 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAlternation and tunable composition in hydrogen bonded supramolecular copolymers†
Thorsten
Felder
,
Tom F. A.
de Greef
,
Marko M. L.
Nieuwenhuizen
and
Rint P.
Sijbesma
*
Laboratory of Macromolecular and Organic Chemistry and Institute for Complex Molecular Systems, Eindhoven University of Technology, P.O. Box 513, 5600 MB Eindhoven, The Netherlands. E-mail: r.p.sijbesma@tue.nl
First published on 10th January 2014
Abstract
Sequence control in supramolecular copolymers is limited by the selectivity of the associating monomer end groups. Here we introduce the use of monomers with aminopyrimidinone and aminohydroxynaphthyridine quadruple hydrogen bonding end groups, which both homodimerize, but form even stronger heterodimers. These features allow the formation of supramolecular copolymers with a tunable composition and a preference for alternating sequences.
More than 20 years after the introduction of the term supramolecular polymers, the field continues to develop rapidly.1,2 Applications such as biomedical materials2 and self-healing materials3 are being explored, and the details of supramolecular polymerization processes have become a subject of detailed study.4 However, in at least one aspect supramolecular polymers remain behind their covalent counterparts, viz. in control over monomer sequences in copolymers. The use of well-designed multiple hydrogen bonding units potentially provides the selectivity and strength to create the desired control. Self-complementary quadruple hydrogen bonding motifs5 have been shown to be useful in the formation of supramolecular homopolymers. In addition, a variety of complementary hydrogen bonding motifs has been reported,6 as well as the possibility to exploit these to form supramolecular copolymers with highly alternating structures in mixtures with a strict 1
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 stoichiometry.7 Another approach toward these highly alternating structures relies on self-sorting in mixtures of two AB-type heteroditopic monomers.8
:1 stoichiometry.7 Another approach toward these highly alternating structures relies on self-sorting in mixtures of two AB-type heteroditopic monomers.8
Multiple hydrogen bonding units that are able to switch from self-complementary to complementary modes of complexation would allow the formation of alternating copolymers with tunable composition and a degree of alternation that depends on the selectivity of heterocomplexation.9 Here we describe the combination of two quadruple hydrogen bonding units, amino-pyrimidinones (AminoUPy) and amino-hydroxy-naphthyridines (NaPyO), in which this capability is inherently present. Monomers functionalized with two of these units form supramolecular polymers. The selectivity for heterocomplexation results in the formation of copolymers with tunable composition and a preference for alternating sequences when the monomers are mixed in solution.
In supramolecular polymers, extensive use has been made of the strong self-association10 (Kdim = 6 × 107 M−1 in CHCl3) of the 2-ureido-4[1H]-pyrimidinone (UPy) unit via a DDAA motif of hydrogen bonding sites. UPy's are also able to form heterodimers with 2,7-diamido-1,8-naphthyridine (NaPy).11 They do so with high selectivity via a complementary ADDA–DAAD motif upon their tautomerization into the 6[1H]-form (Kass = 5 × 106 M−1 in CHCl3). UPy–NaPy hetero-complexation has been used to form supramolecular copolymers from bifunctional bisUPy and bisNaPy monomers.11b Alternating copolymers are formed over a broad composition range. However, the incapability of the NaPy unit to form homodimers means that the NaPy monomers act as endcappers of the supramolecular polymer when they are present in excess to the bis-UPy monomers (Fig. 1a).
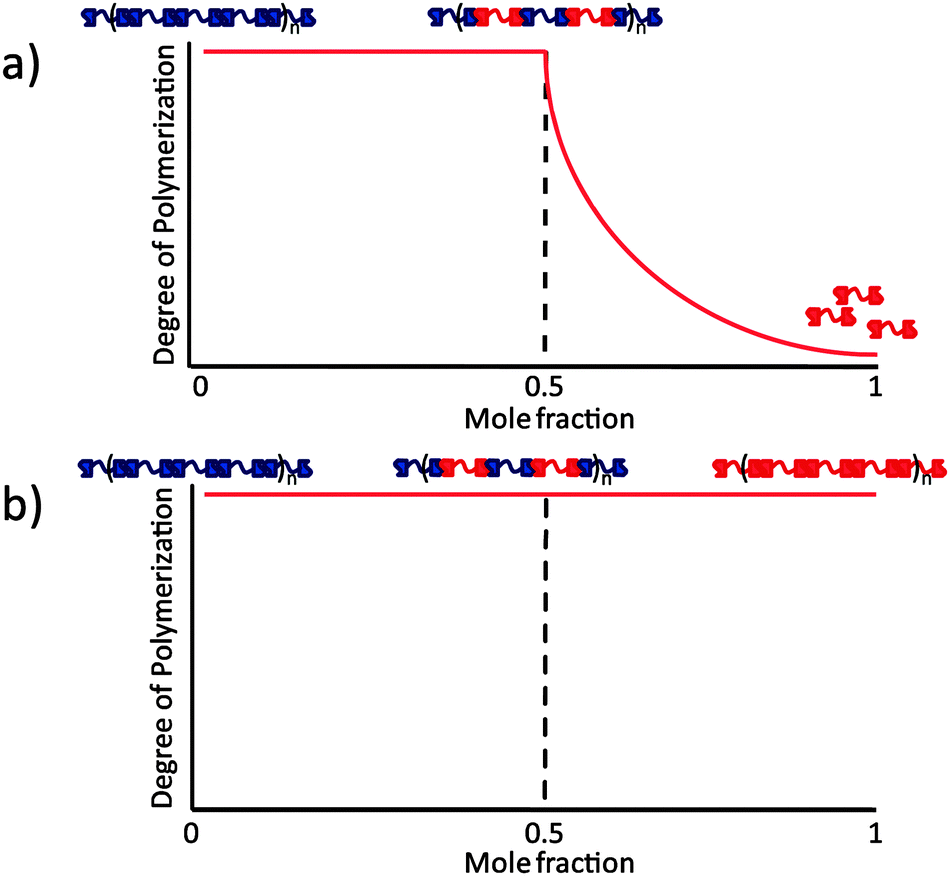 | ||
| Fig. 1 (a) Supramolecular copolymerization of bisUPy and bisNaPy monomers. The NaPy unit, which does not homodimerize, acts as a chain stopper when the bisNaPy monomer is present in excess. (b) Copolymer formation between two monomer species capable of homo- as well as hetero dimerization. Shown is a case in which the binding constants are approximately equal. | ||
In order to obtain a system that maintains a high degree of polymerization (DP) over the whole composition range, both hydrogen bonding units must have the capability to self-associate (Fig. 1b). The NaPyO binding unit is capable of forming a homo-dimeric complex via the formation of quadruple hydrogen bonds via linear acceptor–donor–acceptor–donor (ADAD) arrays.12 UPys with electron donating substituents at the 6-position are present in the enol-tautomeric form and are also able to dimerize using ADAD hydrogen bonding arrays.13 Consequently, a combination of both dimers is expected to result in the formation of a heterodimer (Scheme 1).
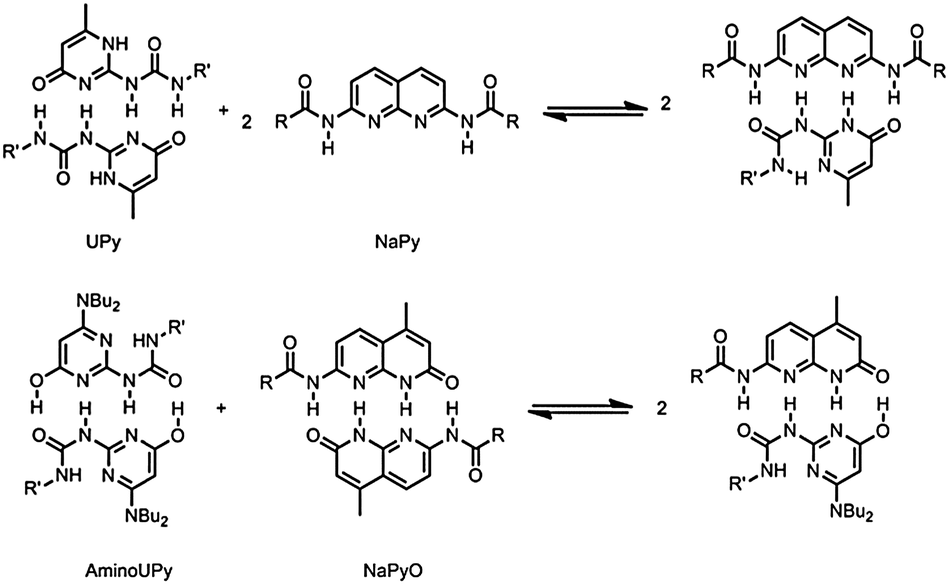 | ||
| Scheme 1 Top: heterodimerization between typical UPy and NaPy groups. Only the UPy unit can self-dimerize. Bottom: heterodimerization between AminoUPy and NaPyO. Both units can form homodimers as well as heterodimers. | ||
Monofunctional NaPyO binding units 1 and 2 and the bifunctional derivative 4 (Chart 1) were prepared on a multigram scale in high yields using a straightforward procedure from 1,8-naphthyridin-2(1H)-one by reaction with aliphatic acyl chlorides (see ESI†). Naphthyridines 1 and 2 and AminoUPy 3 served as model compounds to investigate the strength and selectivity of binding. Bifunctional derivatives NaPyO 4 (NaPyO end-group functionalization >99%) having a polymeric spacer of pTHF2000 (Mn = 2000) and bisUPy 5 containing a short aliphatic spacer group were prepared to study the formation of NaPyO–UPy supramolecular copolymers.
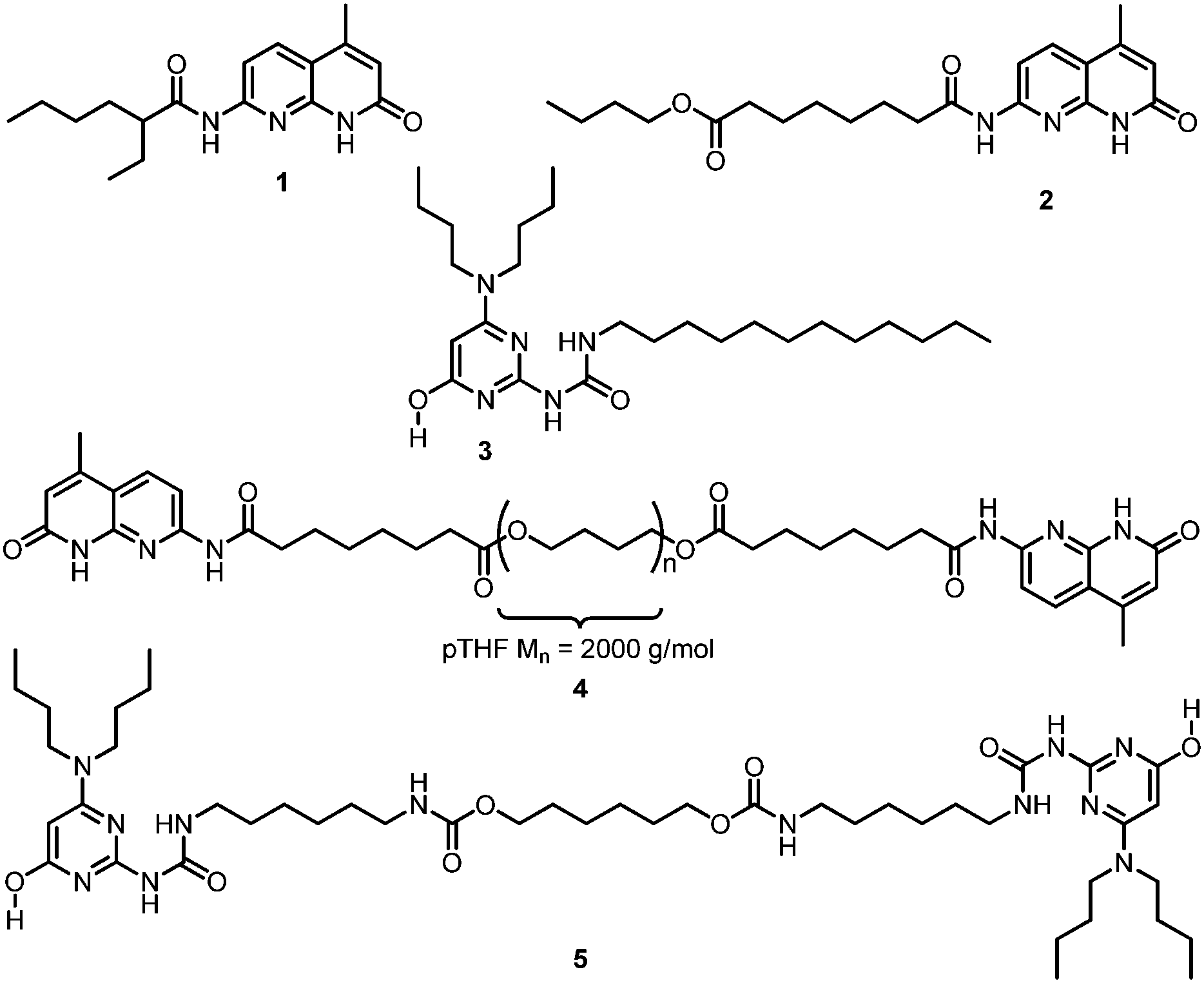 | ||
| Chart 1 | ||
Homodimerization constants Kdim of the NaPyO model compounds 1 and 2 were determined through fitting of concentration dependent NMR chemical shift data (CDCl3, 25 °C) and were determined to be Kdim(1) = 6 ± 1 × 103 M−1 (ΔG° = −21.6 kJ mol−1) and Kdim(2) = 8 ± 4 × 103 M−1 (ΔG° = −22.3 kJ mol−1), respectively. The dimerization constant of AminoUPy 3 (Chart 1) was reported earlier13b and has a value of Kdim(3) = 9.0 × 105 M−1 (ΔG° = −34.0 kJ mol−1). Heterocomplexation of AminoUPy–NaPyO was determined utilizing compounds 1, 2 and 3 by UV/Vis titration experiments in CHCl3 at a constant concentration of naphthyridine, [NaPyO] = 2.5 × 10−4 M (see ESI†). Kass was obtained by appropriate fitting of the received binding curves to a model describing homodimerization of both components as well as heteroassociation between the components. The heteroassociation constants of the complexes were determined to be Kass(1·3) = 5.3 ± 0.3 × 105 M−1 (ΔG° = −32.5 kJ mol−1) and Kass(2·3) = 2.7 ± 0.2 × 105 M−1 (ΔG° = −31.0 kJ mol−1). Based on the free energy change obtained for the individual binding events, the free energy change for the formation of heterocomplexes from homocomplexes is calculated to be negative (ΔG° = −9.4 kJ mol−1 for complex 1·3 and −5.7 kJ mol−1 for complex 2·3) and therefore the heterocomplex is favored over the homocomplexes in equimolar mixtures of the components (Table 1).
| Equilibrium | [Heterodimer]/[homodimer] | |
|---|---|---|
| 298 K (UV) | 248 K (NMR) | |
| 1·1 + 3·3 → 2 (1·3) | 3.6 | 3.8 |
| 2·2 + 3·3 → 2 (2·3) | 3.7 | n.d. |
1H-NMR spectroscopy was also used to study the preference for heterocomplexation. However, spectra from equimolar mixtures (10 mM, CDCl3) of NaPyO 1 and UPy 3 at 298 K exhibit broadened signals particularly in the aromatic region (see ESI†). Therefore, the measurements were also performed at lower temperatures where sharpening and splitting of the peaks occurred. The recorded low temperature NMR spectra showed multiple sets of signals, which were assigned to the homo- and heterodimeric species in solution. Upon integration of the respective signals, the selectivity of the heterocomplex 1·3 was determined to be 73% at 263 K. Further lowering of the temperature led to an increased selectivity of 79% at 248 K.
Bis-functionalized derivatives 4 and 5 (Chart 1) form supramolecular homopolymers in chloroform, as shown by the concentration dependence of the specific viscosities (ηsp) which scale with concentration as ηsp∼C2.16 and ηsp∼C2.31, respectively (Fig. 2a). In contrast to the strong concentration dependence of the viscosity of the bifunctionalized monomers, ηsp of unfunctionalized polytetrahydrofuran with a molecular weight of 2000 g mol−1 (the length of the linker in 4) is low and shows a nearly linear increase of ηsp with concentration. Supramolecular copolymerization was probed by measuring the specific viscosity of a 39 g L−1 NaPyO monomer 4 solution to which increasing amounts of UPy monomer 5 were added. The experiment resulted in a viscosity plot displaying a constant value at ηsp = 8.5 over the whole measured composition range up to 1.6 equiv. of 5. This observation can be understood by a continuous incorporation of the UPy monomer into the preformed NaPyO homo polymer chain, which at a monomer ratio of 1:1 (1 equivalent 5 in 4) results in UPy–NaPyO supramolecular copolymers with preferred alternation and without exhibiting a significant change in solution viscosity. Upon further addition of >1 equiv. of 5 to the solution, no considerable change in ηsp was observed. In contrast, a titration of bisNaPyO 4 with 1.55 equivalents of monoUPy 3 showed a progressive drop in viscosity from the pure solution of 4 (ηsp = 7.6) to ηsp = 2.7 (Fig. 2b).
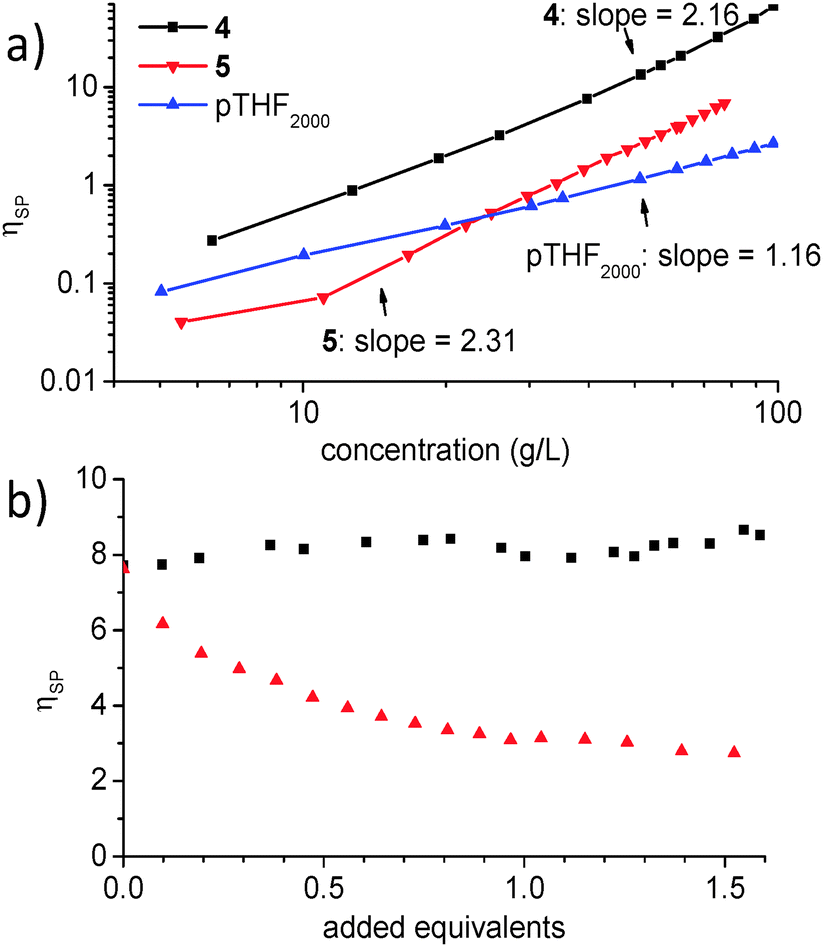 | ||
Fig. 2 (a) Concentration dependent specific viscosities of 4, 5 and pTHF2000 in chloroform. 4 and 5 exhibit a strong concentration dependence of the solution viscosity indicating the formation of supramolecular polymers. (b) Viscosity titration of a 39 g L−1 bisNaPyO 4 solution with bisUPy 5 (■) and with UPy stopper 3 ( ) in chloroform. ) in chloroform. | ||
Heterodimerization of the 1,8-naphthyridin-2(1H)-one derivative to UPy derivatives with high binding strength and modest selectivity has been demonstrated. This allowed formation of supramolecular copolymers with tunable composition from bifunctional monomers over a broad mixing ratio without a notable influence on solution viscosity. Heteroselectivity results in preferred alternation of monomeric units in the copolymer, although the alternation is incomplete. The actual degree of alternation is determined by the heterocomplexation equilibrium. Although the supramolecular copolymer has certain analogies with alternating covalent copolymers obtained by radical copolymerization, an essential difference is that the degree of alternation in radical polymerizations is determined by kinetics, while in the supramolecular polymers it is determined by a the ratio of equilibrium constants.
Further improvement in the system may therefore be achieved by increasing the selectivity of heterocomplexation. The origin of heteroselectivity in the NaPyO–UPy system appears not to be related to improved steric complementarity in the heterodimer, since crystal structures and energy minimized structures of the DADA arrays in both components show that they are linear within 0.1 Å.13b,14 Therefore, we propose that electronic complementarity, i.e. matching of the strongest acceptor and donor sites is responsible for the observed selectivity. Current efforts are focused on improving the selectivity in donor–acceptor arrays optimized for electronic complementarity.
This research was partially supported by NanoNed, a national nanotechnology program coordinated by the Dutch Ministry of Economic Affairs (Project EMM.7126).
Notes and references
- (a) T. F. A. de Greef, M. M. J. Smulders, M. Wolffs, A. P. H. J. Schenning, R. P. Sijbesma and E. W. Meijer, Chem. Rev., 2009, 109, 5687–5754 CrossRef CAS PubMed; (b) T. Aida, E. W. Meijer and S. I. Stupp, Science, 2012, 335, 813–817 CrossRef CAS PubMed.
- P. Y. W. Dankers, M. C. Harmsen, L. A. Brouwer, M. J. A. Van Luyn and E. W. Meijer, Nat. Mater., 2005, 4, 568–574 CrossRef CAS PubMed.
- P. Cordier, F. Tournilhac, C. Soulié-Ziakovic and L. Leibler, Nature, 2008, 451, 977–980 CrossRef CAS PubMed.
- P. A. Korevaar, S. J. George, A. J. Markvoort, M. M. J. Smulders, P. A. J. Hilbers, A. P. H. J. Schenning, T. F. A. De Greef and E. W. Meijer, Nature, 2012, 481, 492–496 CrossRef CAS PubMed.
- (a) R. P. Sijbesma and E. W. Meijer, Chem. Commun., 2003, 5–16 RSC; (b) Y. Hisamatsu, N. Shirai, S. Ikeda and K. Odashima, Org. Lett., 2009, 11, 4342–4345 CrossRef CAS PubMed; (c) V. G. H. Lafitte, A. E. Aliev, E. Greco, K. Bala, P. Golding and H. C. Hailes, New J. Chem., 2011, 35, 1522–1527 RSC; (d) X. Li, Y. Fang, P. Deng, J. Hu, T. Li, W. Feng and L. Yuan, Org. Lett., 2011, 13, 4628–4631 CrossRef CAS PubMed.
- (a) S. K. Chang and A. D. Hamilton, J. Am. Chem. Soc., 1988, 110, 1318–1319 CrossRef CAS; (b) Y. Hisamatsu, N. Shirai, S. Ikeda and K. Odashima, Org. Lett., 2010, 12, 1776–1779 CrossRef CAS PubMed; (c) B. A. Blight, C. A. Hunter, D. A. Leigh, H. McNab and P. I. T. Thomson, Nat. Chem., 2011, 3, 244–248 CrossRef CAS PubMed; (d) M. L. Pellizzaro, S. A. Barrett, J. Fisher and A. J. Wilson, Org. Biomol. Chem., 2012, 10, 4899–4906 RSC.
- (a) V. Berl, M. Schmutz, M. J. Krische, R. G. Khoury and J.-M. Lehn, Chem.–Eur. J., 2002, 8, 1227–1244 CrossRef CAS; (b) S. Sivakova, D. A. Bohnsack, M. E. Mackay, P. Suwanmala and S. J. Rowan, J. Am. Chem. Soc., 2005, 127, 18202–18211 CrossRef CAS PubMed; (c) O. A. Scherman, G. B. W. L. Ligthart, H. Ohkawa, R. P. Sijbesma and E. W. Meijer, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 11850–11855 CrossRef CAS PubMed; (d) T. Park and S. C. Zimmerman, J. Am. Chem. Soc., 2006, 128, 13986–13987 CrossRef CAS PubMed; (e) K. P. Nair, V. Breedveld and M. Weck, Macromolecules, 2008, 41, 3429–3438 CrossRef CAS.
- (a) F. Wang, C. Han, C. He, Q. Zhou, J. Zhang, C. Wang, N. Li and F. Huang, J. Am. Chem. Soc., 2008, 130, 11254–11255 CrossRef CAS PubMed; (b) N. Tomimasu, A. Kanaya, Y. Takashima, H. Yamaguchi and A. Harada, J. Am. Chem. Soc., 2009, 131, 12339–12343 CrossRef CAS PubMed.
- M. L. Pellizzaro, K. A. Houton and A. J. Wilson, Chem. Sci., 2013, 4, 1825–1829 RSC.
- F. H. Beijer, R. P. Sijbesma, H. Kooijman, A. L. Spek and E. W. Meijer, J. Am. Chem. Soc., 1998, 120, 6761–6769 CrossRef CAS.
- (a) X.-Q. Li, X.-K. Jiang, X.-Z. Wang and Z.-T. Li, Tetrahedron, 2004, 60, 2063–2069 CrossRef CAS PubMed; (b) G. W. B. L. Ligthart, H. Ohkawa, R. P. Sijbesma and E. W. Meijer, J. Am. Chem. Soc., 2005, 127, 810–811 CrossRef CAS PubMed.
- S. Goswami, S. Dey, J. F. Gallagher, A. J. Lough, S. García-Granda, L. Torre-Fernández, I. Alkorta and J. Elguero, J. Mol. Struct., 2007, 846, 97–107 CrossRef CAS PubMed.
- (a) V. G. H. Lafitte, A. E. Aliev, H. C. Hailes, K. Bala and P. Golding, J. Org. Chem., 2005, 70, 2701–2707 CrossRef CAS PubMed; (b) T. F. A. de Greef, G. Ercolani, G. B. W. L. Ligthart, E. W. Meijer and R. P. Sijbesma, J. Am. Chem. Soc., 2008, 130, 13755–13764 CrossRef CAS PubMed.
- B. Ośmiałowski, E. Kolehmainen, E. Kalenius, B. Behera, R. Kauppinen and E. Sievänen, Struct. Chem., 2011, 22, 1143–1151 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: General methods; synthetic procedures; determination of the dimerization constants using 1H-NMR; determination of the association constants using UV-Vis titration experiments; curve fitting procedure of the UV-Vis titration data at a single wavelength; variable temperature 1H-NMR experiments; determination of the concentration dependence of the specific viscosity using dilution experiments; viscosity titration experiments. See DOI: 10.1039/c3cc46611f |
| This journal is © The Royal Society of Chemistry 2014 |