two phosphate groups. The inosine ring is slightly tilted and the “southern” ribose does not overlay its crystal structure counterpart, most likely a consequence of the planar triazole ring reducing flexibility in the macrocycle.
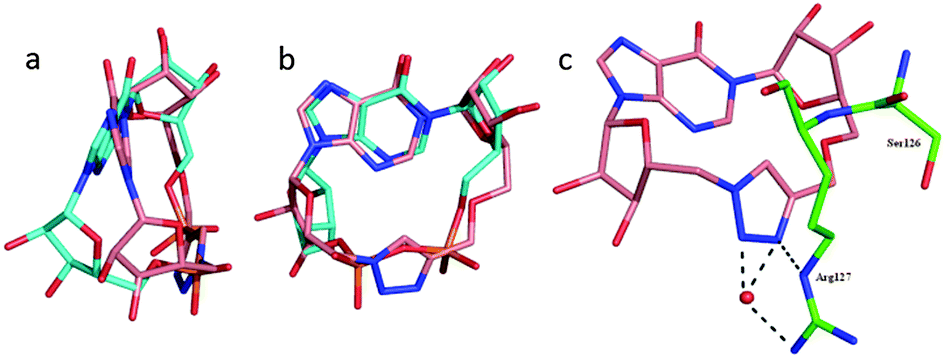 | ||
| Fig. 3 Docking of 3 into the 2PGJ crystal structure. The protein is shown in green, N1-cIDPR in cyan and 3 in pink. | ||
The bioactive conformation of cIDPR bound to CD38 reveals that the hydroxyl group of Ser126 and the ε-NH group of Arg127 form H-bonds to the pyrophosphate (Fig. S2, ESI†). In the simulated protein–ligand complex of 3 the hydroxyl group of Ser126 is too far from the triazole to form an H-bond but the ε-NH of Arg127 forms an H-bond to N3 and the terminal NH2 forms two water-mediated H-bonds to N2 and N3 of the triazole (Fig. 3c). This suggests that the triazole motif is able to mimic the interaction of the pyrophosphate with the binding site to some extent. In addition, the docking of 4 predicts that its 8-NH2 group could H-bond to Asp-155, rationalising the improved inhibitory activity of 4vs.3 observed experimentally. This correlates well with a similar effect in CD38 for 8-NH2-cIDPR vs. cIDPR, recently observed crystallographically in an 8-NH2-cIDPR:CD38 complex29 (Fig. S3, ESI†) and implies a likely similar binding mode for the two types of analogue, supporting the overall concept of pyrophosphate replacement.
Importantly, cIDPR (2), 3 and 4 were also examined for Ca2+ releasing ability in sea urchin egg homogenate (SUH). cIDPR (2) is an agonist, but ca. 1000 fold weaker than cADPR, highlighting differences at this invertebrate receptor, compared to its mammalian T-cell activity.24 Triazole 3 is also substantially weaker than cADPR, but releases Ca2+ only slightly less potently than the more relevant parent ligand 2, demonstrating that 3 acts as an agonist at intracellular Ca2+ stores (Fig. 4). Interestingly, the 8-NH2-substituted 4 showed no Ca2+ releasing activity (data not shown), in keeping with previous observations that the 8-H → 8-NH2 substitution may be a critical agonist/antagonist switch.30 Thus, it may be possible to selectively engineer the activity of analogues towards CD38.
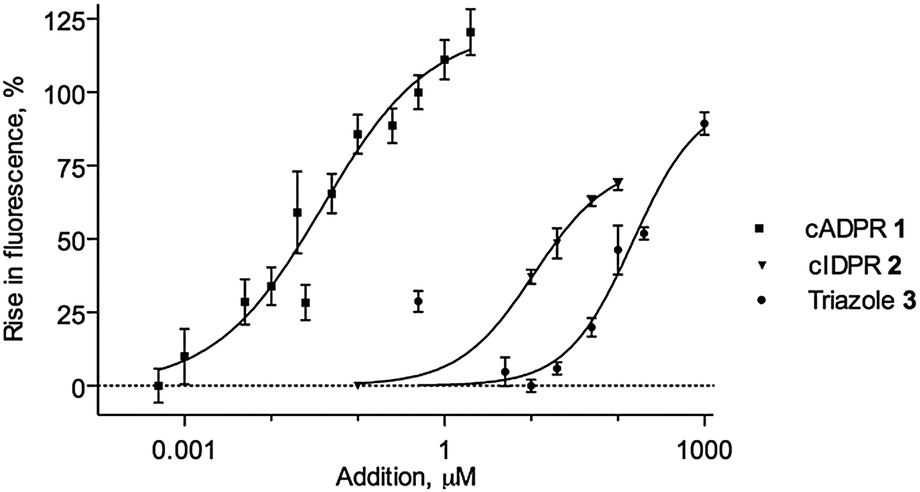 | ||
| Fig. 4 Calcium release by cIDPR (2) and 3 in SUH. | ||
In summary, we have developed a concise synthetic route to a new class of neutral cADPR analogue that is still active at two protein targets. Macrocyclisation was achieved by a clean, high yielding ‘click’ reaction which simultaneously formed the 1,2,3-triazole pyrophosphate bioisostere. The uncharged analogues inhibit CD38 hydrolysis of cADPR with a similar potency to that of the parent molecule, cIDPR. Triazole analogue 3, like 2, is also active at the cADPR receptor and mobilises Ca2+. These are the first active, neutral cADPR analogues with complete pyrophosphate replacement and represent leads for simplified cADPR-based cell signalling modulators. Taken together, their chemical and biological stability, uncharged nature and the surprising retention of activity observed upon complete pyrophosphate substitution point towards CD38 and the cADPR receptor as potentially druggable targets.
We acknowledge the Wellcome Trust, Project Grant 084068, Programme Grants 082837 (BVLP) and 084102 (AG) and RCGAS 201105159001 (RG) for support. BVLP is a Wellcome Trust Senior Investigator (Grant 101010).
Notes and references
- A. H. Guse, Curr. Mol. Med., 2004, 4, 239–248 CrossRef CAS; M. J. Berridge, P. Lipp and M. D. Bootman, Nat. Rev. Mol. Cell Biol., 2000, 1, 11–21 CrossRef PubMed.
- M. J. Berridge, Nature, 1993, 361, 315–325 CrossRef CAS PubMed.
- E. Naylor, A. Arredouani, S. R. Vasudevan, A. M. Lewis, R. Parkesh, A. Mizote, D. Rosen, J. M. Thomas, M. Izumi, A. Ganesan, A. Galione and C. G. Churchill, Nat. Chem. Biol., 2009, 5, 220–226 CrossRef CAS PubMed.
- A. Kannt, K. Sicka, K. Kroll, D. Kadereit and H. Gögelein, Naunyn-Schmiedeberg's Arch. Pharmacol., 2012, 385, 717–727 CrossRef CAS PubMed.
- D. L. Clapper, T. F. Walseth, P. J. Dargie and H. C. Lee, J. Biol. Chem., 1987, 262, 9561–9568 CAS.
- H. Kim, E. L. Jacobson and M. K. Jacobson, Science, 1993, 261, 1330–1333 CAS.
- A. Savarino, T. Bensi, A. Chiocchetti, F. Bottarel, R. Mesturini, E. Ferrero, L. Calosso, S. Deaglio, E. Ortolan, S. Buttò, A. Cafaro, T. Katada, B. Ensoli, F. Malavasi and U. Dianzani, FASEB J., 2003, 17, 461–463 Search PubMed; F. Malavasi, S. Deaglio, R. Damle, G. Cutrona, M. Ferrarini and N. Chiorazzi, Blood, 2011, 118, 3470–3478 CrossRef CAS PubMed; H. C. Lee, Messenger, 2012, 1, 16–33 CrossRef PubMed.
- Y. J. Zhao, C. M. C. Lam and H. C. Lee, Sci. Signaling, 2012, 5, ra67 CrossRef PubMed.
- H. C. Lee, R. Aarhus, R. Graeff, M. E. Gurnack and T. F. Walseth, Nature, 1994, 370, 307–309 CrossRef CAS PubMed; O. A. Ogunbayo, Y. Zhu, D. Rossi, V. Sorrentino, J. Ma, M. X. Zhu and A. M. Evans, J. Biol. Chem., 2011, 286, 9136–9140 CrossRef PubMed.
- G. A. Ashamu, J. K. Sethi, A. Galione and B. V. L. Potter, Biochemistry, 1997, 36, 9509–9517 CrossRef CAS PubMed.
- B. V. L. Potter and T. F. Walseth, Curr. Mol. Med., 2004, 4, 303–311 CrossRef CAS.
- F. J. Zhang and C. J. Sih, Tetrahedron Lett., 1995, 36, 9289–9292 CrossRef CAS; R. M. Graeff, T. F. Walseth, H. K. Hill and H. C. Lee, Biochemistry, 1996, 35, 379–386 CrossRef PubMed.
- V. C. Bailey, S. M. Fortt, R. J. Summerhill, A. Galione and B. V. L. Potter, FEBS Lett., 1996, 379, 227–230 CrossRef CAS; M. Fukuoka, S. Shuto, N. Minakawa, Y. Ueno and A. Matsuda, J. Org. Chem., 2000, 65, 5238–5248 CrossRef; A. Galeone, L. Mayol, G. Oliviero, G. Piccialli and M. Varra, Tetrahedron, 2002, 58, 363–368 CrossRef; X. Gu, Z. Yang, L. Zhang, S. Kunerth, R. Fliegert, K. Weber and A. H. Guse, J. Med. Chem., 2004, 47, 5674–5682 CrossRef PubMed.
- F. J. Zhang, S. Yamada, Q. M. Gu and C. J. Sih, Bioorg. Med. Chem. Lett., 1996, 6, 1203–1208 CrossRef CAS.
- L. Xu, T. F. Walseth and J. T. Slama, J. Med. Chem., 2005, 48, 4177–4181 CrossRef CAS PubMed.
- N. Qi, K. Jung, M. Wang, L. X. Na, Z. J. Yang, L. R. Zhang, A. H. Guse and L. H. Zhang, Chem. Commun., 2011, 47, 9462–9464 RSC.
- R. Aarhus, K. Gee and H. C. Lee, J. Biol. Chem., 1995, 270, 7745–7749 CrossRef CAS PubMed; P.-L. Yu, Z.-H. Zhang, B.-X. Hao, Y.-J. Zhao, L.-H. Zhang, H.-C. Lee, L. Zhang and J. Yue, J. Biol. Chem., 2012, 287, 24774–24783 CrossRef PubMed.
- J. Canales, A. Fernández, J. R. Rodriques, R. Ferreira, J. M. Ribeiro, A. Cabezas, M. J. Costas and J. C. Cameselle, FEBS Lett., 2009, 583, 1593–1598 CrossRef CAS PubMed.
- N. A. Meanwell, J. Med. Chem., 2011, 54, 2529–2591 CrossRef CAS PubMed.
- F. Westheimer, Science, 1987, 235, 1173–1178 CAS.
- T. S. Elliott, A. Slowey, Y. Ye and S. J. Conway, MedChemComm, 2012, 3, 735–751 RSC.
- L. Chen, D. J. Wilson, Y. Xu, C. C. Aldrich, K. Felczak, Y. Y. Sham and K. W. Pankiewicz, J. Med. Chem., 2010, 53, 4768–4778 CrossRef CAS PubMed.
- K. K. Yeoh, T. D. Butters, B. L. Wilkinson and A. J. Fairbanks, Carbohydr. Res., 2009, 344, 586–591 CrossRef CAS PubMed.
- G. K. Wagner, A. H. Guse and B. V. L. Potter, J. Org. Chem., 2005, 70, 4810–4819 CrossRef CAS PubMed.
- Q. Liu, I. A. Kriksunov, C. Moreau, R. Graeff, B. V. L. Potter, H. C. Lee and Q. Hao, J. Biol. Chem., 2007, 282, 24825–24832 CrossRef CAS PubMed.
- J. M. Swarbrick and B. V. L. Potter, J. Org. Chem., 2012, 77, 4191–4197 CrossRef CAS PubMed.
- (a) V. V. Rostovtsev, L. G. Green, V. V. Fokin and K. B. Sharpless, Angew. Chem., Int. Ed., 2002, 41, 2596–2599 CrossRef CAS; (b) A. Isidro-Llobet, T. Murillo, P. Bello, A. Cilibrizzi, J. T. Hodgkinson, W. R. J. D. Galloway, A. Bender, M. Welch and D. R. Spring, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 6793–6798 CrossRef CAS PubMed; (c) R. E. Looper, D. Pizzirani and S. L. Schreiber, Org. Lett., 2006, 8, 2063–2066 CrossRef CAS PubMed.
- F. Liu and D. J. Austin, Tetrahedron Lett., 2001, 42, 3153–3154 CrossRef CAS.
- C. Moreau, Q. Liu, R. Graeff, G. K. Wagner, M. P. Thomas, J. M. Swarbrick, S. Shuto, C. H. Lee, Q. Hao and B. V. L. Potter, PLoS One, 2013, 8, e66247 CAS.
- T. F. Walseth and H. C. Lee, Biochim. Biophys. Acta, 1993, 1178, 235–242 Search PubMed; B. Zhang, G. K. Wagner, K. Weber, C. Garnham, A. J. Morgan, A. Galione, A. H. Guse and B. V. L. Potter, J. Med. Chem., 2008, 51, 1623–1636 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Fig. S1–S3, experimental details, 1H- and 13C-NMR. See DOI: 10.1039/c3cc49249d |
| This journal is © The Royal Society of Chemistry 2014 |