 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Carbon monoxide – physiology, detection and controlled release
Stefan H.
Heinemann
ab,
Toshinori
Hoshi
c,
Matthias
Westerhausen
d and
Alexander
Schiller
*bd
aCenter for Molecular Biomedicine (CMB), Department of Biophysics, Friedrich Schiller University Jena & Jena University Hospital, Hans-Knöll-Straße 2, D-07745 Jena, Germany
bAbbe Center of Photonics, Friedrich Schiller University Jena, Max-Wien-Platz 1, D-07743 Jena, Germany
cDepartment of Physiology, University of Pennsylvania, 415 Curie Boulevard, 605 CRB, Philadelphia, PA 19104-6085, USA
dInstitute for Inorganic and Analytical Chemistry, Friedrich Schiller University Jena, Humboldtstr. 8, D-07743 Jena, Germany. E-mail: alexander.schiller@uni-jena.de
First published on 21st February 2014
Abstract
Carbon monoxide (CO) is increasingly recognized as a cell-signalling molecule akin to nitric oxide (NO). CO has attracted particular attention as a potential therapeutic agent because of its reported anti-hypertensive, anti-inflammatory and cell-protective effects. We discuss recent progress in identifying new effector systems and elucidating the mechanisms of action of CO on, e.g., ion channels, as well as the design of novel methods to monitor CO in cellular environments. We also report on recent developments in the area of CO-releasing molecules (CORMs) and materials for controlled CO application. Novel triggers for CO release, metal carbonyls and degradation mechanisms of CORMs are highlighted. In addition, potential formulations of CORMs for targeted CO release are discussed.
 Stefan H. Heinemann | Stefan H. Heinemann studied physics at the University of Göttingen. Research for his PhD thesis (1990) under the supervision of Nobel Laureate Erwin Neher, performed at Yale University (New Haven) and the Max Planck Institute for Biophysical Chemistry (Göttingen), focused on the biophysics of ion channels. After a post-doctoral period at Stanford University in 1992 he became head of the Max Planck Research Unit “Molecular and Cellular Biophysics” at Jena University Hospital. Since 1998 he has been full professor of biophysics at the Friedrich Schiller University Jena and Jena University Hospital. His research interests include structure, function and pharmacology of ion channels as well as cellular regulation via haem and redox processes. Currently he is the coordinator of the DFG Research Unit “FOR 1738 – Heme and Heme Degradation Products”. |
 Toshinori Hoshi | Toshinori Hoshi received an undergraduate degree from the University of New Hampshire in 1981 and studied ion channel physiology with Prof. Stephen J Smith at Yale University for his PhD dissertation completed in 1985. The postdoctoral work with Prof. Richard W. Aldrich focused on biophysical and molecular mechanisms of Shaker potassium channel function. He is Professor of Physiology at the University of Pennsylvania in Philadelphia. Within the DFG Research Unit “FOR 1738 – Heme and Heme Degradation Products” he investigates the regulation of ion channels by carbon monoxide. |
 Matthias Westerhausen | Matthias Westerhausen obtained his diploma degree in 1983 from the Philipps University in Marburg, Germany, and his PhD in 1987 under the supervision of Professor G. Becker investigating acyl substituted phosphanes and arsanes. In 1987–1988, he worked as a postdoctoral fellow with Professor Robert T. Paine at the University of New Mexico in Albuquerque, USA, on phosphanylboranes. Back in Germany, he finished his habilitation at the University of Stuttgart in 1994. From 1996 to 2004 he was professor at the Ludwig Maximilians University (LMU) in Munich where he was also vice-president from 2001 to 2004. Since 2004 he has been teaching and researching at the Friedrich Schiller University Jena, Germany. His main interests include topics such as the organic chemistry and catalytic application of d0 metals with the focus on heavy alkaline earth metals, cooperative effects in heterobimetallic complexes. He is also enjoying the interdisciplinary research on haem degradation products and CO-releasing molecules within the DFG research unit FOR 1738. |
 Alexander Schiller | Alexander Schiller studied chemistry at the University of Munich (LMU) and completed his PhD in 2006 under the supervision of Prof. K. Severin at the École Polytechnique Fédérale de Lausanne (Switzerland) in the field of bioinorganic and polymer chemistry. For a postdoctoral stay he moved to the University of California, Santa Cruz. Together with Prof. B. Singaram and Dr R. Wessling he worked on fluorescent saccharide sensors. After a stay at Empa (Switzerland) as a project leader and head of the laboratory, he joined the Friedrich Schiller University Jena as a junior professor of the Carl Zeiss foundation. After positive evaluation in 2013, he continues with his research on supramolecular analytical chemistry and molecular logic (FP7 Novosides). As a principal investigator in the DFG Research Unit “FOR 1738 – Heme and Heme Degradation Products” he works on NO and CO releasing materials. |
A. Introduction
Carbon monoxide (CO), a colour- and odourless gas, is typically produced when carbon-containing compounds are only partially oxidized, such as in combustion engines. At high concentrations, CO is toxic to humans and animals. It binds to haemoglobin about 200-times more strongly than O2, and the resulting carboxyhaemoglobin is thus no longer available for the oxygen transport in the body. With 50% of human haemoglobin occupied by CO, seizures and coma may result, sometimes with fatal consequences. The major cause of its toxicity, however, seems to originate from its distribution in the tissue where it affects various targets.1 However, despite this toxicity, CO is increasingly appreciated as a cell-signalling molecule akin to nitric oxide (NO).2 For example, CO relaxes smooth muscles at low concentrations and lowers the blood pressure.3The major endogenous source of CO in the body is breakdown of haem. Physiological degradation of haem occurs in a tightly controlled manner involving the enzyme haem oxygenase (HO; EC 1.14.99.3), which cleaves the porphyrin IX in the presence of NADP and molecular oxygen, resulting in the primary or first-order haem degradation products (HDPs), consisting of CO, Fe2+ and biliverdin IX (Fig. 1).4 Biliverdin reductase catalyzes the next degradation step in the presence of NADPH and H+, yielding the second-order metabolite bilirubin IX, which is then excreted in bile and urine. The bile pigments biliverdin (green) and bilirubin (yellow) are easily recognized in haematomas, which change colour over time as haem degradation proceeds. CO production following this route amounts to about 16 μmol h−1 per human body.5
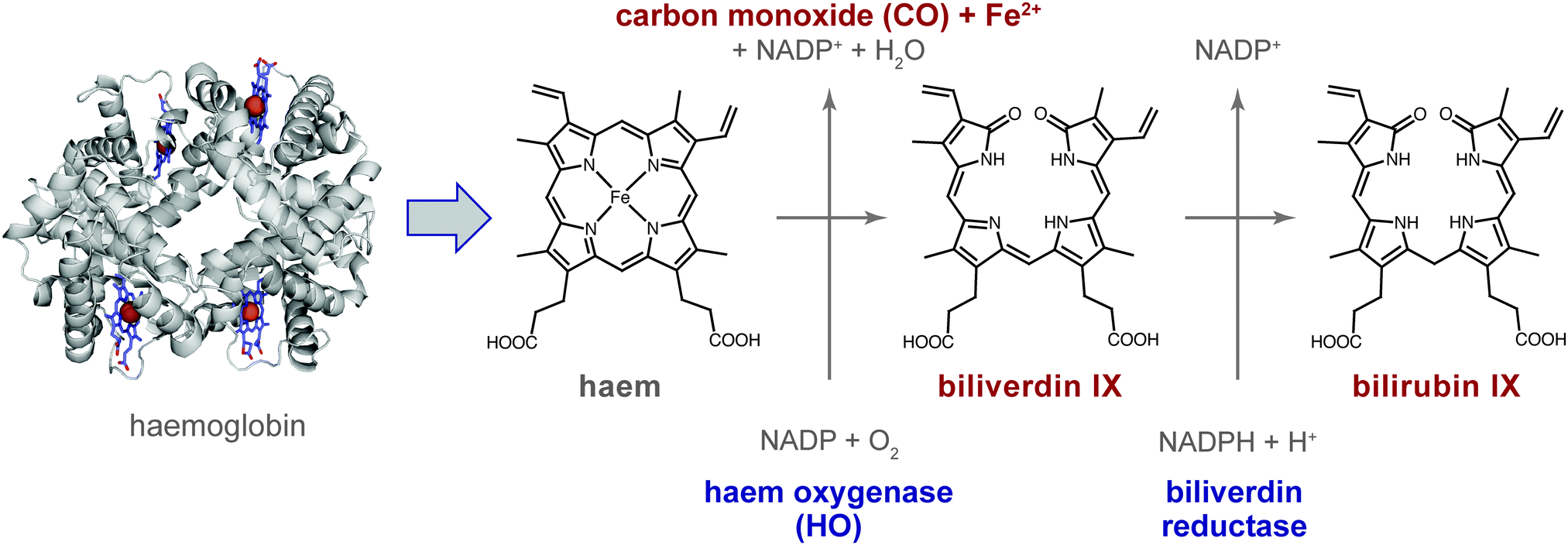 | ||
| Fig. 1 Haem degradation. Haem (Fe2+-protoporphyrin IX), released from haemoglobin (left), is degraded by the aid of haem oxygenase to carbon monoxide (CO), ferrous ions (Fe2+) and biliverdin IX. A subsequent step, catalysed by biliverdin reductase, yields bilirubin IX. | ||
The molecular action of CO, however, is to be considered a local event because of its limited bioavailability and the dependence of its production on haem oxygenases. In humans, there are two isoforms of this enzyme. The expression of HO1 is inducible, triggered by the presence of free haem, thus signalling the need for haem degradation. In contrast, HO2 is constitutively expressed. Since the activity of HOs always results in the clearance of haem and the production of CO, in many cases it is not unambiguously clear whether the removal of haem or the release of the haem degradation products, such as CO, or both, are the primary physiologically relevant end results. In any case, ample evidence has accumulated to demonstrate that haem catabolism and the endogenous production of CO serve a wide array of physiological functions. For example, induction of HO1 in the brain improves outcome after cerebral ischemia6,7 and HO2 was shown to be neuroprotective during intracerebral haemorrhage,8 suggesting that either the clearance of haem or the production of CO (or both) is beneficial. This becomes particularly clear in cerebral malaria, a multifactorial disease induced by cerebral accumulation of haemoglobin via Plasmodium infection, claiming more than 1 million lives annually, most of them being children: HO1 and CO were shown to be beneficial, HO1 because it removes free haem and CO because it binds to haemoglobin and, therefore, inhibits haem release.9,10
CO has attracted particular attention as a potential therapeutic agent because CO is suggested to have anti-hypertensive, anti-inflammatory and cell-protective effects.3,11–14 For example, inhalation of CO gas under controlled conditions alleviates symptoms of human pulmonary hypertension,15 presumably by interacting with the smooth muscle signalling proteins such as guanylyl cyclase and potassium channels. CO inhalation also appears to protect vital organs, including the brain, heart, lung, and liver, during ischemia/hypoxia and organ transplantation,16–19 although the underlying mechanisms remain unknown. Consistent with the postulated beneficial role of CO, higher expression of HO leads to a better outcome for patients after a septic shock.20
However, the practical clinical use of CO gas is currently hampered severely. Owing to the relatively low solubility of CO in water (about 1 mM), its partitioning to body fluids and target tissues is rather limited. To reach appreciable concentration inside the body, high concentrations of CO gas would need to be inhaled. Furthermore, the potential interaction of CO with various molecules in physiological environments complicates a targeted administration as well as research on its physiological functions.3 Therefore, various methods have been devised to deliver CO precisely to the target locations without off-target effect. CO-releasing molecules (CORMs) that can be targeted to specific sites in the body and locally liberate CO are thus urgently needed for research and possibly for clinical applications. Furthermore, in most cases reported beneficial effects of CO on organ function or whole organisms are not understood in molecular detail, specifically lacking insight into the molecular target sites and mechanisms by which CO interacts with biomolecules to alter their function. Here we will therefore review recent advances in studying physiological effector sites of CO as well as novel approaches for the synthesis and use of CO-releasing molecules and materials. Both aspects suffer from a common limitation, namely elucidation of when and where in the body CO is present to exert its activity. Therefore, we will also address new promising methods for the monitoring of CO in living cells.
Several excellent review articles have been published on topics of the physiology and controlled release of CO.21,22 A recent tutorial review discusses features for developing drug molecules for therapy with CO.23 Two novel fluorescent probes for monitoring CO in living cells have been introduced.24 In addition, emerging concepts on the anti-inflammatory actions of CORMs have been shown.3,12 A perspective on CORMs was written by Mann.25,26 Reviews have been published on photoactivated CORMs (photoCORMs).27–29 Potential photoactivated metallopharmaceuticals, such as active molecules and supported drugs, have been featured in this journal.30
B. Effector systems
It is conventionally thought that transduction of CO by biological molecules requires a cofactor. While some prokaryotic oxygenases and oxidases without any prosthetic group or metal cofactor are capable of interacting with O2,31,32 for CO the presence of reduced iron (Fe2+), typically haem iron, is considered to be essential (“haem-based CO sensors”).2 Numerous haem-containing proteins are present in a typical cell,33 rendering the number of potential direct and indirect effectors of CO or CO-sensitive components exceedingly large. The potential effectors include cell-signalling enzymes such as transcription factors, some of which take part in regulation of circadian rhythm, cystathionine β-synthase involved in H2S production, guanylyl cyclase and ion channel proteins.21,22,33 It is not surprising then that experimental application of exogenous CO (see Section A) has been reported to induce multitudes of effects. Selected putative CO effectors – focusing on those in mammalian systems – are discussed below to highlight diverse mechanisms of regulation by CO. Other examples, such as ion channel regulation in mammalian cells, are found in studies of Wilkinson and Kemp34 and Peers.35 For more discussion of CO-sensitive microbial proteins including the CO-sensing transcription factor from Rhodospirillum rubrum (CooA) and CO dehydrogenase (CODH), readers are referred to Roberts et al.,36 Gullotta et al.,22 and Boer et al.37Neuronal PAS domain 2 (NPAS2) transcription factor
Circadian control of cell function involves multiple transcription factors.38 Among them, NPAS2, expressed in the brain, contains a basic helix-loop-helix (bHLH) DNA binding domain and two PAS (Per-Arnt-Sim; PAS-A and PAS-B) domains, and each PAS domain is capable of binding haem.39 NPAS2 forms a heterodimer complex with another protein and binds to its target DNA sequence. An in vitro study suggests that the dimer formation and the DNA binding activity of NPAS2 with haem bound is inhibited by μM levels of CO.39 Raman spectroscopy measurements indicate CO impairs the histidine-mediated ligation of the reduced haem iron.40 How the altered haem-ligation inhibits the dimer formation and DNA binding is not known.Guanylyl cyclase, nitric oxide synthase and CO
The enzyme soluble guanylyl cyclase (sGC; EC 4.6.1.2) is a haem-containing heterodimeric complex (typically α1/β1) that catalyzes conversion of GTP to cGMP, an intracellular messenger that initiates a variety of physiological responses.41–43 Each subunit possesses multiple domains, from the N to C termini: an H-NOX domain (haem-nitric oxide/oxygen binding domain), a PAS domain, a CC (coiled–coiled) domain and a CAT (catalytic) domain.43 The activity of sGC under reducing conditions is markedly enhanced, more than 100-fold, by the gaseous messenger NO44,45 and this regulation accounts for the well-known vasodilatory influence of NO.46 CO has been reported to increase the sGC activity albeit much less effectively than NO,45,47 approx. 3- to 4-fold.44,45 It may be noted that such an increase in the enzymatic activity by CO was not observed in a separate study.48 O2, present at a higher concentration than NO, in contrast, has no effect on sGC.47,49 The postulated CO-mediated increase in the sGC activity may contribute in part to the vasodilatory action of low concentrations of CO.50,51 The structural and physicochemical bases of the interactions of NO and CO with sGC have been studied.52 The H-NOX domain of each β subunit of sGC may contain a haem whose propionate groups are surrounded by the conserved sequence motif Tyr-X-Ser-X-Arg where X is any amino acid (Fig. 2A).53 The reduced haem iron centre is coordinated by a His residue. This results in a pentacoordination mode. A single NO molecule binds to the distal side of the haem iron (at least transiently), creating a hexacoordinated haem (Fig. 2B). The fully activated state of sGC is pentacoordinated44 in which the original proximal His ligand is displaced.52 Like NO, a CO molecule also binds to the distal side of the haem iron (Fig. 2C). The atomic structural changes induced by binding of NO or CO are subtle (Fig. 2); a very slight pivoting of the haem cofactor (<1 Å) has been suggested to account for the enhanced enzymatic activity.52 In addition, a change in the hydrogen bonding network around the haem cofactor has been proposed to account for the contrasting effects of NO and O2 on sGC.49 Additional insights into the conformational changes induced by NO and CO in sGC may be inferred from the results using 3-(5′-hydroxymethyl-2′-furyl)-1-benzylindazole (YC-1).45,54 YC-1 allosterically increases the sGC activity; in particular, in the presence of YC-1, CO is capable of markedly increasing the catalytic activity essentially to the same level as that by NO.45,55 It has been hypothesized that YC-1 binds to the H-NOX domain and alters the interaction between the H-NOX and PAS domains.56 A naturally-occurring compound that acts like YC-1 has not been reported. Albeit much less effective than NO, activation of sGC by CO has potential to cause an increase in cGMP concentration, which in turn activates cGMP-dependent protein kinase (PKG). PKG could then influence a plethora of signalling effectors – hence CO may exert numerous but indirect downstream effects.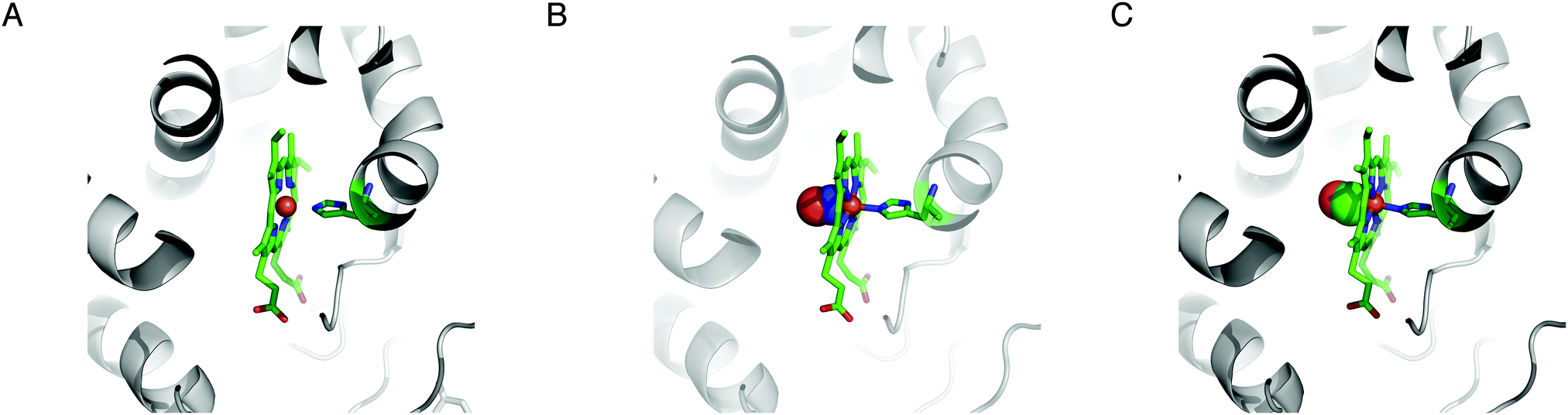 | ||
| Fig. 2 CO binding to soluble guanylyl cyclase. Haem coordination by soluble guanylyl cyclase (A, PDB ID 2O09) with an unoccupied coordination site, in the presence of NO (B, PDB ID 2O0C), and in the presence of CO (C, PDB ID 2O0G). The images were rendered using MacPyMol v0.99. | ||
The synthesis of NO from L-arginine by nitric oxide synthases (NOSs), haem-containing proteins, is also subject to regulation by CO and NO;57 these gaseous regulators inhibit the catalytic activity, potentially providing a negative feedback mechanism. A structural study suggests that CO binds to the distal side of the haem iron centre near the ligand L-arginine.58 While the changes in the overall structure of the enzyme induced by CO binding are again subtle, the presence of a CO near the haem iron centre and the ligand L-arginine is speculated to alter the local hydrogen bonding network, leading to inhibition of the NOS catalytic activity.58
CO binding without haem in microbial enzymes
Selected microbial proteins interact with CO in a haem-independent manner.37,59 For example, CODH (EC 1.2.99.2), which catalyzes oxidation of CO to CO2, contains no haem but each subunit in the dimeric enzyme complex forms multiple metalloclusters with copper, nickel, iron, and/or sulphur.37 Crystal structures of CODH suggest that CO interacts with the nickel ion in one of the metalloclusters (“C-cluster”).60,61 One could speculate that CO binds in particular to iron–sulfur clusters from CODH, which are probably as abundant in cells of higher organism as haem.62 Whether or not similar metallocluster-dependent CO binding occurs in mammals has not been determined. Certainly, the identification of molecular entities capable of binding CO and thus functioning as cellular CO buffer systems will be an important task for future studies.Large-conductance Ca2+- and voltage-gated K+ channels and CO
CO relaxes isolated vascular smooth muscle cells in the absence of endothelial cells.63 A consensus exists that CO (somehow) stimulates large-conductance Ca2+- and voltage-gated K+ channels in vascular smooth muscle cells. These K+ channels, which are also known as BK (“big potassium”), maxi K and Slo1 channels, mediate gated fluxes of K+ ions according to their electrochemical gradient.64 Opening of the “gate” of the channel to allow K+ flux is allosterically facilitated by binding of Ca2+ to its Ca2+ sensors (“RCK1 Ca2+ sensors” and “RCK2 Ca2+ sensors”) in the cytoplasmic domain and/or by depolarization-mediated activation of its transmembrane voltage-sensor domains (VSDs) (Fig. 3A and B). Greater activation of BK channels helps to stabilize the membrane potential at a negative level, which in turn prevents opening of depolarization-activated Ca2+ channels, thereby inhibiting cellular activation. In this manner, stimulation of BK channels in smooth muscle cells by various cellular signalling molecules such as CO exerts a vasodilatory influence.65–67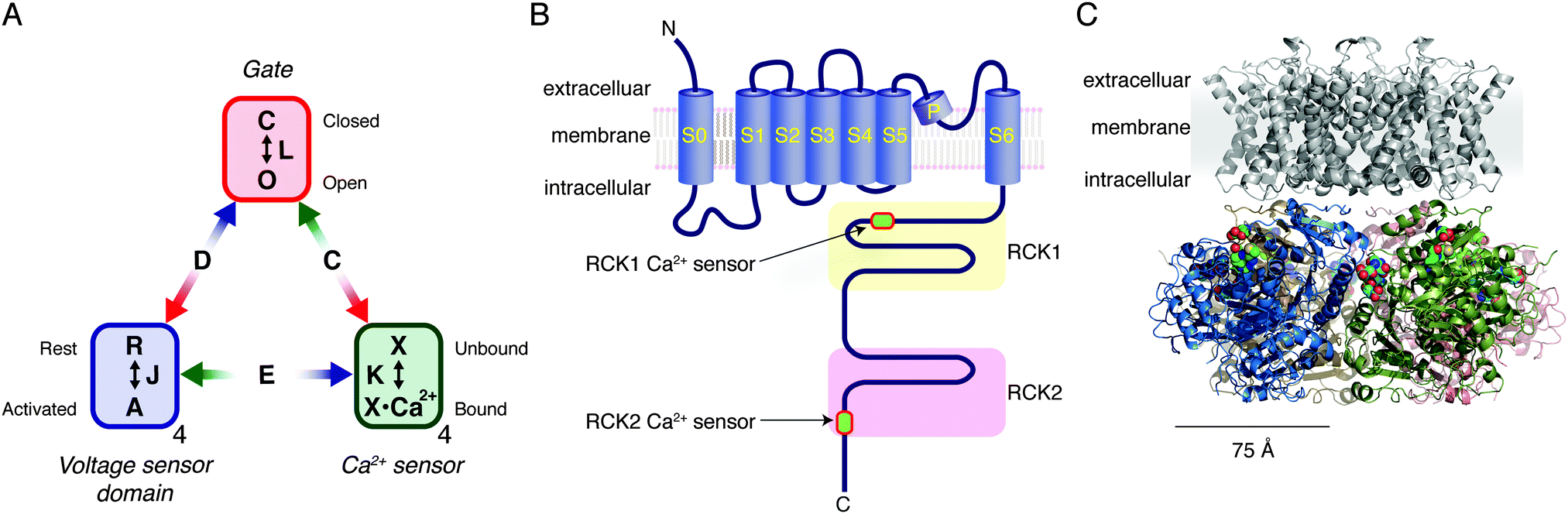 | ||
| Fig. 3 A model of the Slo1 BK channel function and the structure. (A) Allosteric model of the Slo1 BK channel function.74 The ion conduction gate can be closed or open as specified by the equilibrium constant L. Each of the four voltage-sensor domains could be at rest or activated as specified by the equilibrium constant J. Each of the Ca2+ sensors could be unbound or bound as specified by the constant K. The model lumps the two Ca2+ sensors of each Slo1 subunit into one functional unit. The allosteric interactions are specified by the constants D, C and E. (B) Structural organization of each of the four Slo1 subunits in a functional Slo1 BK channel (not drawn to scale). Each polypeptide is about 1100 residues long. (C) Probable structure of a functional Slo1 BK channel. The grey transmembrane domain is a homology model based on PDB ID 2R9R and the cytoplasmic domain is from PDB ID 3NAF. In the cytoplasmic domain, each subunit is shown using a different colour. The Ca2+ ligand residues are displayed using spheres. The images were rendered using MacPyMol v0.99. | ||
In all likelihood, CO activates BK channels in multiple ways. The evidence is consistent with the idea that phosphorylation of the BK channel by PKG at selected cytoplasmic Ser residues (Ser855 and Ser86968 and Ser107269 in human Slo1 AAB65837) increases the overall probability that the channel gate is open. Exactly how phosphorylation of these Ser residues in the cytoplasmic domain alters the energetics of the ion conduction gate in the transmembrane domain located several nanometres away is unclear.70–72 Because CO stimulates sGC, albeit weakly, leading to activation of PKG (see above), it is expected that CO promotes PKG-mediated phosphorylation of BK channels and then increases the channel activity. CO is also reported to stimulate another K+ channel type (TREK1) through activation of the cGMP/PKG pathway.73
Additionally, CO has been postulated to directly alter the BK channel activity in a cell-signalling cascade-independent fashion. Application of CO itself and generation of CO by CORM-2 (Fig. 7) increase the channel open probability in excised membrane patches where the normal intracellular signalling network is disrupted.75–79 According to the idea that the interaction with CO requires a reduced cofactor, the sensitivity of the BK channel to CO and CORM application in cell-free patches suggests the protein itself may harbour a tightly bound cofactor. Structural studies show that some ion channel proteins do contain structurally and functionally important Zn2+.80,81 An atomic structure of the complete BK channel protein is not yet available. However, the structures of the cytoplasmic domain of the BK channel (see Fig. 3C) expressed in insect cells at resolutions of approx. 3.0 Å show no redox-sensitive cofactor.70–72 How could the BK channel apparently without a cofactor respond to CO? This remains an open question.
Some circumstantial clues are available. The enhancement of the channel activity by CO is observed using low Ca2+ solutions chelated by mM concentrations of EGTA or EDTA77 (but see ref. 78). These chelators have higher affinities for multivalent cations like iron, copper, manganese and cobalt than for calcium. Thus CO stimulates the BK channel activity even when the concentrations of these free multivalent cations are negligible. The CO action was also observed following pretreatment of the channel with the oxidizing agent H2O2;77 solvent exposed oxidation-prone cofactors, if present, may not be essential. However, cyanide at μM levels abolished the response to CO (cyano metal complexes often have critical stability constants log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) K > 30).79 This observation may suggest that the BK channel protein contains a metal cofactor. However, as noted above, such a tightly-bound cofactor is not detected in the available structures of the BK channel cytoplasmic domain.70–72 If the BK channel contains a cofactor, it may be present in the unstructured areas of the channel protein such as the cytoplasmic RCK1–RCK2 linker segment and the cytoplasmic distal C terminus unresolved in the available structures. It is also possible that the BK channel protein itself does not contain any cofactor but it may intimately associate with a separate cofactor-containing structural component in select cells, perhaps forming a macromolecular complex. The idea that BK channels participate in formation of macromolecular complexes has received some experimental support.76,82 Clearly, elucidation of the mechanism of CO sensing by the BK and other channels requires further study. Functional eukaryotic channel complexes including BK channels have been difficult to express and purify in large quantities. This limitation has severely hampered the progress in elucidation of the physicochemical mechanism of CO sensing in ion channel proteins, precluding application of more direct metal assay techniques such as inductively coupled plasma mass spectrometry. A high-resolution structure of the whole-channel complex in a native environment will be also helpful.
K > 30).79 This observation may suggest that the BK channel protein contains a metal cofactor. However, as noted above, such a tightly-bound cofactor is not detected in the available structures of the BK channel cytoplasmic domain.70–72 If the BK channel contains a cofactor, it may be present in the unstructured areas of the channel protein such as the cytoplasmic RCK1–RCK2 linker segment and the cytoplasmic distal C terminus unresolved in the available structures. It is also possible that the BK channel protein itself does not contain any cofactor but it may intimately associate with a separate cofactor-containing structural component in select cells, perhaps forming a macromolecular complex. The idea that BK channels participate in formation of macromolecular complexes has received some experimental support.76,82 Clearly, elucidation of the mechanism of CO sensing by the BK and other channels requires further study. Functional eukaryotic channel complexes including BK channels have been difficult to express and purify in large quantities. This limitation has severely hampered the progress in elucidation of the physicochemical mechanism of CO sensing in ion channel proteins, precluding application of more direct metal assay techniques such as inductively coupled plasma mass spectrometry. A high-resolution structure of the whole-channel complex in a native environment will be also helpful.
The stimulatory effect of CO is diminished by high concentrations of intracellular Ca2+77,78 or low intracellular H+.83 Therefore, the conformational changes in the BK channel protein induced by CO may resemble those by Ca2+ and H+;77,83,84 CO increases the gate open probability without requiring activation of the VSDs.83
Chemical modification and mutagenesis studies implicate two specific His residues in the channel's response to CO.75,77 Pretreatment of the channel with the imidazole modifier DEPC75,77 or mutation of His365 and His394 located near one of the two Ca2+ sensors (“RCK1 Ca2+ sensor”; Fig. 3B) in the BK channel protein70–72,77,79 at low intracellular Ca2+ concentrations markedly impairs the channel's response to CO and CORM-2 (50 μM). However, application of high concentrations of CORM-2 (e.g., 300 μM) stimulates the channel even when His365 and His394 are absent,79 suggesting that His365 and His394 may preferentially mediate the channel's sensitivity to low concentrations of CO. The exact functional roles played by His365 and His394 are not yet known. The two His residues could somehow directly interact with CO in a novel way or couple the information regarding the interaction of CO with its sensor located elsewhere to the ion conduction gate. In addition to His365 and His394, Cys911 is involved in the channel's response to CO. Cys911 is located near the RCK2 Ca2+ sensor (Fig. 3B)70–72 and plays an important role in the oxidative sensitivity of the Ca2+ sensing of the channel.85,86 Mutation of Cys911 diminishes the channel's response to CORM-2; the dose dependence results suggest that the mutation preferentially decreases the efficacy of CO without altering the half-maximal effective concentration.79
In summary, the stimulatory effect of CO on the BK channel, not surprisingly, involves multiple amino-acid residues: at least, two His residues (His365 and His394) near the RCK1 Ca2+ sensor and a Cys residue (Cys911) near the RCK2 Ca2+ sensor. Whether these residues comprise the CO sensor(s) of the channel or the coupling mechanism connecting the ultimate effector, the gate of the channel, and the CO sensor elsewhere in the channel or even in a different intimately associated protein remains unclear. Histidine and cysteine can complex with metal ions but the atomic structures of the cytoplasmic domain of the channel70–72 do not reveal any metal bound to His365, His394 or Cys911.
Local action of CO on BK channels
CO is potentially capable of altering functions of numerous haem-containing enzymes. The uncontrolled and global action of CO would be detrimental to normal cell function. How could CO target specific effectors, such as BK channels? The key concept is the local action of CO. CO is primarily generated by HO1 and HO2 from haem in an oxygen- and NADPH-dependent manner (Fig. 1). Thus, the molecules in the close vicinity of HOs are expected to be more readily influenced by CO. In selected cells, Slo1 channels are reported to exist co-localized with HO2 so that small quantities of CO could have a significant and rapid impact on the channel function without altering other proteins.76 Under normoxia, HO2 provides a basal stimulatory influence on nearby BK channels via CO. With a decrease in oxygen tension (hypoxia), the CO generation by HO2 is compromised and the stimulatory influence diminishes. In this manner, CO is an important mediator of hypoxia sensing in carotid body glomus cells76 although other mechanisms are operative.87,88A similar mechanism of O2-dependent regulation involving CO and haem together is observed in voltage-independent trimeric Na+ channels89 expressed widely in epithelial cells (ENaCs).90 Application of haem and NADPH, both of which are required for the HO to produce CO, stimulates the channel. CORM-2 also stimulates the channel activity. In contrast, haem applied under hypoxic conditions inhibits the channel activity. The molecular domain of ENaC responsible for ligation of haem is not known and whether ENaC and HO colocalise has not been established.
As illustrated in the examples discussed earlier, haem is a stable cofactor in numerous proteins, often conferring gas sensitivity to the parent proteins. While the concentration is low, free haem does exist in cells.91 Free haem at nanomolar levels is capable of acutely and directly regulating the BK channel activity.92 The relatively unstructured cytoplasmic RCK1–RCK2 linker region (Fig. 3B) coordinates haem using His and Cys residues.92–95 Addition of exogenous free haem to the channel weakens the allosteric coupling between the VSDs and the ion conduction gate.96 Consequently, the gate open probability is higher at negative voltages and lower at more positive voltages.96 This modulatory action of exogenous free haem is diminished by CO and may represent another mechanism by which CO alters the channel function.93
Voltage-gated Ca2+ channels, CO and ROS
Yet another mechanism by which CO influences its effectors is through generation of reactive oxygen species (ROS). CO binds to the mitochondrial complex IV enzyme cytochrome c oxidase and increases the mitochondrial production of ROS.97,98 ROS can in turn alter many proteins including voltage-gated Ca2+ channels.99Both NO and CO are excellent ligands for haem in sGC with similar binding behaviours (Fig. 2B and C). The high binding affinities to iron porphyrins have been also used in the detection of NO and CO.100 However, elucidation of the physiological roles of CO demands for improved analytical methods of CO sensing.101–103 The following section addresses recent advances in this rapidly developing field.
C. CO detection
Established methods for CO detection include gas chromatography,104 laser infrared absorption105 and electrochemical assays.101,106 In addition, colourimetric CO sensing is an important alternative,103,107 in particular if the aim is to monitor CO in living cells, organs or whole organisms.The standard in vitro test for CO is the carboxy-myoglobin (Mb-CO) assay, where CO is added to a solution of reduced deoxy-Mb and the formation of Mb-CO is followed via absorbance spectroscopy. Optical changes on CO binding in the α or β regions or in the Soret band are detected.108 Using this method, striking discrepancies in the CO release rates of (Ru(CO)3Cl2)2 (CORM-2) and [Ru(CO)3Cl(glycinate)] (CORM-3, see Fig. 7) have been reported.109 Subsequently, it was shown that the reducing agent sodium dithionite is responsible for rapid CO release from Ru-based CORMs in the Mb-CO assay. In addition, dithionite also enhances CO release rates from other CORMs.103 To overcome the problem, two novel protocols have been recently introduced. Oxy-haemoglobin103 and a modified myoglobin assay110 have been used for reliable determination of CO-release rates from organometallic carbonyl complexes.
It was recently shown that CO can be detected selectively with high sensitivity by the binuclear rhodium complex cis-[Rh2(C6H4PPh2)2(O2CCH3)2](HAc)2.111 It contains two cyclometalated phosphine ligands and undergoes a colour change from violet to orange via replacement of an acetic acid (HAc) molecule by CO (Fig. 4). In addition, replacement of the second HAc by CO induces a colour change to yellow.112 A collection of binuclear rhodium complexes displayed high CO selectivity with remarkable detection limits. For example, ([Rh2{(m-CH3C6H3)P(m-CH3C6H4)2}2(O2CCH3)2](HAc)2), adsorbed on silica gel, is capable of CO detection by the “naked eye” at concentrations as low as 0.2 ppm in air.112 Furthermore, the binding of CO in all rhodium complexes was fully reversible.
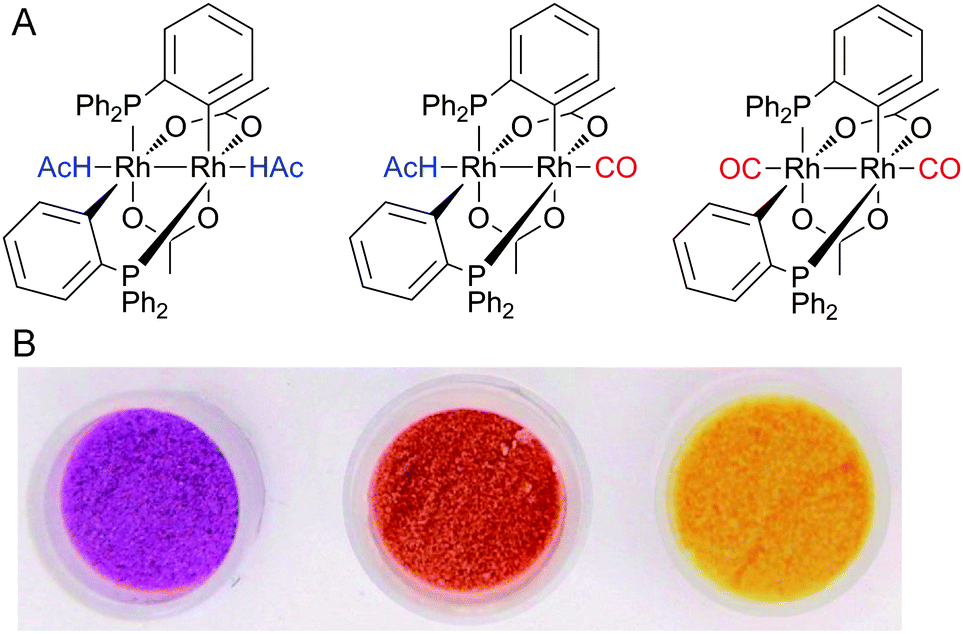 | ||
| Fig. 4 Colourimetric detection of CO via binuclear rhodium complexes. (A) Basic structure of the complexes (HAc = CH3CO2H). (B) The photograph shows ([Rh2{(m-CH3C6H3)P(m-CH3C6H4)2}2(O2CCH3)2]·(CH3CO2H)2) adsorbed on silica gel in the absence (left) and presence of 8 ppm (middle) and 2000 ppm (right) of CO. Adapted and reprinted with permission from ref. 112. Copyright 2011 American Chemical Society. | ||
Unfortunately, the organometallic dirhodium compounds are only soluble in organic solvents, such as chloroform. Thus, these complexes are not useful for the real-time detection of physiological levels of CO inside living cells. In this context, fluorescence sensing and imaging has emerged as one of the most powerful techniques to monitor the concentration, localization and even movements of biomolecules in living systems.113,114 A variety of fluorescent probes for other small signalling molecules, such as NO and hydrogen sulfide,115 have been already deployed in biology.116 Recently, the development of fluorescent probes for CO has experienced a boost with a biosensor117 and an organometallic palladium complex probe.118 Although these two approaches appear distinct, the fundamental design strategy is similar. In both cases the strong binding affinity of CO to transition metal ions is exploited.24
The palladium probe is able to detect CO in living cells based on metal-mediated carbonylation chemistry (Fig. 5).118 The cyclopalladated species COP-1 quenches the fluorescence of the borondipyrromethene difluoride (BODIPY) core via heavy-atom electronic effects. Upon binding of CO, a carbonylation reaction concomitantly releases Pd(0) and a BODIPY dye with high fluorescence intensity. A 10-fold fluorescence enhancement was observed only in the presence of CO compared with biologically relevant reactive oxygen, nitrogen and sulphur species. The fluorescence intensity enhancement is concentration dependent with a detection limit of 1 μM of CO. The nontoxic and biocompatible palladium-based probe allows CO monitoring in living cells. However, the response time is about an hour to reach the highest level of fluorescence enhancement.
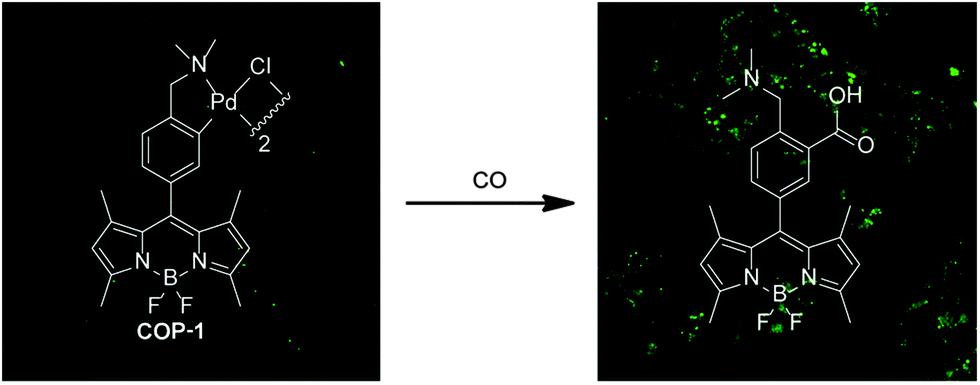 | ||
| Fig. 5 Confocal microscopy images of the turn-on fluorescent probe for selective CO detection based on palladium-mediated carbonylation reactivity (COP-1). The organometallic probe is capable of detecting CO both in aqueous buffer and in living HEK293T cells with high selectivity. Left: cells incubated with COP-1 for 30 min. Right: cells incubated with 50 μM CORM-3 and 1 μM COP-1. Adapted and reprinted with permission from ref. 118. Copyright 2012 American Chemical Society. | ||
The other approach utilizes the interaction of CO with an iron-containing haem protein as exemplified in the detection mechanism of the fluorescent probe COSer (Fig. 6).117 This biosensor uses a yellow fluorescent protein (YFP) as the fluorescent reporter and CooA, a dimeric CO-sensing haem protein from Rhodospirillum rubrum. Upon treatment with 10 μM CO for 10 minutes, the probe exhibits a small two-fold fluorescence intensity enhancement. This observation was attributed to the conformational change of CooA upon binding to CO. The probe showed good selectivity for CO against other relevant haem binding ligands, such as H2S, GSH, NO, O2, CN− and imidazole. The probe COSer is able to monitor CO fluctuations inside living HeLa cells.
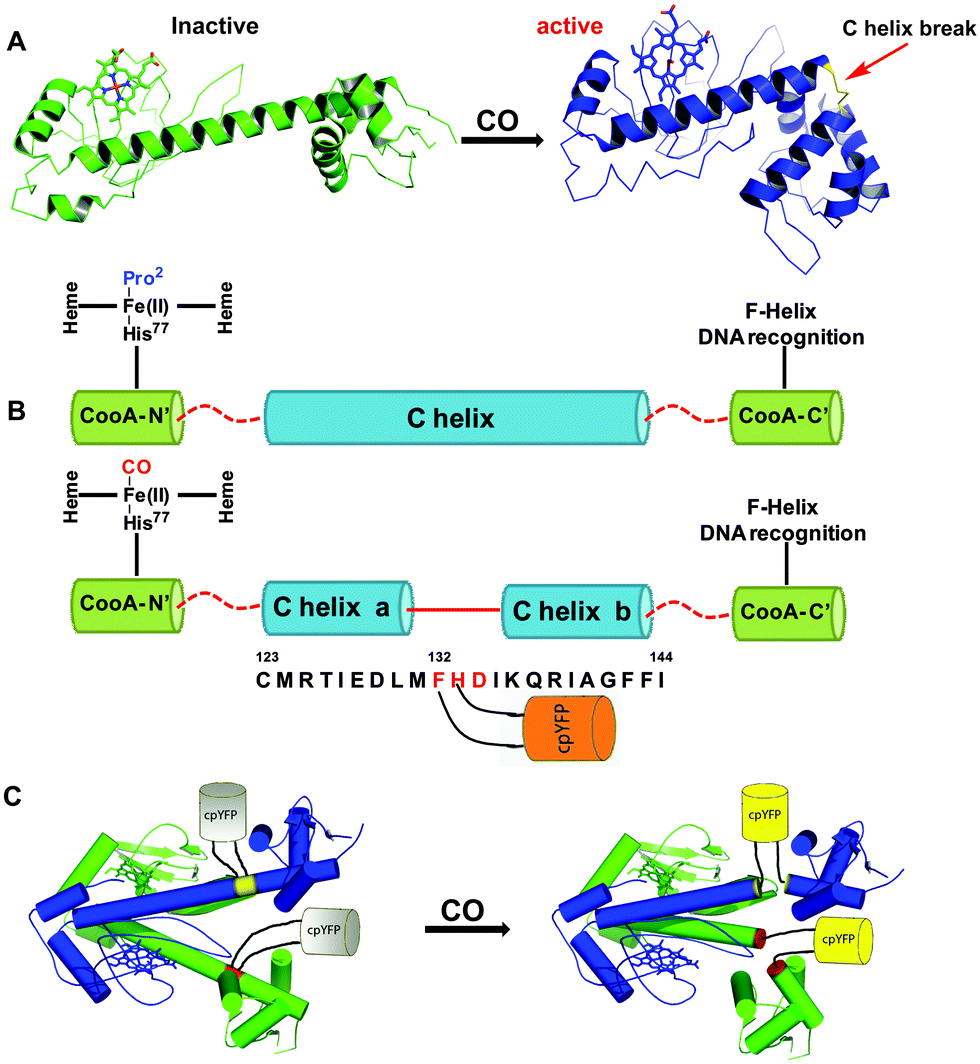 | ||
| Fig. 6 The biosensor COSer is composed of yellow fluorescent protein (YFP) as the fluorescent reporter and CooA, a dimeric haem protein from Rhodospirillum rubrum as the CO recognition unit. (A) Left: inactive monomer CooA. Right: active CO binding to CooA induces a conformational change. (B) Upon CO binding, the C helix of CooA is broken into two parts (“C helix a” and “C helix b”) connected by residues 132 to 134. (C) COSer contains YFP inserted by two short linkers between residues 132 and 133 in each CooA monomer. The resulting structural change upon CO binding to CooA induces an increase in fluorescence of the probe. Reprinted with permission from ref. 117. Copyright 2010 Wiley-VCH. | ||
The properties of the genetically encoded probe COSer and small-molecule probe COP-1 have been critically assessed.24 The COP-1 probe displays a larger fluorescence signal enhancement (10-fold) than the genetically encoded fluorescent probe COSer (2-fold) in vitro. However, the biosensor COSer is faster than COP-1 in the response time. Another striking difference is given by the reversibility of the sensors. While the interaction of the COSer probe with CO is reversible, that of COP-1 is irreversible. Thus, the COSer is better suited for real-time detection of CO, while COP-1 is more appropriate for monitoring low concentrations of CO because of signal accumulation. It is also important to note that the irreversible COP-1 might act as a scavenger of CO from biological systems, much like myoglobin (see beginning of Section C), thereby significantly shifting equilibria of CO exchange.
More robust and more sensitive CO sensors are still needed for the investigation of CO-mediated cellular signalling mechanisms and for the evaluation of potential pharmaceutical CO donors designed to treat human diseases. While pharmaceutical NO donors are widely used, the development of methods for delivering CO has not yet led to clinical trials of promising CO-donating compounds.119 Nevertheless, investigations on the potential role of CO gas and CO-releasing molecules as therapeutics are ongoing.3,12–14
D. Controlled release of CO
CO-releasing molecules (CORMs)
The high toxicity of inhaled CO necessitates sophisticated strategies to administer a defined amount of CO as a therapeutic agent at a predetermined location and time. Consequently, carriers of CO that meet specific requirements, such as solubility in aqueous media, low toxicity of these “small” CO-releasing molecules (CORMs) and their degradation products as well as a triggered CO liberation from these compounds, are clearly needed.Diverse compound classes enable a triggered CO delivery. For example, transition metal-free Na2(H3B-CO2) (CORM-A1) liberates CO upon acidification.120,121 The first protonation step yields Na[H3B-CO2H] and a second leads to split-off of water and formation of H3B-CO, which rapidly loses CO.122 Further, a water-soluble fluorescein analogue 6-hydroxy-3-oxo-3H-xanthene-9-carboxylic acid was recently introduced as the first transition metal-free CORM activated by light (photoCORM) at a wavelength of 500 nm.123 Light-triggered release of a CORM based on micelle-encapsulated unsaturated cyclic α-diketones was also generated. These systems allow the delivery of CO to be monitored by fluorescence imaging techniques.124 Organometallic compounds have recently gained significant attention in potential medicinal treatments.125 Up to now, metal carbonyl complexes represent preferred reagents to deliver CO because they offer manifold advantageous variations, such as nature and oxidation state of the metal centre (size, charge, Pearson hardness, Lewis acidity, structural diversity), number of carbonyl ligands, nature of coligands (charge, Lewis basicity, π-bonding properties, complex stability) and outer coordination sphere (solubility, Brønsted acidity, amphiphilic character, Fig. 7 and 11).23,25,26,126 Current developments also include investigations to enhance the variety of CO-release triggers and to clarify degradation pathways after liberation of CO.
Metal carbonyl complexes have been available for many decades;127 however, their therapeutic use as CO-donating reagents has become appreciated only recently.128 Metal-based CORMs can contain essential trace elements (especially manganese, iron and cobalt) as well as non-physiological metals such as ruthenium, tungsten and rhenium.125,129 Here, only a selection of very recently investigated CORMs of Cr-, Mn-, Fe-, Mo-, W-, Re- and Ir-containing complexes will be discussed, classified by the three most important mechanisms of CO-release: photoCORMs, solvent-induced ligand exchange on CORMs and enzyme-triggered CORMs (ET-CORMs).23,126 CORMs are considered to be possible prodrugs that deliver the signalling molecule CO at the disease site. The vast development of these compounds is based on the first CORM generation consisting of the DMSO- and ethanol-soluble metal carbonyl complexes Mn2(CO)10 (CORM-1, light-triggered CO release) and CORM-2 (ligand exchange-triggered CO release) as well as water-soluble (OC)3RuCl(O2C-CH2-NH2) (CORM-3, ligand substitution-triggered CO release).13
In order to ensure solubility and stability in aqueous media, solvent-separated ions containing non-coordinating anions proved to be advantageous. The recent photoCORM [(OC)3Re{P(CH2OH)3}(bpy)](F3CSO3) was stable and soluble in aerated water and showed no apparent cytotoxicity; irradiation with light initiated the liberation of one CO molecule, which was replaced by a water ligand.130 A similar strategy was applied by the group of Mascharak to deliver CO with photoactive manganese(I) complexes of the type [(OC)3Mn(L)]+ with L being a tripodal ligand such as tris(2-pyridyl)amine or bis(2-pyridylmethyl)amine.131–133 However, [(OC)3Re{P(CH2OH)3}(bpy)](F3CSO3) bears an outstanding feature: the CORM itself and the inactive CORM (iCORM) products are fluorescent and can be detected by fluorescence microscopy in biological medium.130 CORM and iCORM can be discriminated via different emission wavelengths.
CO-containing metal anions also prove to be soluble in aqueous media. Iridates(III) of the type [Cl4Ir(CO)(L)]− with trans-arranged Lewis base L and CO are an impressive example because the nature of L (H2O, pyridine, 1-methyl-imidazole, 4-dimethylamino-pyridine) influences the back donation of charge into the π*(CO) orbital and the M-CO dissociation energies thus allowing predetermining the CO-release properties.134 If the toxicity of the metal comes to the fore, iron-based CORMs seem to be advantageous. This strategy may particularly promising if iron(II) is embedded in a coordination sphere of biogenic ligands also limiting toxic degradation products after CO liberation. Examples of this strategy include photo-labile [(OC)2Fe(SCH2CH2NH2)2] (CORM-S1)135,136 and [(OC)2Fe{SCH2CH(CO2H)NH2}2]137 with bidentate cysteamine and cysteine ligands, respectively, and cis-arranged carbonyl ligands. Homologous [(OC)2Ru(SCH2CH2NH2)2] is not a suitable photoCORM due to the rather short wavelength required for Ru–CO bond activation. However, polypyridyl ruthenium(II) carbonyl complexes allow photoinduced CO liberation.138 In addition, Mascharak et al. demonstrated the role of ancillary ligands in the capacity of CO photorelease of mono- and dicarbonyl ruthenium(II) complexes with an N,N,S-donor ligand.139 Nevertheless, the lack of toxicity led to the development of diverse organoiron complexes as suitable photoCORMs.27–28,30 A recent example was reported by the group of Kodanko. The stable iron carbonyl complex [Fe(CO)(N4Py)](ClO4)2 released CO upon irradiation with 365 nm light and showed photoinitiated growth inhibition of prostate cancer cells.140 Another group of photoCORMs consists of Fe(CO)3 fragments bound to π-systems of unsaturated hydrocarbons, such as norbornadiene,141 cyclohexadiene,142,143 indenyl144 and cyclopentadienyl.145 The substitution patterns of these side-on bound unsaturated hydrocarbons influence solubility in aqueous media and half-lives of CO liberation after irradiation. Photo-activation also initiates CO release from Mn(CO)4 derivatives with 2-pyridylphenyl ligands.146 An elegant method to deliver extremely CO-rich molecular metal complexes can be realized by metallodendrimers. Thus, a metallodendritic photoCORM was built from an organic dendrimer with 2,2′-bipyridyl end groups acting as strong bidentate ligands to multiple Mn(CO)3 fragments.147
Light-triggered CO release is probably not suitable in all therapeutic applications and, therefore, other triggers were studied. In chromium complexes of the type [(OC)5Cr(L)] with L as halide148 or aminoesters149 the rate-determining substitution of L by a solvent molecule (such as water or DMSO) induces the CO release process. Similar CO release mechanisms can be assumed for rhenium(II)-based CORMs also containing varying amounts of bromide anions; an exchange of a bromide by a water molecule in the vicinity of the metal centre also explains the pH dependence of CO liberation.150,151 Further, three iron-based CORMs, [(PaPy3)Fe(CO)](ClO4), [(SBPy3)Fe(CO)](BF4)2, and [(Tpmen)Fe(CO)](ClO4)2, derived from designed polypyridyl ligands, rapidly release CO upon dissolution and caused vasorelaxation in a mouse aorta muscle ring preparation.152 The water soluble [Fe2{μ-SCH2CH(OH)CH2(OH)}2(CO)6] releases CO via substitution by cysteamine with minimal cytotoxicity of the CORM itself on two cell lines QSG-7701 and HepG2.153 In addition, Ford et al. discussed an oxidative cascade leading to the release of further CO from Na3[W(CO)5(tris(sulphonatophenyl)phosphine)] subsequent to the initial photo-activated CO dissociation (Fig. 7).154
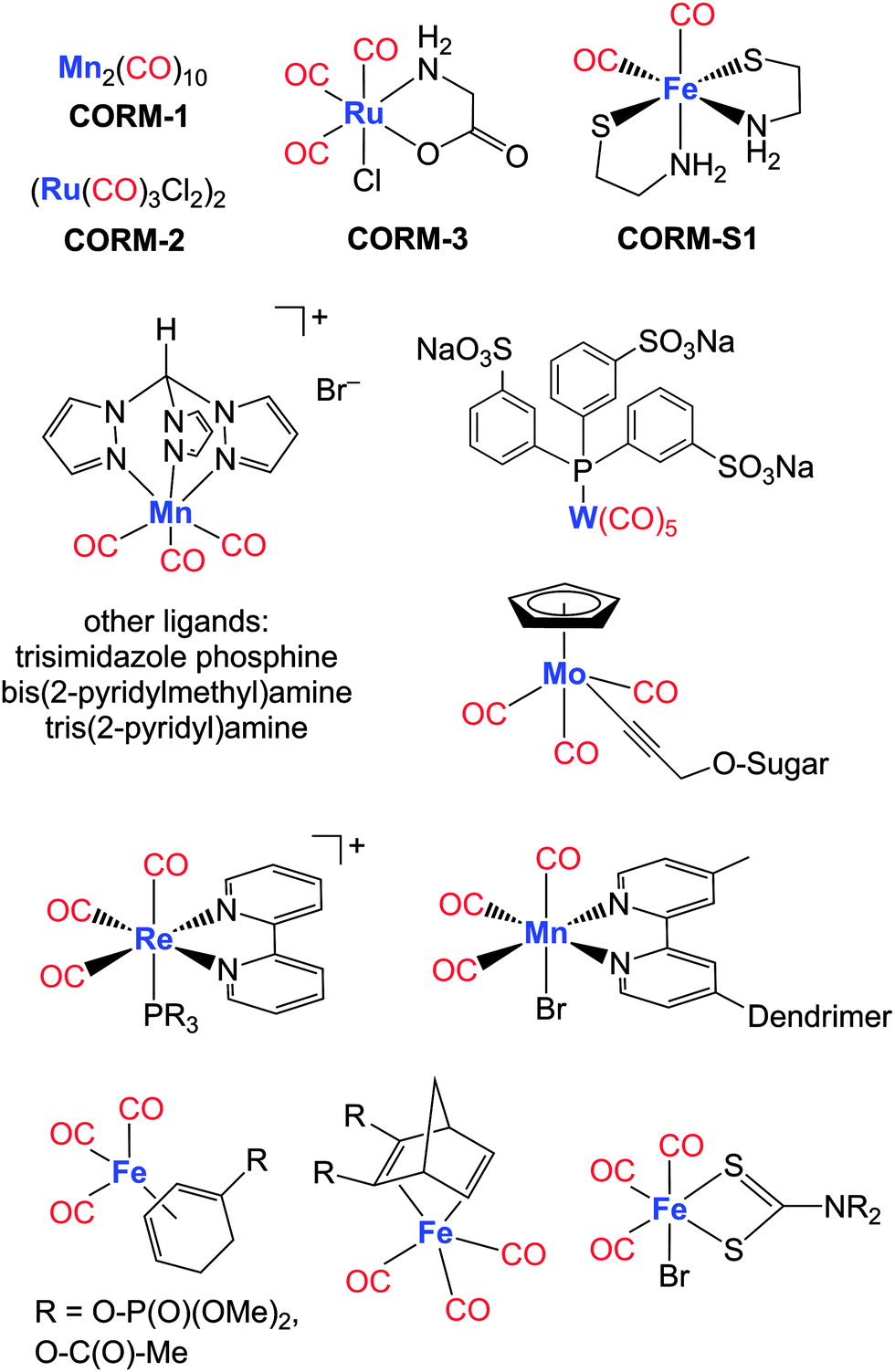 | ||
| Fig. 7 Selection of metal-based CO-releasing molecules (CORMs). | ||
Closely related to the photoCORMs with a Fe(CO)3 moiety bound to an unsaturated hydrocarbon, cyclohexadiene iron tricarbonyl complexes also represent enzyme-triggered CO releasing molecules (ET-CORM) if acyloxy side-arms are bound to the side-on bound unsaturated hydrocarbon.142,143 This substance class has been well studied for many years.155–157 The recently studied use as ET-CORMs is surprisingly convenient. In the first reaction step the ester group is attacked by an esterase leading to a cyclohexadienylalcohol ligand (Fig. 8). Complexes that are modified in such a manner readily decompose under mild oxidative conditions liberating CO. Cytotoxicity and CO-release activity can be tuned by variation of the acyl group of the η4-bound acyloxy-cyclohexadiene ligand158 or by addition of additional substituents at this ligand. CO release from phosphoryloxy-substituted (η4-cyclohexadiene)Fe(CO)3 complexes was induced by a phosphatase and monitored via gas chromatography.159 However, the esterase-triggered oxidative mechanism was monitored with the myoglobin assay under reducing conditions maintained by dithionite. A comparable structural motif, namely a butadiene moiety as part of a six-membered cycle side-on coordinated at a Fe(CO)3 fragment, stabilises the (η4-pyrone)tricarbonyliron(0) complexes that are capable to act as CO transfer reagents for the delivery of controlled amounts of CO.160–162 Stronger π-bases, such as a cyclopentadienide anion, push the electroneutral butadiene base out of the coordination sphere and lead to a η1-coordination of the pyrone ligand via an oxygen base.163
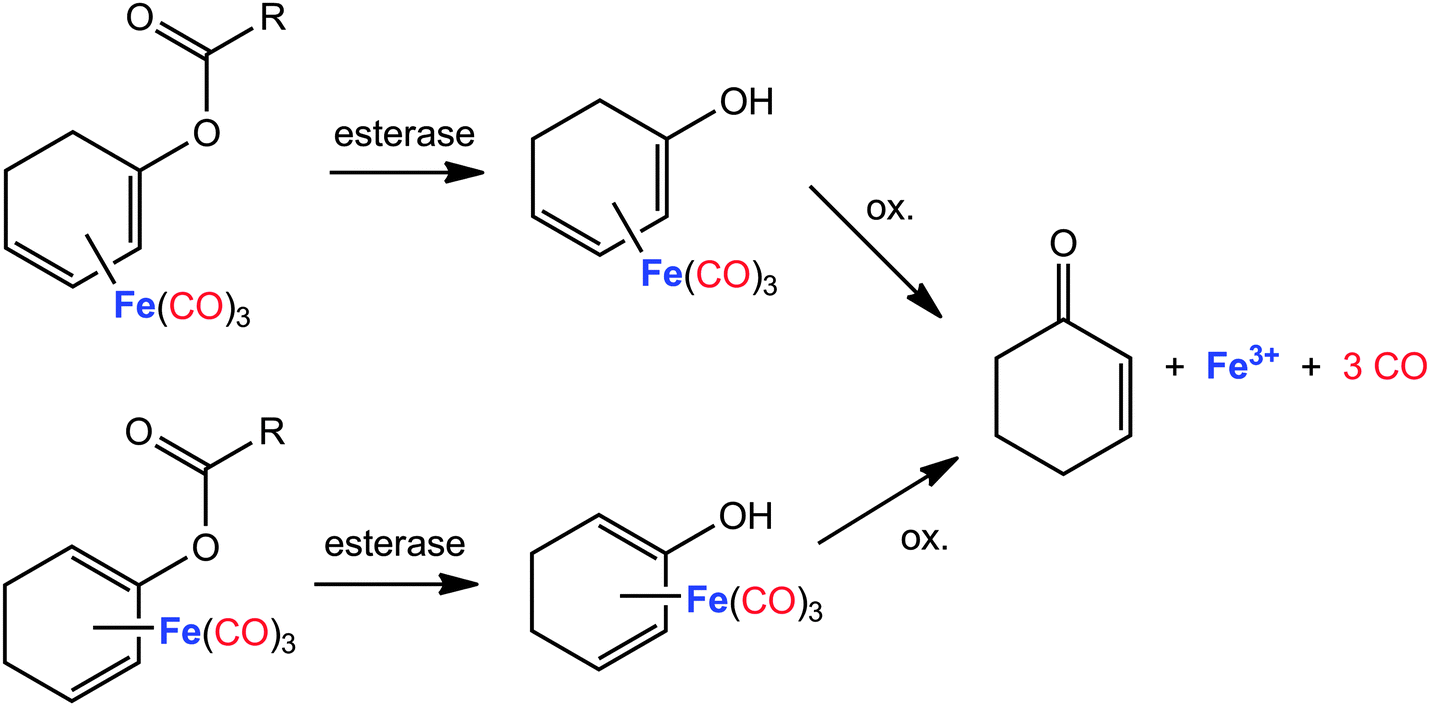 | ||
| Fig. 8 Degradation and CO liberation from acyloxycyclohexadiene tricarbonyliron complexes (enzyme-triggered CORMs, ET-CORMs).158,159 | ||
In order to study the mode of action in biological tissues, the detection of CORMs164 and of liberated CO at the disease site (i.e. at the location of CORM degradation) is of utmost importance (see Section B). Therefore, interactions between CORMs and biologically relevant scaffolds were structurally investigated. L-Histidine can act as a tridentate N,N,O-ligand at a Mn(CO)3 fragment.165 The reaction of a ruthenium-based CORM of the type [(OC)3RuL3] with lysozyme yields the formation of an adduct of five [Ru(CO)(H2O)4]2+ ions with this enzyme, binding to the histidine and aspartate sites; during formation of this complex, the majority of the carbonyl ligands was substituted by water molecules.166,167 In this adduct the histidine moiety acts as a monodentate ligand completing the octahedral coordination sphere of the ruthenium ions (Fig. 9).
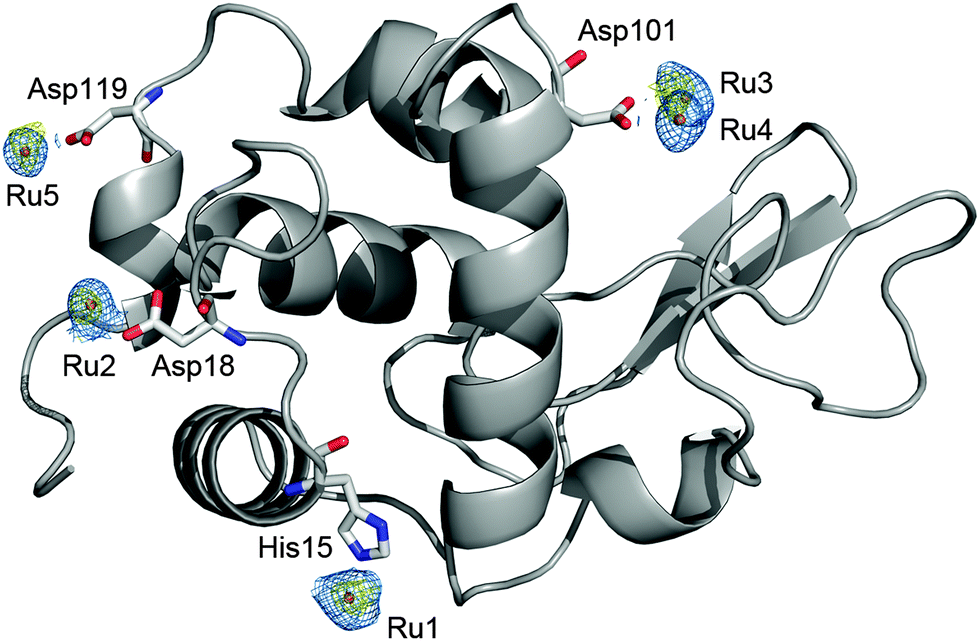 | ||
| Fig. 9 Structure of hen egg white lysozyme bound to Ru fragments derived from fac-[Ru(CO)3Cl2(1,3-thiazole)]. Four amino-acid residues (His15, Asp18, Asp101 and Asp119) interact with five ruthenium atoms. Adapted and reprinted with permission from ref. 167. Copyright 2012 Elsevier. | ||
In light of the potential clinical application of CORMs, the degradation pathway and the nature of the degradation products are of high interest. The first reaction step is the dissociation of at least one CO leaving a vacant coordination site. This remaining metal fragment can either degrade to the metal ions and free coligands as observed for example for [(MeCN)(OC)Fe(H2NCH2CH2PPh2)2]2+ (CORM-P1)168 or attract another ligand to recomplete the coordination sphere as discussed for the dithiocarbamate (dtc) complexes [(OC)4Mn(dtc)]169 and [(OC)3Fe(Br)(dtc)] binding HPO42−, H2PO4−, halide or water at the free coordination site.170 Further degradation steps may proceed via oxidative pathways. Thus, manganese(I)-based mononuclear CORMs were oxidized during step-wise CO release, finally yielding a dinuclear manganese(III) complex with a central Mn–O–Mn unit.171 Further oxidation was observed for CORMs based on heavier transition metals. Thus, the first reaction steps of the degradation of [(OC)2ReBr4]2− (ReCORM-1)172 involve several ligand exchange reactions finally giving the ReO4− anion via intermediate [(OC)2ReII(Br)(H2O)3]+ and [(OC)2ReI(Br)(H2O)2(OH)]−. After CO release and ligand dissociation, cascades followed by oxidation processes, molybdenum-based CORMs end up as phosphomolybdate [PMo12O40]3−; an X-ray structure determination showed the adduct formation of this anion with lysozyme via a hydrogen-bridge network.173
Dependent on the pH value of the aqueous solutions, the carbonyl ligands can be attacked by hydroxide ions yielding M–C(O)OH moieties (Fig. 10). There is evidence that [(OC)3Ru(Cl)(O2CCH2NH2)] acts as a strong acid.174 Thus, this CORM binds OH− from water and the remaining protons lead to a pH value of 3. If the resulting anion is titrated with a base until a nearly neutral pH value of 6 is reached, a doubly charged anion (due to deprotonation of the Ru–CO2H moiety) or a chloride-free anion (via exchange of the chloride ion by a hydroxyl ligand) is formed as shown in the middle row of Fig. 10. In alkaline solution (pH = 10) both reaction patterns are realized yielding the dianion depicted in the bottom row of Fig. 10.174 The mechanisms of CO release from ruthenium(II)-based CORMs with methoxycarbonyl or ethoxycarbonyl ligands are also known.175 Whereas in this example the oxidation state of Ru remained 2+, transition metals can adopt several stable oxidation states and the CO release properties may interfere with the redox chemistry of these metals in aqueous solution. Mann et al. noted that CORMs might be able to catalyze Fenton-type reactions leading to the formation of ROS.25 It is well-known that the pKa values of water molecules in the vicinity of metal cations differ significantly from free water molecules easing the formation of metal-bound hydroxide176 and influencing the redox behaviour of the metals.
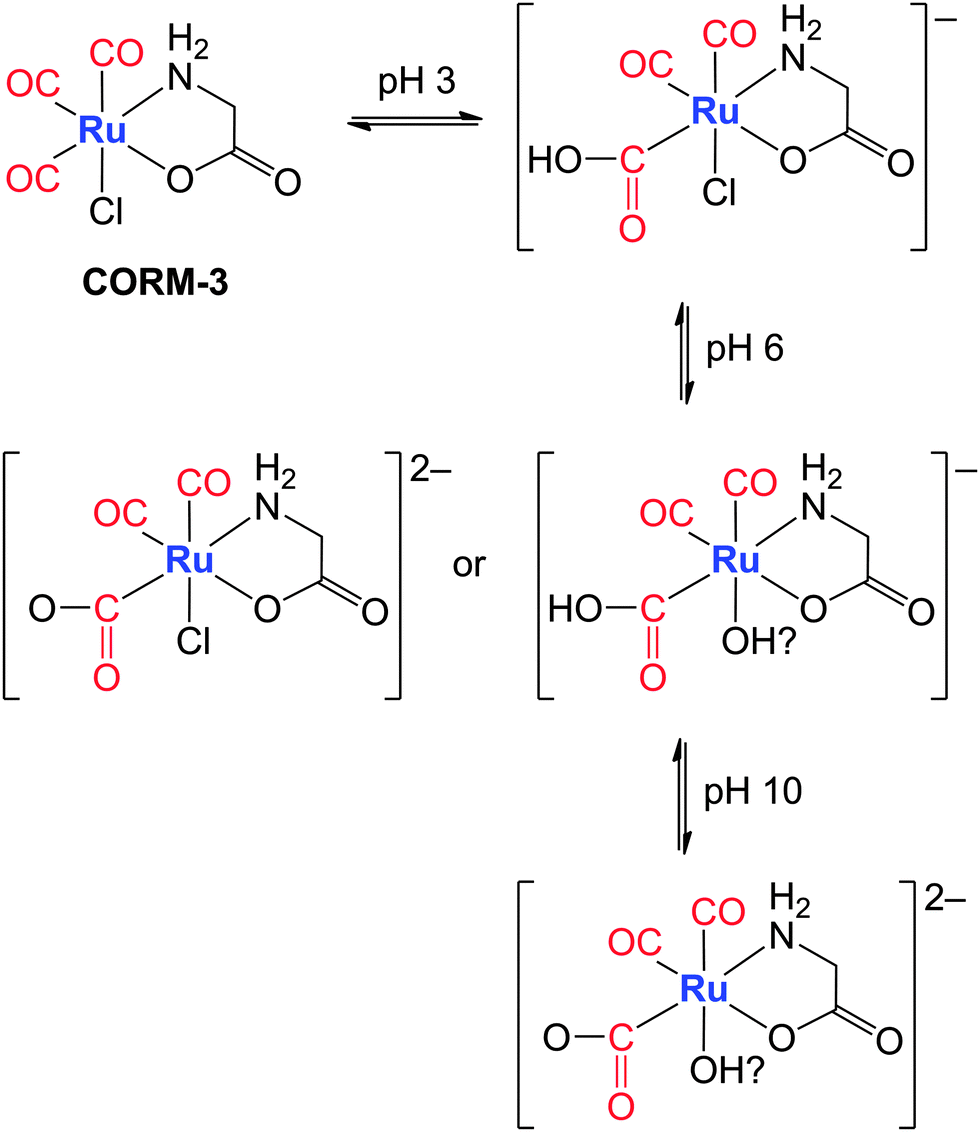 | ||
| Fig. 10 Proposed pH-dependent interaction of CORM-3 with bases. The attack of a hydroxide ion at a carbonyl ligand results in a hydroxycarbonyl moiety; at higher pH values the acyl group can be deprotonated or the chloride ion substituted by an OH group. The suggested final species combines both possible interaction pathways under alkaline conditions.174 | ||
Thus far, CO-release trigger, solubility in aqueous media, kinetics of CO liberation, toxicity of the CORMs itself, and their degradation products played the major role in the development of new CORMs. Recent work focused on the targeted delivery by variation of the outer ligand sphere. Thus, peptides can be part of one ligand in order to support targeted delivery to cellular systems.177–179 In addition, it can be desirable to develop fast and slow CO releasers to suit diverse therapeutic applications. In special cases it can also be advantageous to immobilize the CORMs in order to ease removal of metal-containing degradation products and/or to control the environment of the metal ions (see the section CO-releasing materials). Future developments need to combine strategies for predetermining CO-release properties and for targeted delivery. It is also necessary to prepare and assess iCORMs independently to investigate their physiological properties after CO release.178,180
The recent review by Romão et al. conceptualised elegantly the future CORM design.23 They proposed a model as a tool to help rationalising the design of metal carbonyl CORMs with the appropriate pharmaceutical properties. As an example, an octahedral geometry with six ligands surrounding the metal centre was shown (Fig. 11). At least one CO ligand coordinates to the metal centre. Thermodynamic and kinetic stability of the complex is provided by chelating ligands and 18 electrons in the valence shell of the central metal atom. All ancillary ligands display an influence on the electronic density, oxidation behaviour and CO release at the metal centre. Thus, the coordination sphere of a given CORM drug influences resistance to plasma proteins and responds on a specific CO release trigger. However, a pharmaceutical CORM needs an appropriate pharmacological profile.23 It is very important to control solubility in aqueous solutions, cellular internalisation, as well as the pharmacological ADME characteristics, pharmacokinetic profile and targeting to diseased tissues (ADME is an abbreviation in pharmacokinetics and pharmacology for absorption, distribution, metabolism, and excretion, and describes the disposition of a pharmaceutical compound within an organism). The resulting “drug sphere” can be obtained by modifying the coordinating ligands at their distal sites (Fig. 11). Further, the pharmaceutical formulation determines which different chemical substances, including the active CORM, are combined to produce a final medicinal product.181 For example, a tablet contains a variety of other substances apart from the drug itself, and studies have to be carried out to ensure that the drug is compatible with these other substances. Extending the model of Romão et al., we postulate five different substituents in the coordination sphere. Carbohydrates and peptides can enhance water solubility,182 biocompatibility and even biodistribution to certain tissues.177,183,184 Morpholino groups may provide an amphiphilic character to the CORM. Solubility, membrane permeation and the pharmacokinetic profile may be controlled by terminal groups, such as amino, carboxylate groups and fluorine moieties. In addition, trackable dyes could help to investigate the metabolism of the CORM in vitro and in vivo.130 Finally, we emphasize that the coordination sphere, drug sphere and the pharmaceutical formulation “will play a decisive role in the generation of novel CORM drugs”.23
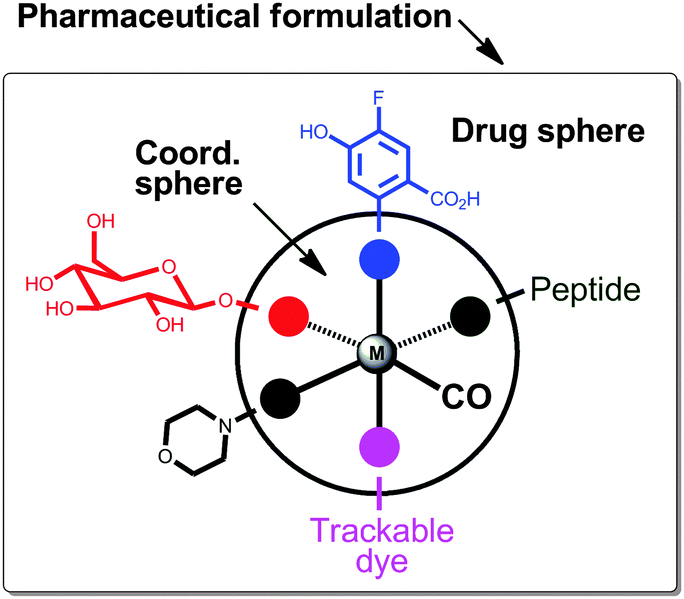 | ||
| Fig. 11 Conceptual model for the development of pharmaceutical CORMs (adapted from Romão et al.).23 | ||
CO-releasing materials (CORMAs)
As previously shown, metal carbonyl complexes are the most appropriate and successful class of (soluble) CORMs; however, it is also important to evaluate their possible shortcomings. In fact, very few pharmaceutical drugs are organometallic compounds,125,129 mostly due to side reactivity of metals with biological substances (e.g., nucleophilic and electrophilic side chains of proteins) and the toxicity of many heavy metals. Systemic application of water-soluble CORMs results in distribution throughout the body, which can lead to increased toxicities against healthy tissues. The spatially- and time-controlled release in the tissue still remains a great challenge.119 Moreover, the CO release process inevitably generates a metal–coligand fragment, which can potentially have biological activity. These fragments could be retained in insoluble matrices. Thus, development of solid-storage forms of CO in combination with a specific trigger for the gas release is an important research goal. In addition, macromolecular and nanoscale carrier systems185 can be utilized to achieve tissue-specific enrichment and delivery of CORMs.30,186Hubbell et al. developed CO-releasing micelles with reduced diffusion in tissues and better ability to target distal tissue draining sites.106 The micelles were prepared from triblock copolymers composed of a hydrophilic poly(ethylene glycol) block, a poly(ornithine acrylamide) block bearing [Ru(CO)3Cl(ornithinate)] moieties and a hydrophobic poly(n-butylacrylamide) block.187 CO release from the micelles was induced via addition of cysteine. It was slower than that of [Ru(CO)3Cl(glycinate)] (CORM-3, Fig. 7); however, the micelles attenuated successfully the lipopolysaccharide-induced inflammatory response of human monocytes. In addition, the toxicity of [Ru(CO)3Cl(amino acidate)] moieties was significantly reduced by the “stealth” feature of poly(ethylene glycol).106 Ru(CO)3Cl(glycinate) was also covalently attached to an amphiphilic peptide. The small peptide self-assembled into nanofiber gels and spontaneously released CO with prolonged release kinetics compared with CORM-3.179
Recently, a novel concept of triggering CORMAs was presented. Biocompatible magnetic iron oxide nanoparticles have been used as carriers for CORMs. In the proof-of-concept study the rate of CO release from [RuCl(CO3)(μ-DOPA)]@maghemite nanoparticles was doubled upon exposure to an external alternating magnetic field (31.7 kAm−1, 247 kHz, 25 °C, 39.9 mTesla, DOPA = dioxyphenyl-alaninato, Fig. 12).188
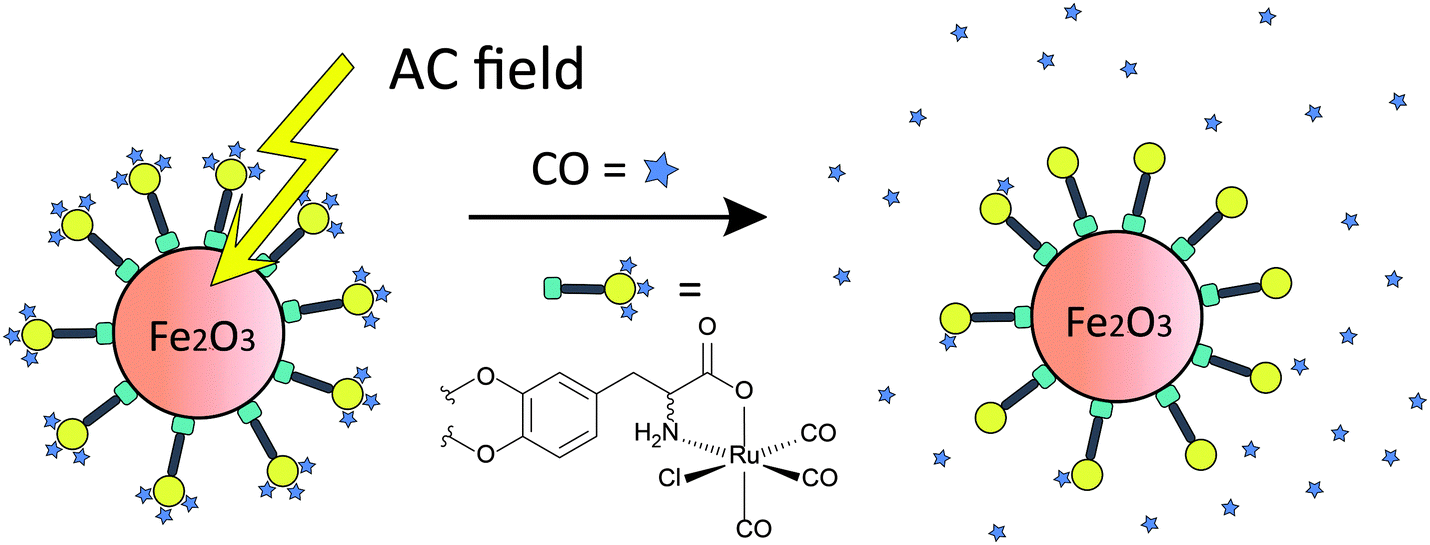 | ||
| Fig. 12 Schematic presentation of induced CO release from CORM-functionalized iron oxide nanoparticles through an alternating magnetic (AC) field. Tri(carbonyl)-chlorido-dihydroxyphenylalaninato-ruthenium(II) displays an immobilised analogue of CORM-3. Adapted and reprinted with permission from ref. 188. Copyright 2013 RSC Publishing. | ||
Porous coordination polymers, also known as metal–organic frameworks (MOFs), form structures with very high inner surface areas and ordered pore channels with various sizes.189,190 These features make MOFs highly attractive materials for gas-storage, especially for small gaseous molecules, such as H2, CH4 and CO2.191,192 Very recently, iron-based MOFs have been generated for the loading and delivery of CO.193 The materials are rapidly synthesized in the microwave from iron(III)chloride and terephthalic acid and derivatives thereof (Fig. 13). CO loading occurs via unsaturated coordination sites shown as empty circles in Fig. 13C. CO coordination was verified by infrared and Mössbauer spectroscopy. This novel type of CORMA shows good biocompatibility and releases CO with t1/2 from 38 to 76 min via degradation of the material under physiological conditions.
 | ||
| Fig. 13 Metal–organic frameworks (MOFs) with iron for the loading and delivery of CO. (A and B) Scanning electron microscope pictures of crystals of Fe MOF with terephthalic acid. (C) Structure of Fe MOF, as viewed along the c axis. Empty circles represent the coordination sites for CO, iron atoms: light-gray spheres, oxygen atoms: gray spheres, and carbon atoms: black spheres. Adapted and reprinted with permission from ref. 193. Copyright 2013 Wiley-VCH. | ||
Protocols for the covalent immobilization of photoCORMs28 on nanoparticles have also been established. These nanocarriers have many potential benefits for diagnosing164 and treating local and metastatic cancer, following the enhanced permeation and retention effect.185 [Mn(CO)3(tpm)]+ (tpm = tris(pyrazolyl)methane) complexes containing alkyne-functionalized tpm ligands were used. These complexes were covalently linked to silicon dioxide nanoparticles and dopable nanodiamonds via the copper-catalyzed azide-alkyne 1,3-dipolar cycloaddition (Fig. 14).194,195 The myoglobin assay103 demonstrated that the CORM-functionalized nanoparticles have photoinducible CO-release properties very similar to the free complexes.28,29
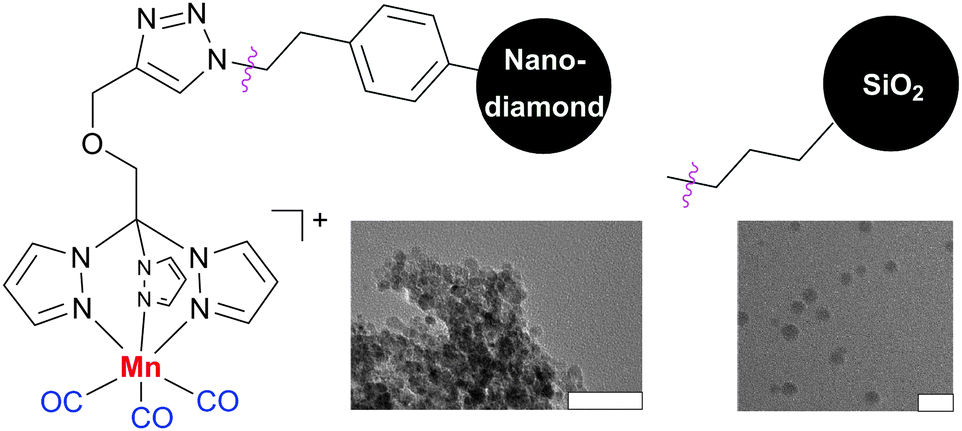 | ||
| Fig. 14 [Mn(CO)3(tpm)]+ (tpm = tris(pyrazolyl)methane) complexes containing alkyne-functionalized tpm ligands covalently attached to dopable nanodiamonds194 and silica nanoparticles.195 The corresponding transmission electron microscope pictures are also shown (bars indicate 50 nm). Adapted and reprinted with permission from ref. 194 and 195. Copyright 2011 American Chemical Society and 2012 RSC Publishing. | ||
The organometallic fac-Mn(CO)3 fragment was also bound to a methacrylate or methacrylamide polymer backbone via bis(pyridylmethyl)amine-type ligands. The resulting Mn(CO)3–polymer conjugates were investigated as photoinducible CO-releasing materials (photoCORMAs).196 In general, NO- and CO-releasing materials (NORMAs & CORMAs) with NO and CO photodonors can retain toxic metabolites after gas release in the biocompatible polymer matrix.197 The concept of embedding water-insoluble, photoactive NO metal complexes into nanoparticles198 and fibrous polymer non-wovens199 has been transferred to phototriggerable metal carbonyls (Fig. 15). Effective NO or CO release into the surrounding medium is initiated by light stimulation of the high surface area materials. For the generation of NORMAs, novel biscarboxamide rutheniumnitrosyl complexes {RuNO}6 have been synthesised.200,201 For a photoCORMA, Mn2(CO)10 (CORM-1) was used.202,203 The metal complexes were non-covalently embedded into the polymer matrices via miniemulsion technique204 or electrospinning.205 Leaching of the metal complexes out of the polymeric matrices into water was negligible due to their water insolubility. Irradiation with λ = 366–480 nm in water showed a significant phototriggered NO/CO release from the nanoparticles or non-wovens. Cytotoxicity tests of the CORMA with 3T3 mouse fibroblast cells in the dark revealed very low cell death. After illumination, CO bubbled out of the nanofibres thereby eradicating the fibroblast cell culture.202,203
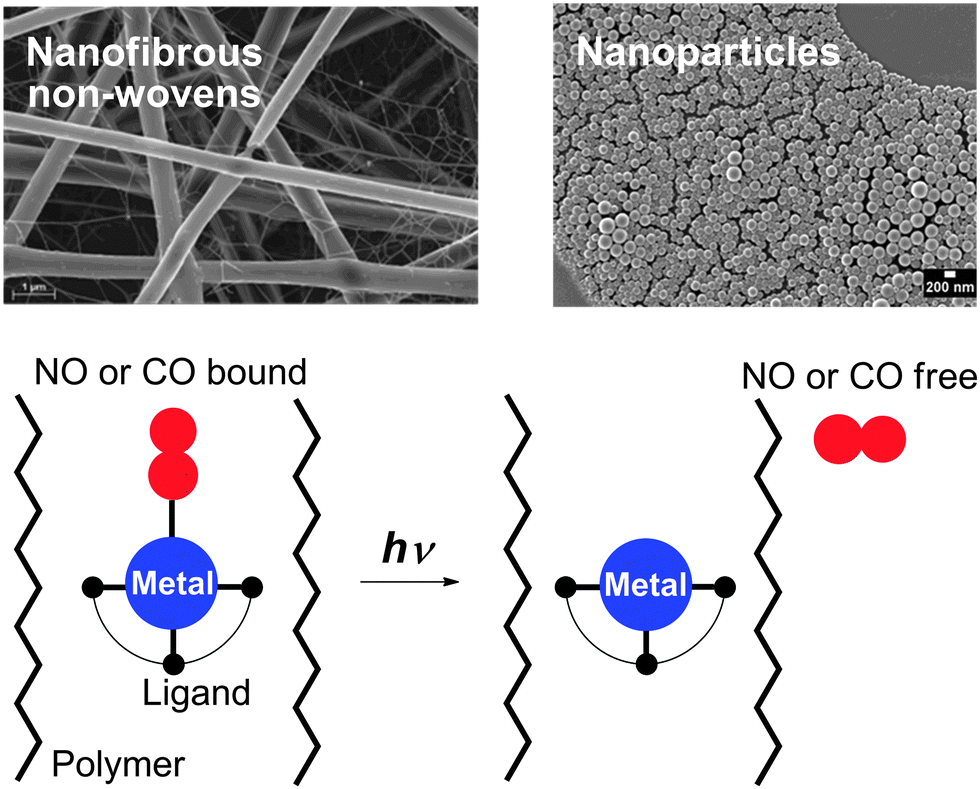 | ||
| Fig. 15 Concept of embedding water-insoluble, photoactive NO and CO metal complexes into fibrous polymer non-wovens and nanoparticles. The NORMAs and CORMAs are important for the development of safe NO and CO delivering devices for therapeutic purposes; toxic metabolites after gas release are retained in the biocompatible polymer matrix.200–203 | ||
E. CO – where does it go?
The discovery of CO as an endogenous gaseous messenger has triggered intensive research regarding cellular CO signalling and the design of carrier systems that provide a controlled release of CO. Despite substantial progress on various levels, there are quite a number of open questions that need to be addressed, in particular if clinical applications of CO or CO-releasing molecules and materials are envisioned. Some of such open questions are discussed in the following.For understanding the CO-related physiology it will be mandatory to profile the expression and targeting of haem oxygenases. Because CO signalling is most probably a “local” event, we need to know in detail (a) the localization of HO, (b) the availability of haem and (c) the availability of NADPH. Once released via HO activity, what is the fate of the CO molecule? It is often assumed that CO will immediately find the desired target system, but it is not yet clear what CO buffer capacity the cellular cytosol provides and, hence, what is the effective sphere of action of a cytosolic CO molecule? In addition, what is the ultimate destination of CO? How much becomes covalently bound, which fraction only undergoes loose interactions with other molecules, and how much is finally cleared from the body via the lungs? Quantitative data are required to facilitate predictions about CO-related physiological processes.
Except for some haemoproteins, our knowledge on the molecular mechanisms by which CO affects protein function is still very much limited. Recent examples showing that CO modulates the function of ion channels, for example, still lack a clear mechanistic insight. Does an action of CO on a protein always require the presence of a haem group or a transition metal or are there other modes of CO-protein interactions feasible?
The necessity of CORMs to target specific disease sites and to release CO at a predetermined time point is obvious. In order to effectively initiate liberation of CO from metal carbonyl complexes, a variety of CO release triggers are required to fulfil boundary conditions such as governed CO release via irradiation, enzymatic activation, pH changes, ligand substitution, temperature, redox reactions, and others. Therefore, in some cases it might be beneficial if CORMs are either hydrophobic or amphiphilic in order to have them enter the cells or enrich in the membranes, respectively. In addition, different therapeutic applications might need slow or fast liberation of CO. Specific applications call for particular CORMs with respect to delivery of the carbonyl complexes at the disease site, initiation of CO release, interference of the tissues with the metal complexes themselves or with their degradation products. The performance of those CORMs can be further adapted with a suitable carrier system (e.g. micelle, nanoparticle, fibre, non-woven etc.). Smart materials that release CO by a trigger and retain degradation products can reach their target via specific interactions of materials and cells. In future investigations, the interaction of these complexes and their degradation products with reactive oxygen species (occasionally causing Fenton-type chemistry), peptides, amino acids and other biological environments deserves particular attention.
Another issue involves inactivated CORM (iCORM) products. Unfortunately, the potential biological activity of such metal–coligand fragments, inevitably remaining after CO release from the metal coordination sphere, has been often neglected. In the future, novel CORMs with biological activity should include a detailed characterisation of corresponding iCORMs.
With the development of specific CORMs it is also necessary to intensify research on real-time detection of physiological levels of CO inside living cells. New CO sensors must be robust, selective and sensitive in the lower μM range. Furthermore, rapid response times and a good signal-to-noise-ratio will be required.
Understanding the CO-related physiology is still in its infancy; the currently available CORMs and CORMAs described are not yet optimized for clinical or experimental applications. Therefore, physiologists, physicians and chemists must collaborate for a better understanding of CO in the body and how to utilize CO as a drug.
Acknowledgements
We thank the Deutsche Forschungsgemeinschaft (DFG) for supporting FOR 1738. A.S. is grateful to the Carl Zeiss foundation for a Junior Professor fellowship and the EC for financial support through the FP7 project ‘Novosides’ (grant agreement no. KBBE-4-265854). We also acknowledge the support of the Center of Medical Optics and Photonics (CEMOP), the Abbe Center of Photonics (ACP) and the Jena Center of Soft Matter (JCSM) at the Friedrich Schiller University Jena. T.H. was supported in part through the National Institutes of Health (R01GM057654). We also thank C. Romão, N. Metzler-Nolte, C. Janiak, U. Schatzschneider, C. Chang, C. He and R. Martínez-Máñez for providing figures from their original publications.References
- L. R. Goldbaum, R. G. Ramirez and K. B. Absalon, Aviat., Space Environ. Med., 1975, 46, 1289–1291 CAS.
- H. P. Kim, S. W. Ryter and A. M. Choi, Annu. Rev. Pharmacol. Toxicol., 2006, 46, 411–449 CrossRef CAS PubMed.
- R. Motterlini and L. E. Otterbein, Nat. Rev. Drug Discovery, 2010, 9, 728–743 CrossRef CAS PubMed.
- M. D. Maines, FASEB J., 1988, 2, 2557–2568 CAS.
- R. F. Coburn, Ann. N. Y. Acad. Sci., 1970, 174, 11–22 CrossRef CAS.
- E. Aztatzi-Santillán, F. E. Nares-López, B. Márquez-Valadez, P. Aguilera and M. E. Chánez-Cárdenas, Cent. Nerv. Syst. Agents Med. Chem., 2010, 10, 310–316 CrossRef.
- E. Zeynalov, Z. A. Shah, R. C. Li and S. Doré, Neurobiol. Dis., 2009, 35, 264–269 CrossRef CAS PubMed.
- J. Wang, H. Zhuang and S. Doré, Neurobiol. Dis., 2006, 22, 473–476 CrossRef CAS PubMed.
- A. Ferreira, J. Balla, V. Jeney, G. Balla and M. P. Soares, J. Mol. Med., 2008, 86, 1097–1111 CrossRef CAS PubMed.
- A. Pamplona, A. Ferreira, J. Balla, V. Jeney, G. Balla, S. Epiphanio, A. Chora, C. D. Rodrigues, I. P. Gregoire, M. Cunha-Rodrigues, S. Portugal, M. P. Soares and M. M. Mota, Nat. Med., 2007, 13, 703–710 CrossRef CAS PubMed.
- S. W. Ryter and A. M. K. Choi, Am. J. Respir. Cell Mol. Biol., 2009, 41, 251–260 CrossRef CAS PubMed.
- R. Motterlini, B. Haas and R. Foresti, Med. Gas Res., 2012, 2, 28 CrossRef CAS PubMed.
- R. Foresti, M. G. Bani-Hani and R. Motterlini, Intensive Care Med., 2008, 34, 649–658 CrossRef CAS PubMed.
- B. E. Mann and R. Motterlini, Chem. Commun., 2007, 4197–4208 RSC.
- B. S. Zuckerbraun, B. Y. Chin, B. Wegiel, T. R. Billiar, E. Czsimadia, J. Rao, L. Shimoda, E. Ifedigbo, S. Kanno and L. E. Otterbein, J. Exp. Med., 2006, 203, 2109–2119 CrossRef CAS PubMed.
- J. Kohmoto, A. Nakao, T. Kaizu, A. Tsung, A. Ikeda, K. Tomiyama, T. R. Billiar, A. M. K. Choi, N. Murase and K. R. McCurry, Surgery, 2006, 140, 179–185 CrossRef PubMed.
- G. L. Volti, L. F. Rodella, C. D. Giacomo, R. Rezzani, R. Bianchi, E. Borsani, D. Gazzolo and R. Motterlini, Nephron Exp. Nephrol., 2006, 104, e135–e139 CrossRef PubMed.
- R. Motterlini, A. Gonzales, R. Foresti, J. E. Clark, C. J. Green and R. M. Winslow, Circ. Res., 1998, 83, 568–577 CrossRef CAS.
- A. Nakao, D. J. Kaczorowski, Y. Wang, J. S. Cardinal, B. M. Buchholz, R. Sugimoto, K. Tobita, S. Lee, Y. Toyoda, T. R. Billiar and K. R. McCurry, J. Heart Lung Transplant., 2010, 29, 544–553 CrossRef PubMed.
- S. Takaki, N. Takeyama, Y. Kajita, T. Yabuki, H. Noguchi, Y. Miki, Y. Inoue, T. Nakagawa and H. Noguchi, Intensive Care Med., 2010, 36, 42–48 CrossRef CAS PubMed.
- C. A. Piantadosi, Free Radicals Biol. Med., 2008, 45, 562–569 CrossRef CAS PubMed.
- F. Gullotta, A. d. Masi, M. Coletta and P. Ascenzi, BioFactors, 2012, 38, 1–13 CrossRef CAS PubMed.
- C. C. Romão, W. A. Blattler, J. D. Seixas and G. J. L. Bernardes, Chem. Soc. Rev., 2012, 41, 3571–3583 RSC.
- L. Yuan, W. Lin, L. Tan, K. Zheng and W. Huang, Angew. Chem., Int. Ed., 2013, 52, 1628–1630 CrossRef CAS PubMed.
- B. E. Mann, Organometallics, 2012, 31, 5728–5735 CrossRef CAS.
- B. Mann, in Medicinal Organometallic Chemistry, ed. G. Jaouen and N. Metzler-Nolte, Springer, Berlin, Heidelberg, 2010, vol. 32, ch. 10, pp. 247–285 Search PubMed.
- R. D. Rimmer, A. E. Pierri and P. C. Ford, Coord. Chem. Rev., 2012, 256, 1509–1519 CrossRef CAS PubMed.
- U. Schatzschneider, Inorg. Chim. Acta, 2011, 374, 19–23 CrossRef CAS PubMed.
- U. Schatzschneider, Eur. J. Inorg. Chem., 2010, 1451–1467 CrossRef CAS.
- D. Crespy, K. Landfester, U. S. Schubert and A. Schiller, Chem. Commun., 2010, 46, 6651–6662 RSC.
- S. Fetzner and R. A. Steiner, Appl. Microbiol. Biotechnol., 2010, 86, 791–804 CrossRef CAS PubMed.
- S. Fetzner, Appl. Microbiol. Biotechnol., 2002, 60, 243–257 CrossRef CAS PubMed.
- H. M. Girvan and A. W. Munro, J. Biol. Chem., 2013, 288, 13194–13203 CrossRef CAS PubMed.
- W. J. Wilkinson and P. J. Kemp, J. Physiol., 2011, 589, 3055–3062 CrossRef CAS PubMed.
- C. Peers, Exp. Physiol., 2011, 96, 836–839 CrossRef CAS PubMed.
- G. P. Roberts, H. Youn and R. L. Kerby, Microbiol. Mol. Biol. Rev., 2004, 68, 453–473 CrossRef CAS PubMed.
- J. L. Boer, S. B. Mulrooney and R. P. Hausinger, Arch. Biochem. Biophys., 2014, 544, 142–152 CrossRef CAS PubMed.
- S. M. Siepka, S. H. Yoo, J. Park, C. Lee and J. S. Takahashi, Cold Spring Harbor Symp. Quant. Biol., 2007, 72, 251–259 CrossRef CAS PubMed.
- E. M. Dioum, J. Rutter, J. R. Tuckerman, G. Gonzalez, M. A. Gilles-Gonzalez and S. L. McKnight, Science, 2002, 298, 2385–2387 CrossRef CAS PubMed.
- T. Uchida, E. Sato, A. Sato, I. Sagami, T. Shimizu and T. Kitagawa, J. Biol. Chem., 2005, 280, 21358–21368 CrossRef CAS PubMed.
- T. Ingi, J. Cheng and G. V. Ronnett, Neuron, 1996, 16, 835–842 CrossRef CAS.
- R. Feil and B. Kemp-Harper, EMBO Rep., 2006, 7, 149–153 CrossRef CAS PubMed.
- E. R. Derbyshire and M. A. Marletta, Annu. Rev. Biochem., 2012, 81, 533–559 CrossRef CAS PubMed.
- J. R. Stone and M. A. Marletta, Biochemistry, 1994, 33, 5636–5640 CrossRef CAS.
- A. Friebe, G. Schultz and D. Koesling, EMBO J., 1996, 15, 6863–6868 CAS.
- L. J. Ignarro, Nitric Oxide - Biology and Pathobiology, Elsevier Inc., London, 2010 Search PubMed.
- E. Martin, V. Berka, E. Bogatenkova, F. Murad and A. L. Tsai, J. Biol. Chem., 2006, 281, 27836–27845 CrossRef CAS PubMed.
- J. N. Burstyn, A. E. Yu, E. A. Dierks, B. K. Hawkins and J. H. Dawson, Biochemistry, 1995, 34, 5896–5903 CrossRef CAS.
- E. M. Boon, S. H. Huang and M. A. Marletta, Nat. Chem. Biol., 2005, 1, 53–59 CrossRef CAS PubMed.
- E. Dubuis, M. Potier, R. Wang and C. Vandier, Cardiovasc. Res., 2005, 65, 751–761 CrossRef CAS PubMed.
- K. Decaluwe, B. Pauwels, S. Verpoest and J. V. d. Voorde, Eur. J. Pharmacol., 2012, 674, 370–377 CrossRef CAS PubMed.
- X. Ma, N. Sayed, A. Beuve and F. van den Akker, EMBO J., 2007, 26, 578–588 CrossRef CAS PubMed.
- P. M. Schmidt, C. Rothkegel, F. Wunder, H. Schroder and J. P. Stasch, Eur. J. Pharmacol., 2005, 513, 67–74 CrossRef CAS PubMed.
- F. Ko, C. Wu, S. Kuo, F. Lee and C. Teng, Blood, 1994, 84, 4226–4233 CAS.
- E. Martin, Y. C. Lee and F. Murad, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 12938–12942 CrossRef CAS PubMed.
- R. Purohit, A. Weichsel and W. R. Montfort, Protein Sci., 2013, 22, 1439–1444 CAS.
- K. A. White and M. A. Marletta, Biochemistry, 1992, 31, 6627–6631 CrossRef CAS.
- H. Li, J. Igarashi, J. Jamal, W. Yang and T. L. Poulos, JBIC, J. Biol. Inorg. Chem., 2006, 11, 753–768 CrossRef CAS PubMed.
- S. W. Ragsdale, J. Inorg. Biochem., 2007, 101, 1657–1666 CrossRef CAS PubMed.
- Y. Kung, T. I. Doukov, J. Seravalli, S. W. Ragsdale and C. L. Drennan, Biochemistry, 2009, 48, 7432–7440 CrossRef CAS PubMed.
- J. H. Jeoung and H. Dobbek, J. Am. Chem. Soc., 2009, 131, 9922–9923 CrossRef CAS PubMed.
- Anorganische Chemie, ed. R. Steudel, De Gruyter, Berlin, 2012 Search PubMed.
- H. Lin and J. J. McGrath, Drug Chem. Toxicol., 1988, 11, 371–385 CrossRef CAS PubMed.
- T. Hoshi, A. Pantazis and R. Olcese, Physiology, 2013, 28, 172–189 CrossRef CAS PubMed.
- M. T. Nelson and J. M. Quayle, Am. J. Physiol., 1995, 268, C799–C822 CAS.
- R. Brenner, G. J. Perez, A. D. Bonev, D. M. Eckman, J. C. Kosek, S. W. Wiler, A. J. Patterson, M. T. Nelson and R. W. Aldrich, Nature, 2000, 407, 870–876 CrossRef CAS PubMed.
- A. J. Patterson, J. Henrie-Olson and R. Brenner, Trends Cardiovasc. Med., 2002, 12, 78–82 CrossRef CAS.
- M. Nara, P. D. Dhulipala, G. J. Ji, U. R. Kamasani, Y. X. Wang, S. Matalon and M. I. Kotlikoff, Am. J. Physiol.: Cell Physiol., 2000, 279, C1938–C1945 CAS.
- M. Fukao, H. S. Mason, F. C. Britton, J. L. Kenyon, B. Horowitz and K. D. Keef, J. Biol. Chem., 1999, 274, 10927–10935 CrossRef CAS PubMed.
- Y. Wu, Y. Yang, S. Ye and Y. Jiang, Nature, 2010, 466, 393–397 CrossRef CAS PubMed.
- P. Yuan, M. D. Leonetti, A. R. Pico, Y. Hsiung and R. Mackinnon, Science, 2010, 329, 182–186 CrossRef CAS PubMed.
- P. Yuan, M. D. Leonetti, Y. Hsiung and R. MacKinnon, Nature, 2012, 481, 94–97 CrossRef CAS PubMed.
- M. L. Dallas, J. L. Scragg and C. Peers, Neuroreport, 2008, 19, 345–348 CrossRef CAS PubMed.
- F. T. Horrigan and R. W. Aldrich, J. Gen. Physiol., 2002, 120, 267–305 CrossRef CAS.
- R. Wang and L. Wu, J. Biol. Chem., 1997, 272, 8222–8226 CrossRef CAS PubMed.
- S. E. Williams, P. Wootton, H. S. Mason, J. Bould, D. E. Iles, D. Riccardi, C. Peers and P. J. Kemp, Science, 2004, 306, 2093–2097 CrossRef CAS PubMed.
- S. Hou, R. Xu, S. H. Heinemann and T. Hoshi, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 4039–4043 CrossRef CAS PubMed.
- S. E. Williams, S. P. Brazier, N. Baban, V. Telezhkin, C. T. Muller, D. Riccardi and P. J. Kemp, Pflugers Arch., 2008, 456, 561–572 CrossRef CAS PubMed.
- V. Telezhkin, S. P. Brazier, R. Mears, C. T. Muller, D. Riccardi and P. J. Kemp, Pflugers Arch., 2011, 461, 665–675 CrossRef CAS PubMed.
- K. A. Bixby, M. H. Nanao, N. V. Shen, A. Kreusch, H. Bellamy, P. J. Pfaffinger and S. Choe, Nat. Struct. Biol., 1999, 6, 38–43 CrossRef CAS PubMed.
- G. Wang, C. Strang, P. J. Pfaffinger and M. Covarrubias, J. Biol. Chem., 2007, 282, 13637–13647 CrossRef CAS PubMed.
- G. Liu, J. Shi, L. Yang, L. Cao, S. M. Park, J. Cui and S. O. Marx, EMBO J., 2004, 23, 2196–2205 CrossRef CAS PubMed.
- S. Hou, R. Xu, S. H. Heinemann and T. Hoshi, Nat. Struct. Mol. Biol., 2008, 15, 403–410 CAS.
- S. Hou, F. T. Horrigan, R. Xu, S. H. Heinemann and T. Hoshi, Channels, 2009, 3, 249–258 CrossRef.
- X. D. Tang, M. L. Garcia, S. H. Heinemann and T. Hoshi, Nat. Struct. Mol. Biol., 2004, 11, 171–178 Search PubMed.
- G. Zhang, R. Xu, S. H. Heinemann and T. Hoshi, Biochem. Biophys. Res. Commun., 2006, 342, 1389–1395 CrossRef CAS PubMed.
- Y. J. Peng, J. Nanduri, G. Raghuraman, D. Souvannakitti, M. M. Gadalla, G. K. Kumar, S. H. Snyder and N. R. Prabhakar, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 10719–10724 CrossRef CAS.
- N. R. Prabhakar, J. Physiol., 2013, 591, 2245–2257 CAS.
- S. Wang, S. Publicover and Y. Gu, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 2957–2962 CrossRef CAS PubMed.
- A. L. Eastwood and M. B. Goodman, Physiology, 2012, 27, 282–290 CrossRef CAS PubMed.
- P. Ponka, Am. J. Med. Sci., 1999, 318, 241–256 CrossRef CAS PubMed.
- X. D. Tang, R. Xu, M. F. Reynolds, M. L. Garcia, S. H. Heinemann and T. Hoshi, Nature, 2003, 425, 531–535 CrossRef CAS PubMed.
- J. H. Jaggar, A. Li, H. Parfenova, J. Liu, E. S. Umstot, A. M. Dopico and C. W. Leffler, Circ. Res., 2005, 97, 805–812 CrossRef CAS PubMed.
- L. Yi, J. T. Morgan and S. W. Ragsdale, J. Biol. Chem., 2010, 285, 20117–20127 CrossRef CAS PubMed.
- T. Kuhl, N. Sahoo, M. Nikolajski, B. Schlott, S. H. Heinemann and D. Imhof, ChemBioChem, 2011, 12, 2846–2855 CrossRef PubMed.
- F. T. Horrigan, S. H. Heinemann and T. Hoshi, J. Gen. Physiol., 2005, 126, 7–21 CrossRef CAS PubMed.
- J. R. Alonso, F. Cardellach, S. Lopez, J. Casademont and O. Miro, Pharmacol. Toxicol., 2003, 93, 142–146 CAS.
- Y. K. Choi, E. D. Por, Y. G. Kwon and Y. M. Kim, Oxid. Med. Cell. Longevity, 2012, 794237 Search PubMed.
- M. L. Dallas, J. L. Scragg and C. Peers, Adv. Exp. Med. Biol., 2009, 648, 89–95 CrossRef CAS.
- E. M. Hetrick and M. H. Schoenfisch, Annu. Rev. Anal. Chem., 2009, 2, 409–433 CrossRef CAS PubMed.
- S. S. Park, J. Kim and Y. Lee, Anal. Chem., 2012, 84, 1792–1796 CrossRef CAS PubMed.
- R. A. Hunter, W. L. Storm, P. N. Coneski and M. H. Schoenfisch, Anal. Chem., 2013, 85, 1957–1963 CrossRef CAS PubMed.
- S. McLean, B. E. Mann and R. K. Poole, Anal. Biochem., 2012, 427, 36–40 CrossRef CAS PubMed.
- G. S. Marks, H. J. Vreman, B. E. McLaughlin, J. F. Brien and K. Nakatsu, Antioxid. Redox Signaling, 2002, 4, 271–277 CrossRef CAS PubMed.
- Y. Morimoto, W. Durante, D. G. Lancaster, J. Klattenhoff and F. K. Tittel, Am. J. Physiol.: Heart Circ. Physiol., 2001, 280, H483 CAS.
- U. Hasegawa, A. J. van der Vlies, E. Simeoni, C. Wandrey and J. A. Hubbell, J. Am. Chem. Soc., 2010, 132, 18273–18280 CrossRef CAS PubMed.
- J.-M. Barbe, G. Canard, S. Brandès and R. Guilard, Chem.–Eur. J., 2007, 13, 2118–2129 CrossRef CAS PubMed.
- K. S. Davidge, G. Sanguinetti, C. H. Yee, A. G. Cox, C. W. McLeod, C. E. Monk, B. E. Mann, R. Motterlini and R. K. Poole, J. Biol. Chem., 2009, 284, 4516–4524 CrossRef CAS PubMed.
- M. Desmard, R. Foresti, D. Morin, M. Dagouassat, A. Berdeaux, E. Denamur, S. H. Crook, B. E. Mann, D. Scapens, P. Montravers, J. Boczkowski and R. Motterlini, Antioxid. Redox Signaling, 2012, 16, 153–163 CrossRef CAS PubMed.
- A. J. Atkin, J. M. Lynam, B. E. Moulton, P. Sawle, R. Motterlini, N. M. Boyle, M. T. Pryce and I. J. S. Fairlamb, Dalton Trans., 2011, 40, 5755–5761 RSC.
- J. Esteban, J. Ros-Lis, R. Martínez-Máñez, M. Marcos, M. Moragues, J. Soto and F. Sancenón, Angew. Chem., Int. Ed., 2010, 49, 4934–4937 CrossRef CAS PubMed.
- M. E. Moragues, J. Esteban, J. V. Ros-Lis, R. Martínez-Máñez, M. D. Marcos, M. Martínez, J. Soto and F. Sancenón, J. Am. Chem. Soc., 2011, 133, 15762–15772 CrossRef CAS PubMed.
- J. R. Lakowicz, Principles of Fluorescence Spectroscopy, Springer, New York, 2006 Search PubMed.
- B. Wang and E. V. Anslyn, Chemosensors - Principles, Strategies, and Applications, John Wiley & Sons, Hoboken, New Jersey, 2011 Search PubMed.
- V. S. Lin and C. J. Chang, Curr. Opin. Chem. Biol., 2012, 16, 595–601 CrossRef CAS PubMed.
- N. Kumar, V. Bhalla and M. Kumar, Coord. Chem. Rev., 2013, 257, 2335–2347 CrossRef CAS PubMed.
- J. Wang, J. Karpus, B. S. Zhao, Z. Luo, P. R. Chen and C. He, Angew. Chem., Int. Ed., 2012, 51, 9652–9656 CrossRef CAS PubMed.
- B. W. Michel, A. R. Lippert and C. J. Chang, J. Am. Chem. Soc., 2012, 134, 15668–15671 CrossRef CAS PubMed.
- A. R. Marques, L. Kromer, D. J. Gallo, N. Penacho, S. S. Rodrigues, J. D. Seixas, G. J. L. Bernardes, P. M. Reis, S. L. Otterbein, R. A. Ruggieri, A. S. G. Gonçalves, A. M. L. Gonçalves, M. N. D. Matos, I. Bento, L. E. Otterbein, W. A. Blättler and C. C. Romão, Organometallics, 2012, 31, 5810–5822 CrossRef CAS.
- R. Motterlini, P. Sawle, J. Hammad, S. Bains, R. Alberto, R. Foresti and C. J. Green, FASEB J., 2005, 19, 284–286 CAS.
- T. S. Pitchumony, B. Spingler, R. Motterlini and R. Alberto, Chimia, 2008, 62, 277–279 CrossRef CAS.
- T. S. Pitchumony, B. Spingler, R. Motterlini and R. Alberto, Org. Biomol. Chem., 2010, 8, 4849–4854 CAS.
- L. A. P. Antony, T. Slanina, P. Šebej, T. Šolomek and P. Klán, Org. Lett., 2013, 15, 4552–4555 CrossRef CAS PubMed.
- P. Peng, C. Wang, Z. Shi, V. K. Johns, L. Ma, J. Oyer, A. Copik, R. Igarashi and Y. Liao, Org. Biomol. Chem., 2013, 11, 6671–6674 CAS.
- G. Gasser and N. Metzler-Nolte, Curr. Opin. Chem. Biol., 2012, 16, 84–91 CrossRef CAS PubMed.
- F. Zobi, Future Med. Chem., 2013, 5, 175–188 CrossRef CAS PubMed.
- W. A. Herrmann, J. Organomet. Chem., 1990, 383, 21–44 CrossRef.
- R. Motterlini, J. E. Clark, R. Foresti, P. Sarathchandra, B. E. Mann and C. J. Green, Circ. Res., 2002, 90, e17–e24 CrossRef CAS.
- N. P. E. Barry and P. J. Sadler, Chem. Commun., 2013, 49, 5106–5131 RSC.
- A. E. Pierri, A. Pallaoro, G. Wu and P. C. Ford, J. Am. Chem. Soc., 2012, 134, 18197–18200 CrossRef CAS PubMed.
- M. A. Gonzalez, S. J. Carrington, N. L. Fry, J. L. Martinez and P. K. Mascharak, Inorg. Chem., 2012, 51, 11930–11940 CrossRef CAS PubMed.
- M. A. Gonzalez, M. A. Yim, S. Cheng, A. Moyes, A. J. Hobbs and P. K. Mascharak, Inorg. Chem., 2011, 51, 601–608 CrossRef PubMed.
- S. J. Carrington, I. Chakraborty and P. K. Mascharak, Chem. Commun., 2013, 49, 11254–11256 RSC.
- D. E. Bikiel, E. G. Solveyra, F. D. Salvo, H. M. S. Milagre, M. N. Eberlin, R. S. Corréa, J. Ellena, D. A. Estrin and F. Doctorovich, Inorg. Chem., 2011, 50, 2334–2345 CrossRef CAS PubMed.
- R. Kretschmer, G. Gessner, H. Görls, S. H. Heinemann and M. Westerhausen, J. Inorg. Biochem., 2011, 105, 6–9 CrossRef CAS PubMed.
- V. P. L. Velásquez, T. M. A. Jazzazi, A. Malassa, H. Görls, G. Gessner, S. H. Heinemann and M. Westerhausen, Eur. J. Inorg. Chem., 2012, 1072–1078 CrossRef.
- L. Hewison, T. R. Johnson, B. E. Mann, A. J. H. M. Meijer, P. Sawle and R. Motterlini, Dalton Trans., 2011, 40, 8328–8334 RSC.
- C. Bischof, T. Joshi, A. Dimri, L. Spiccia and U. Schatzschneider, Inorg. Chem., 2013, 52, 9297–9308 CrossRef CAS PubMed.
- M. A. Gonzalez, S. J. Carrington, I. Chakraborty, M. M. Olmstead and P. K. Mascharak, Inorg. Chem., 2013, 52, 11320–11331 CrossRef CAS PubMed.
- C. S. Jackson, S. Schmitt, Q. P. Dou and J. J. Kodanko, Inorg. Chem., 2011, 50, 5336–5338 CrossRef CAS PubMed.
- A. J. Atkin, I. J. S. Fairlamb, J. S. Ward and J. M. Lynam, Organometallics, 2012, 31, 5894–5902 CrossRef CAS.
- S. Romanski, H. Rücker, E. Stamellou, M. Guttentag, J.-M. Neudörfl, R. Alberto, S. Amslinger, B. Yard and H.-G. Schmalz, Organometallics, 2012, 31, 5800–5809 CrossRef CAS.
- S. Romanski, B. Kraus, U. Schatzschneider, J.-M. Neudörfl, S. Amslinger and H.-G. Schmalz, Angew. Chem., Int. Ed., 2011, 50, 2392–2396 CrossRef CAS PubMed.
- L. Hewison, S. H. Crook, T. R. Johnson, B. E. Mann, H. Adams, S. E. Plant, P. Sawle and R. Motterlini, Dalton Trans., 2010, 39, 8967–8975 RSC.
- D. Scapens, H. Adams, T. R. Johnson, B. E. Mann, P. Sawle, R. Aqil, T. Perrior and R. Motterlini, Dalton Trans., 2007, 4962–4973 RSC.
- J. S. Ward, J. M. Lynam, J. W. B. Moir, D. E. Sanin, A. P. Mountford and I. J. S. Fairlamb, Dalton Trans., 2012, 41, 10514–10517 RSC.
- P. Govender, S. Pai, U. Schatzschneider and G. S. Smith, Inorg. Chem., 2013, 52, 5470–5478 CrossRef CAS PubMed.
- W.-Q. Zhang, A. J. Atkin, R. J. Thatcher, A. C. Whitwood, I. J. S. Fairlamb and J. M. Lynam, Dalton Trans., 2009, 4351–4358 RSC.
- W.-Q. Zhang, A. C. Whitwood, I. J. S. Fairlamb and J. M. Lynam, Inorg. Chem., 2010, 49, 8941–8952 CrossRef CAS PubMed.
- F. Zobi, A. Degonda, M. C. Schaub and A. Y. Bogdanova, Inorg. Chem., 2010, 49, 7313–7322 CrossRef CAS PubMed.
- F. Zobi and O. Blacque, Dalton Trans., 2011, 40, 4994–5001 RSC.
- M. A. Gonzalez, N. L. Fry, R. Burt, R. Davda, A. Hobbs and P. K. Mascharak, Inorg. Chem., 2011, 50, 3127–3134 CrossRef CAS PubMed.
- L. Long, X. Jiang, X. Wang, Z. Xiao and X. Liu, Dalton Trans., 2013, 42, 15663–15669 RSC.
- R. D. Rimmer, H. Richter and P. C. Ford, Inorg. Chem., 2010, 49, 1180–1185 CrossRef CAS PubMed.
- G. R. Stephenson, P. W. Howard, D. A. Owen and A. J. Whitehead, J. Chem. Soc., Chem. Commun., 1991, 641–642 RSC.
- A. Bohac, M. Lettrichova, P. Hrnciar and M. Hutta, J. Organomet. Chem., 1996, 507, 23–29 CrossRef CAS.
- S. Ban, H. Sakurai, Y. Hayashi and K. Narasaka, Chem. Lett., 1997, 699–700 CrossRef CAS.
- S. Botov, E. Stamellou, S. Romanski, M. Guttentag, R. Alberto, J.-M. Neudorfl, B. Yard and H.-G. Schmalz, Organometallics, 2013, 32, 3587–3594 CrossRef CAS.
- S. Romanski, B. Kraus, M. Guttentag, W. Schlundt, H. Rücker, A. Adler, J.-M. Neudörfl, R. Alberto, S. Amslinger and H.-G. Schmalz, Dalton Trans., 2012, 41, 13862–13875 RSC.
- I. J. S. Fairlamb, S. M. Syvänne and A. C. Whitwood, Synlett, 2003, 1693–1697 CrossRef CAS PubMed.
- I. J. S. Fairlamb, A.-K. Duhme-Klair, J. M. Lynam, B. E. Moulton, C. T. O'Brien, P. Sawle, J. Hammad and R. Motterlini, Bioorg. Med. Chem. Lett., 2006, 16, 995–998 CrossRef CAS PubMed.
- P. Sawle, J. Hammad, I. J. S. Fairlamb, B. Moulton, C. T. O'Brien, J. M. Lynam, A. K. Duhme-Klair, R. Foresti and R. Motterlini, J. Pharmacol. Exp. Ther., 2006, 318, 403–410 CrossRef CAS PubMed.
- I. J. S. Fairlamb, J. M. Lynam, B. E. Moulton, I. E. Taylor, A. K. Duhme-Klair, P. Sawle and R. Motterlini, Dalton Trans., 2007, 3603–3605 RSC.
- K. Meister, J. Niesel, U. Schatzschneider, N. Metzler-Nolte, D. A. Schmidt and M. Havenith, Angew. Chem., Int. Ed., 2010, 49, 3310–3312 CrossRef CAS PubMed.
- F. Mohr, J. Niesel, U. Schatzschneider and C. W. Lehmann, Z. Anorg. Allg. Chem., 2012, 638, 543–546 CrossRef CAS.
- T. Santos-Silva, A. Mukhopadhyay, J. D. Seixas, G. J. L. Bernardes, C. C. Romão and M. J. Romão, J. Am. Chem. Soc., 2011, 133, 1192–1195 CrossRef CAS PubMed.
- M. F. A. Santos, J. D. Seixas, A. C. Coelho, A. Mukhopadhyay, P. M. Reis, M. J. Romão, C. C. Romão and T. Santos-Silva, J. Inorg. Biochem., 2012, 117, 285–291 CrossRef CAS PubMed.
- T. M. A. Jazzazi, H. Görls, G. Gessner, S. H. Heinemann and M. Westerhausen, J. Organomet. Chem., 2013, 733, 63–70 CrossRef CAS PubMed.
- S. V. C. Vummaleti, D. Branduardi, M. Masetti, M. De Vivo, R. Motterlini and A. Cavalli, Chem.–Eur. J., 2012, 18, 9267–9275 CrossRef CAS PubMed.
- L. Hewison, S. H. Crook, B. E. Mann, A. J. H. M. Meijer, H. Adams, P. Sawle and R. A. Motterlini, Organometallics, 2012, 31, 5823–5834 CrossRef CAS.
- H.-M. Berends and P. Kurz, Inorg. Chim. Acta, 2012, 380, 141–147 CrossRef CAS PubMed.
- F. Zobi, O. Blacque, R. A. Jacobs, M. C. Schaub and A. Y. Bogdanova, Dalton Trans., 2012, 41, 370–378 RSC.
- J. D. Seixas, A. Mukhopadhyay, T. Santos-Silva, L. E. Otterbein, D. J. Gallo, S. S. Rodrigues, B. H. Guerreiro, A. M. L. Goncalves, N. Penacho, A. R. Marques, A. C. Coelho, P. M. Reis, M. J. Romão and C. C. Romão, Dalton Trans., 2013, 42, 5985–5998 RSC.
- T. R. Johnson, B. E. Mann, I. P. Teasdale, H. Adams, R. Foresti, C. J. Green and R. Motterlini, Dalton Trans., 2007, 1500–1508 RSC.
- L. Oresmaa, H. Tarvainen, K. Machal and M. Haukka, Dalton Trans., 2012, 41, 11170–11175 RSC.
- G. Parkin, Chem. Rev., 2004, 104, 699–768 CrossRef CAS PubMed.
- H. Pfeiffer, A. Rojas, J. Niesel and U. Schatzschneider, Dalton Trans., 2009, 4292–4298 RSC.
- H. Pfeiffer, T. Sowik and U. Schatzschneider, J. Organomet. Chem., 2013, 734, 17–24 CrossRef CAS PubMed.
- J. B. Matson, M. J. Webber, V. K. Tamboli, B. Weber and S. I. Stupp, Soft Matter, 2012, 8, 6689–6692 RSC.
- J. L. Wilson, H. E. Jesse, B. Hughes, V. Lund, K. Naylor, K. S. Davidge, G. M. Cook, B. E. Mann and R. K. Poole, Antioxid. Redox Signaling, 2013, 19, 497–509 CrossRef CAS PubMed.
- G. L. Patrick, An Introduction to Medicinal Chemistry, Oxford University Press, 2013 Search PubMed.
- T. Desmet, W. Soetaert, P. Bojarová, V. Kren, L. Dijkhuizen, V. Eastwick-Field and A. Schiller, Chem.–Eur. J., 2012, 18, 10786–10801 CrossRef CAS PubMed.
- I. Neundorf, J. Hoyer, K. Splith, R. Rennert, H. W. P. N'Dongo and U. Schatzschneider, Chem. Commun., 2008, 5604–5606 RSC.
- K. Splith, I. Neundorf, W. Hu, H. W. P. N'Dongo, V. Vasylyeva, K. Merz and U. Schatzschneider, Dalton Trans., 2010, 39, 2536–2545 RSC.
- A. Schroeder, D. A. Heller, M. M. Winslow, J. E. Dahlman, G. W. Pratt, R. Langer, T. Jacks and D. G. Anderson, Nat. Rev. Cancer, 2012, 12, 39–50 CrossRef CAS PubMed.
- L. Y. T. Chou, K. Ming and W. C. W. Chan, Chem. Soc. Rev., 2011, 40, 233–245 RSC.
- F. H. Schacher, P. A. Rupar and I. Manners, Angew. Chem., Int. Ed., 2012, 51, 7898–7921 CrossRef CAS PubMed.
- P. C. Kunz, H. Meyer, J. Barthel, S. Sollazzo, A. M. Schmidt and C. Janiak, Chem. Commun., 2013, 49, 4896–4898 RSC.
- T. R. Cook, Y.-R. Zheng and P. J. Stang, Chem. Rev., 2013, 113, 734–777 CrossRef CAS PubMed.
- M. Eddaoudi, D. B. Moler, H. Li, B. Chen, T. M. Reineke, M. O'Keeffe and O. M. Yaghi, Acc. Chem. Res., 2001, 34, 319–330 CrossRef CAS PubMed.
- S. K. Henninger, F. Jeremias, H. Kummer and C. Janiak, Eur. J. Inorg. Chem., 2012, 2625–2634 CrossRef CAS.
- C. Janiak and J. K. Vieth, New J. Chem., 2010, 34, 2366–2388 RSC.
- M. Ma, H. Noei, B. Mienert, J. Niesel, E. Bill, M. Muhler, R. A. Fischer, Y. Wang, U. Schatzschneider and N. Metzler-Nolte, Chem.–Eur. J., 2013, 19, 6785–6790 CrossRef CAS PubMed.
- G. Dördelmann, T. Meinhardt, T. Sowik, A. Krueger and U. Schatzschneider, Chem. Commun., 2012, 48, 11528–11530 RSC.
- G. Dördelmann, H. Pfeiffer, A. Birkner and U. Schatzschneider, Inorg. Chem., 2011, 50, 4362–4367 CrossRef PubMed.
- N. E. Brückmann, M. Wahl, G. J. Reiß, M. Kohns, W. Wätjen and P. C. Kunz, Eur. J. Inorg. Chem., 2011, 4571–4577 CrossRef.
- E. Tfouni, F. G. Doro, A. J. Gomes, R. S. d. Silva, G. Metzker, P. G. Z. Benini and D. W. Franco, Coord. Chem. Rev., 2010, 254, 355–371 CrossRef CAS PubMed.
- D. Bechet, P. Couleaud, C. Frochot, M.-L. Viriot, F. Guillemin and M. Barberi-Heyob, Trends Biotechnol., 2008, 26, 612–621 CAS.
- K. A. Wold, V. B. Damodaran, L. A. Suazo, R. A. Bowen and M. M. Reynolds, ACS Appl. Mater. Interfaces, 2012, 4, 3022–3030 CAS.
- C. Bohlender, M. Wolfram, H. Goerls, W. Imhof, R. Menzel, A. Baumgaertel, U. S. Schubert, U. Mueller, M. Frigge, M. Schnabelrauch, R. Wyrwa and A. Schiller, J. Mater. Chem., 2012, 22, 8785–8792 RSC.
- C. Bohlender, K. Landfester, D. Crespy and A. Schiller, Part. Part. Syst. Charact., 2013, 30, 138–142 CrossRef CAS.
- R. Wyrwa, M. Schnabelrauch, C. Altmann and A. Schiller, Kohlenstoffmonoxid freisetzende Materialien und deren Verwendung, German Pat., DE10 2012 04 132.2, 2012 Search PubMed.
- C. Bohlender, S. Gläser, M. Klein, J. Weisser, S. Thein, U. Neugebauer, J. Popp, R. Wyrwa and A. Schiller, J. Mater. Chem. B, 2014 10.1039/C3TB21649G.
- K. Landfester, Annu. Rev. Mater. Res., 2006, 36, 231–279 CrossRef CAS.
- A. Greiner and J. Wendorff, Angew. Chem., Int. Ed., 2007, 46, 5670–5703 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2014 |