 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Diastereoselective synthesis of a bicyclic β-lactam with penicillin G-like spectrum of activity by carbonylation of an acyclic diaminocarbene†
Tim
Schulz
a,
Christian
Färber
a,
Michael
Leibold
a,
Clemens
Bruhn
a,
Pascal
Prochnow
b,
Julia E.
Bandow
b,
Tanja
Schneider
c,
Timo
Porsch
d,
Max C.
Holthausen
*d and
Ulrich
Siemeling
*a
aInstitute of Chemistry, University of Kassel, Heinrich-Plett-Str. 40, D-34132 Kassel, Germany. E-mail: siemeling@uni-kassel.de
bBiology of Microorganisms, Ruhr University Bochum, Universitätsstr. 150, D-44801 Bochum, Germany
cPharmaceutical Microbiology, University of Bonn, Meckenheimer Allee 168, D-53115 Bonn, Germany
dInstitut für Anorganische und Analytische Chemie, Johann Wolfgang Goethe-Universität Frankfurt, Max-von-Laue-Str. 7, D-60438 Frankfurt am Main, Germany. E-mail: max.holthausen@chemie.uni-frankfurt.de
First published on 10th January 2014
Abstract
Diisopropylamino-cis-2,6-dimethylpiperidinocarbene reacts regio- and diastereoselectively with CO to afford a bicyclic β-lactam with 100% atom efficiency, whose spectrum of activity resembles that of penicillin G or amoxicillin.
The chemistry of carbenes and ketenes has been intertwined for a century now.1 In 1913 Staudinger described the thermal decarbonylation of ketenes, together with an analysis of the follow-up products of the resulting transient carbenes.2 The decomposition of the parent ketene H2C
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CO to CO and CH2 is one of the most extensively studied reactions in physical chemistry.3 Conversely, a classic method for the detection of transient carbenes is their trapping by carbonylation.4
CO to CO and CH2 is one of the most extensively studied reactions in physical chemistry.3 Conversely, a classic method for the detection of transient carbenes is their trapping by carbonylation.4
The advent of isolable N-heterocyclic carbenes in 19915 triggered the development of these and related persistent diaminocarbenes from laboratory curiosities to reliable workhorses in synthesis and catalysis.6 Such carbenes are usually inert towards CO,7 but exceptions occur with particularly electrophilic representatives such as, for example, acyclic diaminocarbenes (ADACs) 1.8 (iPr2N)2C (1a) was reported in 1996 as the first ADAC to be isolated and structurally characterised.9 We found that its primary carbonylation product (iPr2N)2CCO (2a) undergoes a remarkable intramolecular follow-up reaction (Scheme 1).8b,e A retro-Wolff rearrangement leads to the (amino)(carboxamido)carbene 3a, which subsequently affords the β-lactam 4a by a C–H insertion. Bona fide examples of this reaction type are rare. Previously studied cases exhibit considerably higher calculated activation barriers (≥37 kcal mol−1).10 The reaction 1a + CO → 4a represents a new entry to the important β-lactam ring system11 and proceeds with 100% atom efficiency.12 As a first milestone of a systematic study to probe the limitations of this new synthetic method, we have shown that β-lactam formation requires very bulky ADACs.8b We here address the question whether this reaction can be applied to the synthesis of bicyclic β-lactams, using bulky ADACs with cyclic amino groups. This is important in view of the bicyclic nature of the penicillins and cephalosporins, which are the most widely used β-lactam antibiotics.13 Diisopropylamino-cis-2,6-dimethylpiperidinocarbene (iPr2N)C(PipMe2) (1b)14 is the only ADAC known to date which meets the requirements for this investigation. Just like 1a, it is very bulky. In addition, it contains a cyclic amino group (PipMe2), which incidentally may be viewed as a conformationally constrained version of the iPr2N group.
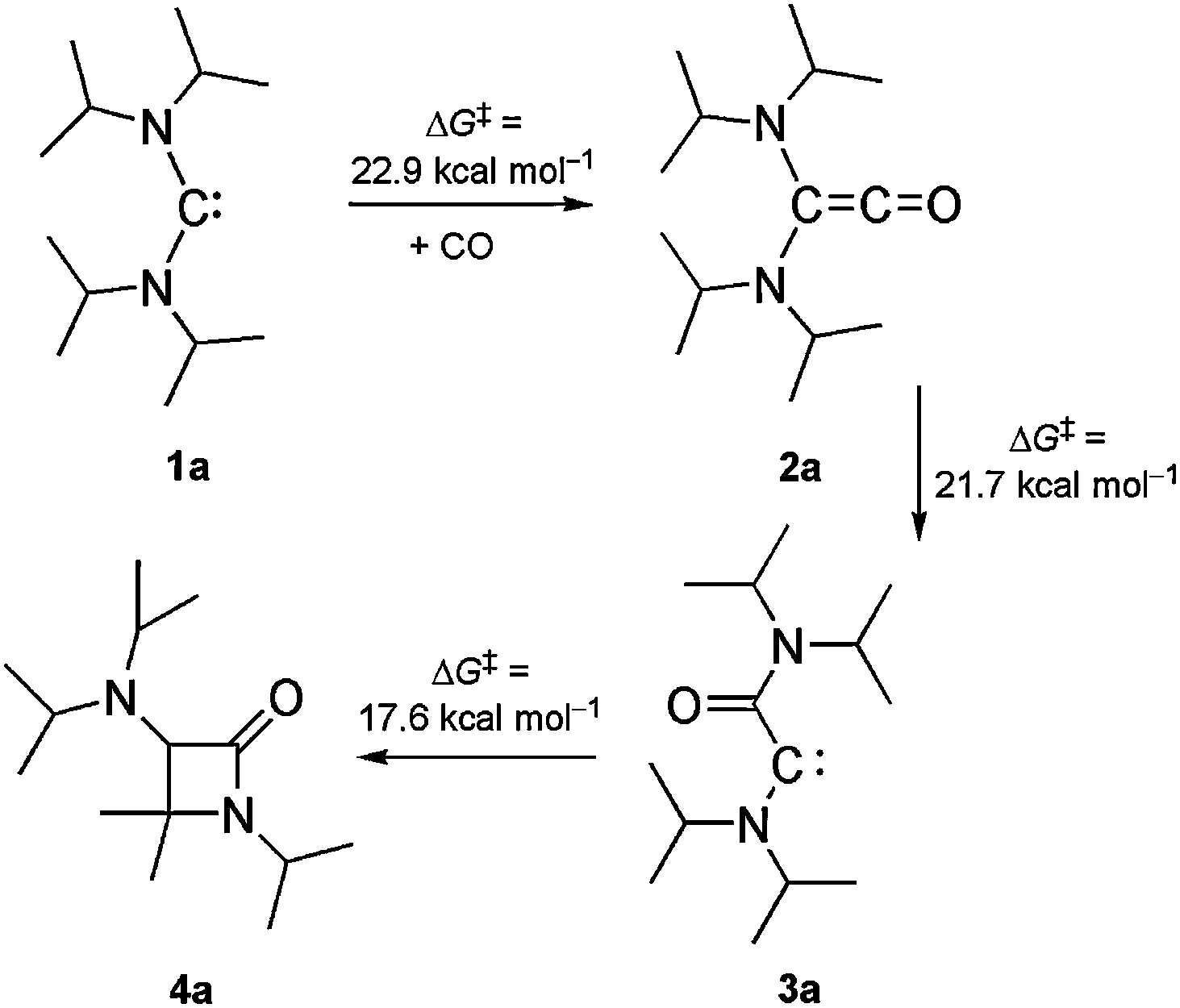 | ||
Scheme 1 Carbonylation of 1a, leading to β-lactam 4a (racemic mixture) as the final product. 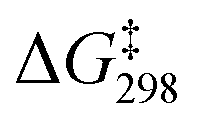 values were calculated by DFT methods. values were calculated by DFT methods. | ||
We have shown previously that 1a and 1b are very similar in terms of stability, both undergoing a slow β-fragmentation reaction in solution.15 Indeed, we have found such a chemical similarity also in their carbonylation. The reaction of 1b with CO proceeds smoothly and swiftly at room temperature, cleanly affording the bicyclic β-lactam derivative cis-4b (Scheme 2). This process is regioselective, since only the PipMe2 unit undergoes the rearrangement and concomitant C–H insertion. The monocyclic β-lactam 4c, which contains an intact PipMe2 unit, is not observed. Equally remarkable is the diastereoselectivity of the reaction. The diastereomer of 4b, which exhibits a trans orientation of the iPr2N group with respect to the methyl substituents (trans-4b), is not observed.
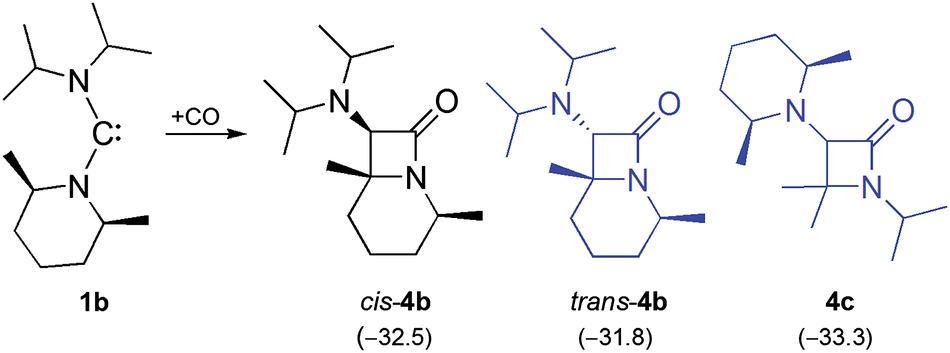 | ||
| Scheme 2 Carbonylation of 1b, leading to the bicyclic β-lactam cis-4b as the final product. The isomers trans-4b and 4c shown in grey are not observed. Only one enantiomer is shown in each case. Calculated ΔG298 values (kcal mol−1) are given in parentheses. | ||
The all-cis arrangement of the substituents at the bicyclic core of the final product is unequivocally demonstrated by the structure of the hydrochloride [cis-4bH]Cl, which we were able to determine by single-crystal X-ray diffraction (Fig. 1).
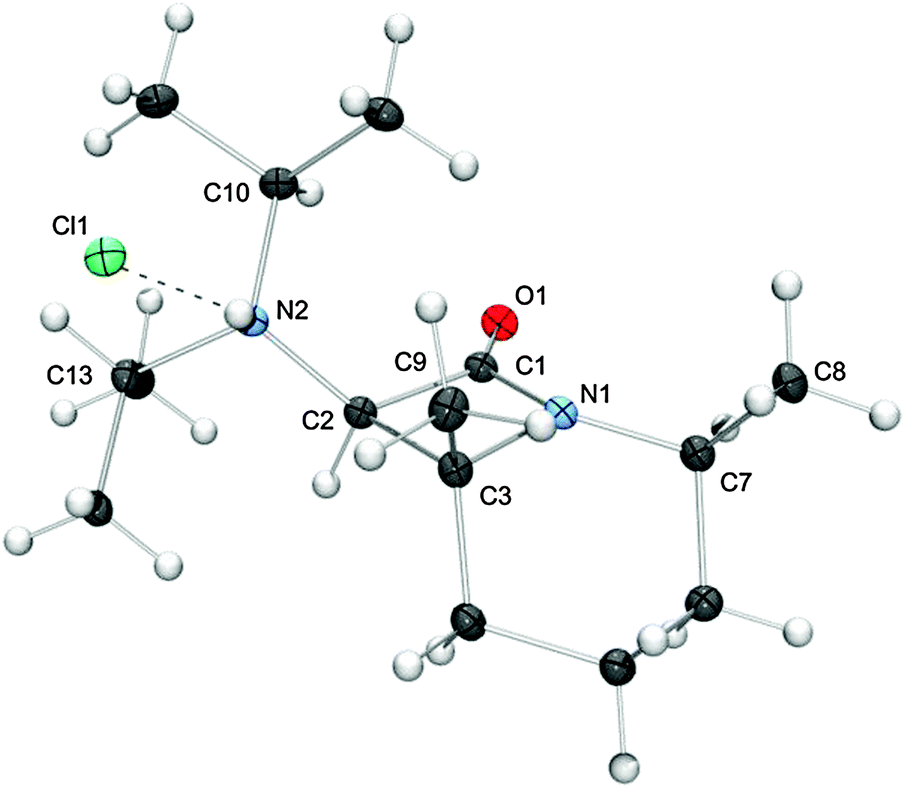 | ||
| Fig. 1 Molecular structure of [cis-4bH]Cl in the crystal (ellipsoids drawn at the 30% probability level). The broken line indicates a hydrogen bond between the chloride anion and the cationic iPr2HN substituent. | ||
We have investigated the formation of cis-4b using high-level DFT calculations (B2GP-PLYP-D/def2-QZVP//B97-D/SVP, see ESI† for details), with special attention to the axial (ax) and the equatorial conformation (eq) of the PipMe2 group, including in our analysis also possible boat (b) conformations of the six-membered ring (Fig. 2). First of all, we address thermodynamic aspects concerning the final carbonylation product. The energy of the experimentally observed bicyclic β-lactam cis-4b is 0.7 kcal mol−1 lower than that of its diastereomer trans-4b and 0.8 kcal mol−1 higher than that of the monocyclic β-lactam 4c (Scheme 2). These small energy differences indicate that the intramolecular follow-up reaction of 2b, which exclusively gives rise to cis-4b, is governed by kinetic, rather than thermodynamic, factors. Therefore, we now turn our attention to the course of the reaction. The axial carbene conformer 1bax is more stable than the equatorial one (1beq) by 6.8 kcal mol−1 (not shown in Fig. 2; see ESI†). This significant contrasteric bias is due to the anomeric effect.16 It is less pronounced for the ketene 2b, whose axial conformer is only 0.9 kcal mol−1 lower in energy than the equatorial one, which corresponds to a value of the equilibrium constant of ca. 5 at room temperature. For each calculated reaction pathway, the first step, viz. the retro-Wolff rearrangement, is rate-determining. This rearrangement can involve either the PipMe2 or the iPr2N group. For the dominant conformer 2bax the activation barrier has a value of 21.3 kcal mol−1 for the process which involves the PipMe2 group, leading to the transient carbene 3bax. The corresponding process which involves the iPr2N group and would finally lead to 4c has a significantly higher barrier (ΔG‡ = 25.6 kcal mol−1, see ESI† for details). In the case of the less abundant conformer 2beq too, the retro-Wolff rearrangement involving the PipMe2 unit is kinetically favoured over the alternative process involving the iPr2N group (ΔG‡ = 20.1 vs. 23.7 kcal mol−1). For both ketene conformers the activation energy differences (ΔΔG‡ = 4.3 kcal mol−1 and 3.6 kcal mol−1 for 2bax and 2beq, respectively) are sufficiently large to be compatible with an essentially exclusive formation of cis-4bvia the kinetically favoured carbene 3b. At the same time, the energy barrier differences for the various calculated pathways are small enough to suggest that subtle changes in the periphery of a bulky ADAC may have a dramatic influence on the outcome of its carbonylation. A comprehensive study will be required to develop a rationale for the reactivity of the primary carbonylation product, viz. the diaminoketene 2.
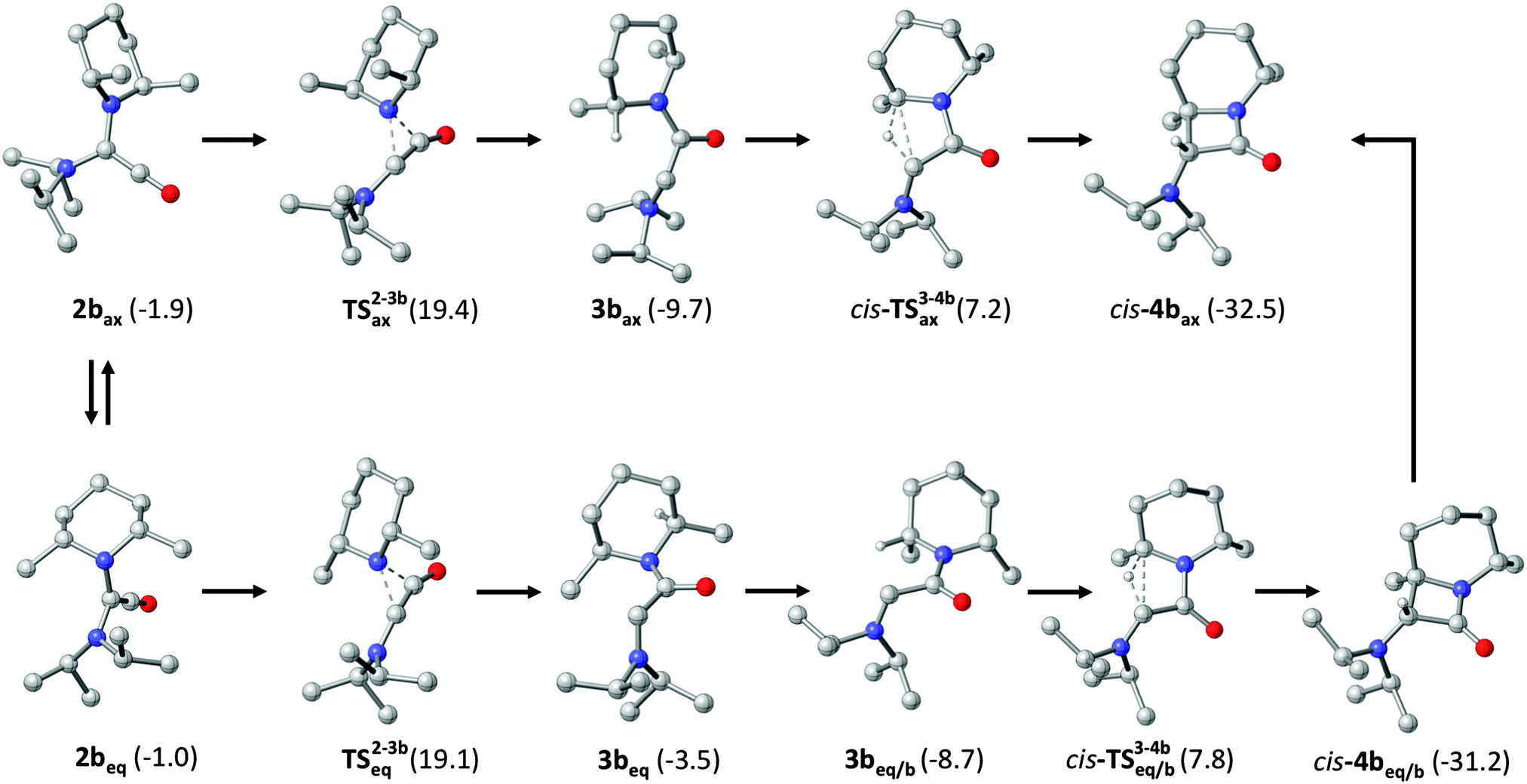 | ||
| Fig. 2 Computed kinetically favourable reaction pathways for diaminoketene 2b (shown for one enantiomer only). Pathways potentially leading to trans-4b and 4c (Scheme 2) are kinetically less favourable and have been omitted for clarity (see ESI† for details).‡ | ||
We have investigated the antimicrobial activity of the monocyclic β-lactam 4a and its bicyclic relative cis-4b against Gram-positive and Gram-negative bacteria by determining their minimal inhibitory concentrations (MICs) (see ESI†). The bicyclic β-lactam cis-4b exhibits significant activity against the Gram-positive bacteria B. subtilis and S. aureus. MIC values are 64–128 μg mL−1 for the S. aureus type strain and 128 μg mL−1 for B. subtilis 168 and a methicillin-resistant S. aureus (MRSA) strain. The antibiotic activity of 4a is lower by a factor of 2 (MIC = 256 μg mL−1 in all cases). Both compounds are inactive against Gram-negative bacteria. Their spectrum of activity resembles that of penicillin G or amoxicillin, whose activities, however, are higher than that of cis-4b by ca. two orders of magnitude.
In conclusion, the efficient and highly selective synthesis of cis-4b from 1b and CO under mild conditions opens up new possibilities to access unprecedented bicyclic β-lactams with useful antibiotic properties. We will continue our study with new ADACs containing bulky cyclic amino substituents, which we are currently developing.
We thank the DFG for generous funding (grant SI 429/19-1). T. S. is grateful to the Studienstiftung des deutschen Volkes for a doctoral fellowship. J. E. B. and P. P. are financially supported by a grant from the German federal state of North Rhine-Westphalia and the European Union (European Regional Development Fund, Investing in your future). Quantum-chemical calculations were performed at the Center for Scientific Computing (CSC) Frankfurt on the LOEWE-CSC high-performance computing cluster.
Notes and references
- (a) A. D. Allen and T. T. Tidwell, Chem. Rev., 2013, 113, 7287 CrossRef CAS PubMed; (b) A. D. Allen and T. T. Tidwell, Eur. J. Org. Chem., 2012, 1081 CrossRef CAS; (c) T. T. Tidwell, Ketenes, Wiley, Hoboken, 2nd edn., 2006 Search PubMed.
- H. Staudinger and R. Endle, Ber. Dtsch. Chem. Ges., 1913, 46, 1437 CrossRef CAS.
- (a) H. Xiao, S. Maeda and K. Morokuma, J. Phys. Chem. A, 2013, 117, 7001 CrossRef CAS PubMed; (b) Y. Ogihara, T. Yamamoto and S. Kato, J. Phys. Chem. A, 2010, 114, 9981 CrossRef CAS PubMed; (c) A. L. Kaledin, J. Seong and K. Morokuma, J. Phys. Chem. A, 2001, 105, 2731 CrossRef CAS; (d) C. G. Morgan, M. Drabbels and A. M. Wodtke, J. Phys. Chem., 1996, 105, 4550 CrossRef CAS PubMed; (e) K. Knox, R. G. W. Norrish and G. Porter, J. Chem. Soc., 1952, 1477 RSC; (f) W. F. Ross and G. B. Kistiakowski, J. Am. Chem. Soc., 1934, 56, 1112 CrossRef CAS; (g) R. W. G. Norrish, H. G. Crone and O. Saltmarsh, J. Chem. Soc., 1933, 1533 RSC.
- (a) S. C. Reed, G. J. Capitosti, Z. Zhu and D. A. Modarelli, J. Org. Chem., 2011, 66, 287 CrossRef; (b) W. Sander, R. Hübert, E. Kraka, J. Gräfenstein and D. Cremer, Chem.–Eur. J., 2000, 6, 4567 CrossRef CAS; (c) P. Visser, R. Zuhse, M. W. Wong and C. Wentrup, J. Am. Chem. Soc., 1996, 118, 12598 CrossRef CAS; (d) J. R. Ammann, R. Subramanian and R. S. Sheridan, J. Am. Chem. Soc., 1992, 114, 7592 CrossRef CAS; (e) W. W. Sander, J. Org. Chem., 1988, 53, 121 CrossRef CAS; (f) M. S. Baird, I. R. Dunkin, N. Hacker, M. Poliakoff and J. J. Turner, J. Am. Chem. Soc., 1981, 103, 5190 CrossRef CAS PubMed.
- A. J. Arduengo III, R. L. Harlow and M. Kline, J. Am. Chem. Soc., 1991, 113, 361 CrossRef.
- (a) M. Fèvre, J. Pinaud, Y. Gnanou, J. Vignolle and D. Taton, Chem. Soc. Rev., 2013, 42, 2142 RSC; (b) in N-Heterocyclic Carbenes, ed. S. Díez-González, RSC, Cambridge, 2011; (c) T. Dröge and F. Glorius, Angew. Chem., Int. Ed., 2010, 49, 6940 CrossRef PubMed; (d) J. Vignolle, X. Cattoën and D. Bourissou, Chem. Rev., 2009, 109, 3333 CrossRef CAS PubMed; (e) F. E. Hahn and M. C. Jahnke, Angew. Chem., Int. Ed., 2008, 47, 3122 CrossRef CAS PubMed; (f) D. Enders, O. Niemeier and A. Henseler, Chem. Rev., 2007, 107, 5606 CrossRef CAS PubMed.
- D. Martin, M. Soleilhavoup and G. Bertrand, Chem. Sci., 2011, 2, 389 RSC.
- (a) D. Martin, C. E. Moore, A. L. Rheingold and G. Bertrand, Angew. Chem., Int. Ed., 2013, 52, 7014 CrossRef CAS PubMed; (b) T. Schulz, C. Färber, M. Leibold, C. Bruhn, W. Baumann, D. Selent, T. Porsch, M. C. Holthausen and U. Siemeling, Chem. Commun., 2013, 49, 6834 RSC; (c) U. Siemeling, Aust. J. Chem., 2011, 64, 1109 CrossRef CAS; (d) C. Goedecke, M. Leibold, U. Siemeling and G. Frenking, J. Am. Chem. Soc., 2011, 133, 3557 CrossRef CAS PubMed; (e) U. Siemeling, C. Färber, C. Bruhn, M. Leibold, D. Selent, W. Baumann, M. von Hopffgarten, C. Goedecke and G. Frenking, Chem. Sci., 2010, 1, 697 RSC.
- R. W. Alder, P. R. Allen, M. Murray and A. G. Orpen, Angew. Chem., Int. Ed. Engl., 1996, 35, 1121 CrossRef CAS.
- (a) C. Wentrup, H. Bibas, A. Kuhn, U. Mitschke and M. C. MsMills, J. Org. Chem., 2013, 78, 10705 CrossRef CAS PubMed; (b) G. G. Qiao, W. Meutermans, M. W. Wong, M. Träubel and C. Wentrup, J. Am. Chem. Soc., 1996, 118, 3852 CrossRef CAS; (c) M. T. Nguyen, M. R. Hajnal and L. G. Vanquickenborne, J. Chem. Soc., Perkin Trans. 2, 1994, 169 RSC; (d) M. T. Nguyen, M. R. Hajnal, T.-K. Ha, L. G. Vanquickenborne and C. Wentrup, J. Am. Chem. Soc., 1992, 114, 4387 CrossRef.
- (a) β-Lactams: Unique Structures of Distinction for Novel Molecules, Top. Heterocycl. Chem., ed. B. K. Banik, Springer, Berlin, 2013, vol. 30 Search PubMed; (b) Heterocyclic Scaffolds I: β-Lactams, Top. Heterocycl. Chem., ed. B. K. Banik, Springer, Berlin, 2010, vol. 22 Search PubMed; (c) T. T. Tidwell, Angew. Chem., Int. Ed., 2008, 47, 1016 CrossRef CAS PubMed.
- (a) R. A. Sheldon, Pure Appl. Chem., 2000, 72, 1233 CrossRef CAS; (b) B. M. Trost, Angew. Chem., Int. Ed. Engl., 1995, 34, 259 CrossRef CAS; (c) B. M. Trost, Science, 1991, 254, 1471 CAS.
- (a) J. F. Fisher, S. O. Meroueh and S. Mobashery, Chem. Rev., 2005, 105, 395 CrossRef CAS PubMed; (b) R. P. Elander, Appl. Microbiol. Biotechnol., 2003, 61, 385 CAS.
- G. D. Frey and W. A. Herrmann, J. Organomet. Chem., 2005, 690, 5876 CrossRef CAS PubMed.
- T. Schulz, M. Leibold, C. Färber, M. Maurer, T. Porsch, M. C. Holthausen and U. Siemeling, Chem. Commun., 2012, 48, 9123 RSC.
- E. V. Anslyn and D. A. Dougherty, Modern Physical Organic Chemistry, University Science Books, Sausalito, 2006, p. 123 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental, crystallographic and computational details, Fig. S1–S3 and Table S1. CCDC 951678. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c3cc48538b |
| ‡ In the same vein, the nucleophilic addition of 1b to 2b, affording the oxyallyl species (1b)2CO, turned out to be kinetically unfavourable and has therefore not been incorporated in Fig. S2 (see ESI† for details). |
| This journal is © The Royal Society of Chemistry 2014 |