Reactions of a Zn(I) complex with group 14 azides – formation of zinc azide and zinc hexazene complexes†‡
S.
Gondzik
,
S.
Schulz
*,
D.
Bläser
,
C.
Wölper
,
R.
Haack
and
G.
Jansen
University of Duisburg-Essen, Universitätsstr. 5-7, S07 S03 C30, 45117 Essen, Germany. E-mail: stephan.schulz@uni-due.de; Fax: +49 201 1833830; Tel: +49 201 1834635
First published on 1st November 2013
Abstract
Two zinc hexazene complexes L2Zn2(μ-1,6-R2-N6) (L = HC[C(Me)N(2,4,6-Me3C6H2)]2; R = Ph (3), Dipp = 2,6-i-Pr2C6H3 (4)), were synthesized by reaction of the Zn(I) complex L2Zn2 (1) with phenyl azide and 2,6-diisopropylphenyl azide, respectively. 3 represents the second structurally characterized transition metal hexazene complex. In contrast, reactions of 1 with Me3MN3 (M = Si, Sn) yielded the azido complex [LZn(μ-N3)]2 (2) and Me3M-MMe3.
Metal-to-metal bonds in clusters and molecules are of fundamental interest in natural science.1 Since the pioneering work in the mid-1960s,2 main group metal complexes containing group 2,3 13,4 14,5 and 15 metals6 as well as transition metal complexes with multiple bonding including quintuple bonding7 have received steadily growing interest. Two major stabilizing strategies have been developed, i.e. kinetic stabilization by use of sterically demanding substituents8 and electronic stabilization by coordination of a Lewis base.9 Aside from their synthesis, the reactivity of such metal complexes has received increasing interest.10
Since the landmark discovery of [η5-Cp*2Zn2] (Cp* = C5Me5) by Carmona et al. in 200411 roughly 25 compounds containing Zn in the formal oxidation state +I have been structurally characterized12 and their chemical reactivity has been investigated. Lewis acid–base reactions, protonation reactions,13 oxidative addition reactions of PhCCPh and alkyl halides14 as well as reactions with transition metal complexes were reported.15 In addition, the electrochemical behavior of Cp*2Zn2 was studied by cyclic voltammetry16 and its use as a precatalyst for the intermolecular hydroamination reaction was studied.17
Our general interest in the reactivity of low-valent organometallic complexes18 prompted us to study reactions of L2Zn21 (L = HC[C(Me)N(2,4,6-Me3C6H2)]2) with group 14 azides RN3 (R = Ph, Dipp) and Me3MN3 (M = Si, Sn). We report herein on the structural characterization of the first zinc hexazene complex. Theoretical calculations were performed to identify N6 vibrations.
The reaction of 2.0 equiv. of Me3MN3 (M = Si, Sn) with 1 yielded hexamethyldisilane and -distannane Me6M2 as well as the previously reported zinc azido complex [LZnN3]22,19 while the reactions with two equiv. of PhN3 and DippN3 (Dipp = 2,6-i-Pr2C6H3) yielded orange crystalline solids, whose 1H NMR spectra showed resonances of both the β-diketiminato (L) group and the Ph/Dipp ligands. Since no gas evolution (N2) was observed, the formation of hexazene complexes became likely. The IR spectra of 3 and 4 are similar to those of 1 (ESI‡). The most prominent differences are the stronger intensities of two absorption bands at 1200 and 1259 cm−1 in the spectrum of 3 as well as an additional absorption band at 953 cm−1 (3).
Orange crystals of 3 were obtained upon storage of a solution of 3 in fluorobenzene at 4 °C (Fig. 1).20
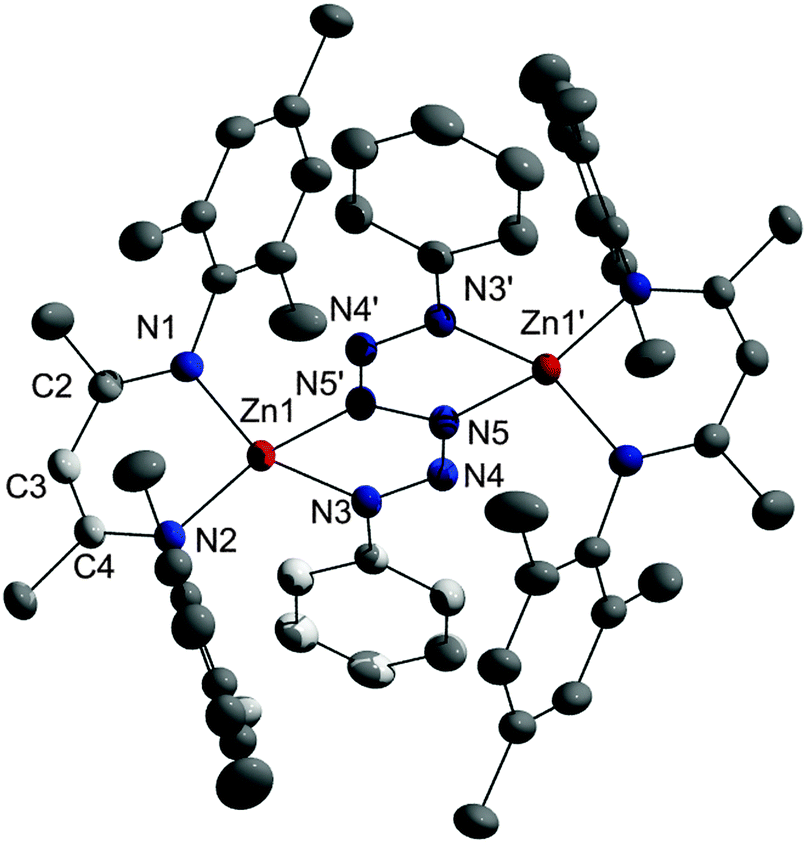 | ||
| Fig. 1 Solid state structure of 3; inversion center between N5 and N5′. Non-H-atoms are shown as thermal ellipsoids at 50% probability levels. H atoms are omitted for clarity. | ||
A single crystal X-ray diffraction study revealed the formation of the hexazene complex [(LZn)2(μ-η2:η2-PhN6Ph)] 3, which crystallizes in the monoclinic space group P21/n with two molecules in the unit cell. The Zn atoms in 3 are slightly out of the plane of the C3N2Zn rings (deviation from the least-squares plane 0.4249(17) Å, r.m.s. deviation of the ligand atoms from the plane 0.0368 Å) as was observed in L2Zn21,21 whereas the N4Zn metallacycle is almost perfectly planar (r.m.s. deviation from the least-squares plane 0.0057 Å). The central PhN6Ph hexazene unit adopts a bridging position and is connected to two four-coordinate Zn atoms. The Zn–N bond lengths within the six-membered C3N2Zn ring (Zn1–N1 1.9519(12), Zn1–N2 1.9364(12) Å) are slightly shorter than those within the five-membered N4Zn ring (Zn1–N3 2.0166(12), Zn1–N5′ 1.9801(12) Å). The different N–N bond lengths within the hexazene unit give insight into the electron localization within the complex. The central N5–N5′ bond (1.403(2) Å) is significantly elongated compared to the almost identical N3–N4 (1.3041(17) Å) and N4–N5 (1.3012(16) Å) bonds, hence the hexazene unit is best described as a dianionic ligand with a central N5–N5′ single bond and two allyl-like, monoanionic N3 moieties containing delocalized π-electrons between N3, N4 and N5. The N3–N4–N5 bond angle of 117.37(11)° is comparable to those previously observed in hexazene complexes by Holland et al. and Jones et al.24–26 The hexazene unit serves as a dianionic ligand, which agrees with the description of the Zn atoms in the formal oxidation state +II.
Four resonance structures of the dianionic hexazene unit can be drawn, out of which C and D are identical. A is less reasonable due to the neighbouring negatively charged N-atoms. The resonance structures with a central N–N single bond and delocalized π-electrons over the terminal N3 units perfectly agree with the experimentally observed N–N bond distances (Scheme 1).
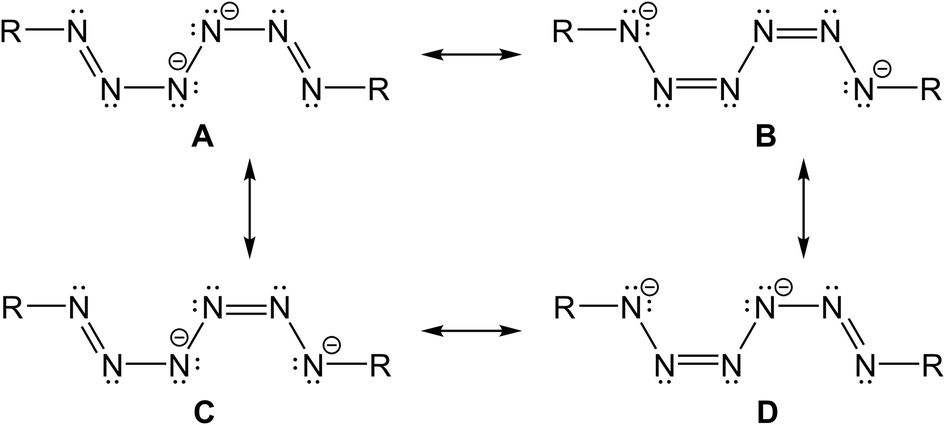 | ||
| Scheme 1 Resonance structures of a dianionic hexazene. | ||
Theoretical computations with dispersion corrected density functional theory confirm this picture.22 Starting from the crystal structure, first geometry optimizations of a single molecule of compound 3 were carried out by enforcing Ci symmetry. The most relevant optimized equilibrium bond lengths are those of the Zn1–N3 (2.028 Å), Zn1–N5′ (2.001 Å), N5–N5′ (1.391 Å), N3–N4 (1.310 Å) and N4–N5 (1.301 Å) bonds. They agree within 0.02 Å with the experimental X-ray structure values. Yet, a harmonic frequency computation for this structure showed two small imaginary frequencies, thus revealing it as a second order saddle point. An optimization of the molecular geometry without symmetry constrains resulted in a very similar structure that displayed no imaginary frequencies (4.8 kJ mol−1 lower in energy). Although the N6 unit is slightly twisted, the bond lengths deviate by less than 0.004 Å from the values given before, with the exception of one of the (now symmetry inequivalent) Zn1–N5′ distances, which deviates by 0.022 Å. According to a natural population analysis23 the total charge on the N6 unit is −1.76e (N3 −0.50e, N5 −0.35e) and that of the zinc atoms in 3 is 1.62e. The hexazene moiety in 3 was formed by reductive coupling of two azides. In contrast, the reaction of a Zn(I) diimine complex with phenyl acetylene did not proceed with C–C bond formation but with elimination of H2 and subsequent formation of the Zn acetylides.14a
Hexazene complexes are still rare and only five complexes have been reported to date. Holland et al. synthesized a Fe(II)–hexazene complex, which was obtained from the N–N coupling reaction of the Fe(I) complex (L′Fe)2N2 with adamantyl azide AdN3.24 In addition, Jones et al. synthesized three Mg hexazene complexes by reaction of Mg(I) complexes with AdN3.25,26 These complexes and 3 adopt similar structures, in which the metal centers are stabilized by N,N′-chelating, sterically demanding β-diketiminato ligands, and almost identical bonding parameters of the hexazene units. However, while Holland and Jones exclusively used adamantyl azide, we successfully used two different azides with either sterically less (R = Ph) or more (R = Dipp) demanding organic substituents. Moreover, the hexazene complexes 3 and 4 are neither shock nor thermally sensitive, in contrast to related neutral hexaazadienes.27
In order to prove that hexazene complexes can be identified by their IR spectra, we calculated the IR spectrum of 3. The second most intensive line in the calculated infrared spectrum of 3 in C1 symmetry at 1278 cm−1 corresponds to the antisymmetric linear combination of N3–C (phenyl) and N3′–C′(phenyl') stretching vibrations (with contributions from phenyl ring deformations). It is likely that this mode is responsible for the strong enhancement of the absorption band at 1260 cm−1 as observed in the experimental IR spectrum of 1 to the line observed at 1259 cm−1 in the experimental IR spectrum of 3. The calculated IR spectrum of 3 also shows a relatively intense absorption band at 1209 cm−1 due to an antisymmetric linear combination of N3–N4 and N3′–N4′ stretching vibrations (combined with a phenyl ring in-plane C–C–H bend), which likely explains the strong enhancement of the intensity of the absorption band at 1200 cm−1 in 1 to 1198 cm−1 in 3. Furthermore the experimental IR spectrum of 3 shows an absorption band at 953 cm−1 which is absent in 1. In the theoretical IR spectrum of 3 a relatively intense line is found at 949 cm−1, which corresponds to an antisymmetric linear combination of N3–N4–N5 and N3′–N4′–N5′ bending (with further contributions from phenyl ring deformations).
The hexazene complexes previously reported also show absorption bands close to the values observed and calculated for 3. However, further work is necessary in order to verify the assignment of the hexazene moiety.
In summary, the first zinc–hexazene complexes were synthesized by reaction of L2Zn2 containing the zinc atoms in the formal oxidation state I with organic group 14 azides RN3. In contrast, the reactions with trimethylsilyl- and stannyl azide gave the zinc azido complex [LZnN3]2. 3 represents the first non-adamantyl substituted hexazene complex. The identification of the reaction mechanism leading to the formation of the hexazene unit is currently investigated by detailed computational studies.
Notes and references
- F. A. Cotton, L. A. Murillo and R. A. Walton, Multiple Bonds Between Metal Atoms, Springer, Berlin, 3rd edn, 2005 RSC; G. Schmid, M. Baumle, M. Geerkens, I. Heim, C. Osemann and T. Sawitowski, Chem. Soc. Rev., 1999, 28, 179 RSC.
- F. A. Cotton, N. F. Curtis, C. B. Harris, B. F. G. Johnson, S. J. Lippard, J. T. Mague, W. R. Robinson and J. S. Wood, Science, 1964, 145, 1305 CAS.
- S. P. Green, C. Jones and A. Stasch, Science, 2007, 318, 1754 CrossRef CAS PubMed.
- H. Schnöckel, Chem. Rev., 2010, 110, 4125 CrossRef PubMed.
- T. Sasamori and N. Tokitoh, Group 14 Multiple Bonding, Encycl. Inorg. Chem., 2006 Search PubMed; A. Schnepf, Chem. Soc. Rev., 2007, 36, 745 RSC.
- T. Sasamori and N. Tokitoh, Dalton Trans., 2008, 1395 RSC.
- T. Nguyen, A. D. Sutton, M. Brynda, J. C. Fettinger, G. J. Long and P. P. Power, Science, 2005, 310, 844–847 CrossRef CAS PubMed; G. Frenking, Science, 2005, 310, 796 CrossRef PubMed; F. R. Wagner, A. Noor and R. Kempe, Nat. Chem., 2009, 1, 529 CrossRef PubMed; L. J. Clouston, R. B. Siedschlag, P. A. Rudd, N. Planas, S. Hu, A. D. Miller, L. Gagliardi and C. C. Lu, J. Am. Chem. Soc., 2013, 135, 13142 CrossRef PubMed.
- R. C. Fischer and P. P. Power, Chem. Rev., 2010, 110, 3877 CrossRef CAS PubMed.
- Y. Wang and G. H. Robinson, Inorg. Chem., 2011, 50, 12326 CrossRef CAS PubMed; Y. Wang and G. H. Robinson, Dalton Trans., 2012, 41, 337 RSC.
- C. A. Caputo and P. P. Power, Organometallics, 2013, 32, 2278 CrossRef CAS.
- I. Resa, E. Carmona, E. Gutierrez-Puebla and A. Monge, Science, 2004, 305, 1136 CrossRef CAS PubMed; A. Grirrane, I. Resa, A. Rodriguez, E. Carmona, E. Alvarez, E. Gutierrez-Puebla, A. Monge, A. Galindo, D. del Rio and R. A. Andersen, J. Am. Chem. Soc., 2007, 129, 693 CrossRef PubMed.
- For review articles see: T. Li, S. Schulz and P. W. Roesky, Chem. Soc. Rev., 2012, 41, 3759 RSC; S. Schulz, Chem.–Eur. J., 2010, 16, 6416 CrossRef CAS PubMed; A. Grirrane, I. Resa, A. Rodríguez and E. Carmona, Coord. Chem. Rev., 2008, 252, 1532 CrossRef; E. Carmona and A. Galindo, Angew. Chem., Int. Ed., 2008, 47, 6526 CrossRef PubMed.
- D. Schuchmann, U. Westphal, S. Schulz, U. Flörke, D. Bläser and R. Boese, Angew. Chem., Int. Ed., 2009, 48, 807 CrossRef CAS PubMed; S. Schulz, D. Schuchmann, I. Krossing, D. Himmel, D. Bläser and R. Boese, Angew. Chem., Int. Ed., 2009, 48, 5748 CrossRef PubMed; S. Schulz, S. Gondzik, D. Schuchmann, U. Westphal, L. Dobrzycki, R. Boese and S. Harder, Chem. Commun., 2010, 46, 7757 RSC; S. Gondzik, D. Bläser, C. Wölper and S. Schulz, Chem.–Eur. J., 2010, 16, 13599 CrossRef PubMed; M. Carrasco, R. Peloso, A. Rodríguez, E. Álvarez, C. Maya and E. Carmona, Chem.–Eur. J., 2010, 16, 9754 CrossRef PubMed; K. Freitag, H. Banh, C. Ganesamoorthy, C. Gemel, R. W. Seidel and R. A. Fischer, Dalton Trans., 2013, 42, 10540 RSC.
- (a) J. Gao, S. Li, Y. Zhao, B. Wu and X.-J. Yang, Organometallics, 2012, 31, 2978 CrossRef CAS; (b) A. Stasch, Chem.–Eur. J., 2012, 18, 15105 CrossRef CAS PubMed.
- T. Bollermann, K. Freitag, C. Gemel, R. W. Seidel and R. A. Fischer, Organometallics, 2011, 30, 4123 CrossRef CAS; T. Bollermann, K. Freitag, C. Gemel, M. Molon, R. W. Seidel, M. von Hopffgarten, P. Jerabek, G. Frenking and R. A. Fischer, Inorg. Chem., 2011, 50, 10486 CrossRef PubMed; T. Bollermann, C. Gemel and R. A. Fischer, Coord. Chem. Rev., 2012, 256, 537 CrossRef.
- M. Carrasco, R. Peloso, I. Resa, A. Rodríguez, L. Sánchez, E. Álvarez, C. Maya, R. Andreu, J. J. Calvente, A. Galindo and E. Carmona, Inorg. Chem., 2011, 50, 6361 CrossRef CAS PubMed.
- (a) A. Lühl, H. P. Nayek, S. Blechert and P. W. Roesky, Chem. Commun., 2011, 47, 8280 RSC; (b) A. Lühl, L. Hartenstein, S. Blechert and P. W. Roesky, Organometallics, 2012, 31, 7109 CrossRef.
- S. Schulz, L. Häming, R. Herbst-Irmer, H. W. Roesky and G. M. Sheldrick, Angew. Chem., 1994, 106, 1052 CrossRef CAS; S. Schulz, A. Voigt, H. W. Roesky, L. Häming and R. Herbst-Irmer, Organometallics, 1996, 15, 5252 CrossRef; S. Schulz, F. Thomas, W. Priesmann and M. Nieger, Organometallics, 2006, 25, 1392 CrossRef.
- G. Bendt, S. Schulz, J. Spielmann, S. Schmidt, D. Bläser and C. Wölper, Eur. J. Inorg. Chem., 2012, 3725 CrossRef CAS.
- Crystal data for 3: C64H72FN10Zn2, M = 1131.05, monoclinic, a = 12.8569(9) Å, b = 17.7885(13) Å, c = 13.5948(10) Å, β = 104.691(4)°, V = 3007.6(4) Å3, T = 180(1) K, space group P21/n, Z = 2, μ(MoKα) = 0.848 mm−1, 55
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 228 reflections measured, 11500 independent reflections (Rint = 0.0277). The final R1 values were 0.0395 (I > 2σ(I)) and 0.0618 (all data). The final wR(F2) values were 0.1043 (I > 2σ(I)) and 0.1178 (all data). The goodness of fit on F2 was 1.031.
228 reflections measured, 11500 independent reflections (Rint = 0.0277). The final R1 values were 0.0395 (I > 2σ(I)) and 0.0618 (all data). The final wR(F2) values were 0.1043 (I > 2σ(I)) and 0.1178 (all data). The goodness of fit on F2 was 1.031. - S. Schulz, D. Schuchmann, U. Westphal and M. Bolte, Organometallics, 2009, 28, 1590 CrossRef CAS.
- Computational details are given in the ESI‡.
- (a) A. E. Reed, R. B. Weinstock and F. Weinhold, J. Chem. Phys., 1985, 83, 735 CrossRef CAS; (b) A. E. Reed, L. A. Curtis and F. Weinhold, Chem. Rev., 1988, 88, 899 CrossRef CAS.
- R. E. Cowley, J. Elhaïk, N. A. Eckert, W. W. Brennessel, E. Bill and P. L. Holland, J. Am. Chem. Soc., 2008, 130, 6074 CrossRef CAS PubMed.
- S. J. Bonyhady, S. P. Green, C. Jones, S. Nembenna and A. Stasch, Angew. Chem., Int. Ed., 2009, 48, 2973 CrossRef CAS PubMed.
- S. J. Bonyhady, C. Jones, S. Nembenna, A. Stasch, A. J. Edwards and G. J. McIntyre, Chem.–Eur. J., 2010, 16, 938 CrossRef CAS PubMed.
- F. R. Berson, The High Nitrogen Compounds, Wiley, New York, 1984 CrossRef CAS; O. Nuyken, C. Scherer, A. Baindl, A. R. Brenner, U. Dahn, R. Gärtner, S. Kaiser-Röhrich, R. Kollefrath, P. Matusche and B. Voit, Prog. Polym. Sci., 1997, 22, 93 CrossRef CAS; K. A. Hofmann and H. Hock, Chem. Ber., 1911, 44, 2946 CrossRef; D. Mackay, D. D. McIntyre and N. J. Taylor, J. Org. Chem., 1982, 47, 532 CrossRef; C. M. Fitchett, C. Richardson and P. Steel, Org. Biomol. Chem., 2005, 3, 498 Search PubMed.
Footnotes |
| † Dedicated to Prof. Peter Jutzi on the occasion of his 75th birthday. |
| ‡ Electronic supplementary information (ESI) available: Experimental procedure and characterization of 3. CCDC 959097 (3). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c3cc47687a |
| This journal is © The Royal Society of Chemistry 2014 |