Diverse reactions of N-heterocyclic carbenes with an alkynylborane and isolation of a reactive zwitterionic borataallene†
Rüdiger
Bertermann
,
Holger
Braunschweig
*,
Christopher K. L.
Brown
,
Alexander
Damme
,
Rian D.
Dewhurst
,
Christian
Hörl
,
Thomas
Kramer
,
Ivo
Krummenacher
,
Bernd
Pfaffinger
and
Krzysztof
Radacki
Institut für Anorganische Chemie, Julius-Maximilians-Universität Würzburg, Am Hubland, 97074 Würzburg, Germany. E-mail: h.braunschweig@mail.uni-wuerzburg.de; Web: http://www-anorganik.chemie.uni-wuerzburg.de/Braunschweig/
First published on 22nd October 2013
Abstract
Ethynyldimesitylborane (1) is synthesised via salt elimination and its reactivity towards NHCs is studied. Depending on their size, NHCs attack either at the boron atom or at the β-alkynyl carbon atom. Steric control over the reaction was probed by reactions with N-heterocyclic carbenes yielding a carbene–borane adduct (2), a 1-boraindane (3), and the first structurally characterised borataallene (4).
N-heterocyclic carbenes (NHCs) are active species in many organocatalytic reactions including the benzoin condensation, Stetter, Diels–Alder and acyl-transfer reactions.1–6 Stoichiometric reactions of NHCs with organic substrates7 include insertion into X–H (X = O, N, S) bonds,8,9 while with organohalides NHCs were reported to react either as bases or nucleophiles.10 The nucleophilic attack of NHCs at unsaturated organic and inorganic compounds has also been reported, but this is rare and only possible for electron-poor systems. Enders9 studied the reactivity of NHCs towards electron-poor alkynes, which led to formation of a spirocyclic compound composed of a 1,3,4-triphenyl-1,2,4-triazol-5-ylidene unit and two alkyne units (Scheme 1, top), while reaction with electron-poor alkenes yielded 5-methylene-triazolines (Scheme 1, bottom). The proposed mechanism for this reaction includes the formation of a cyclopropane system via a [2+1] cycloaddition. Upon the subsequent ring opening a zwitterion is formed, which then undergoes a 1,2-proton shift yielding the respective 5-methylene-triazoline. Alternatively, the carbene can attack at the β-carbon atom similar to a Michael addition, directly leading to the zwitterion.
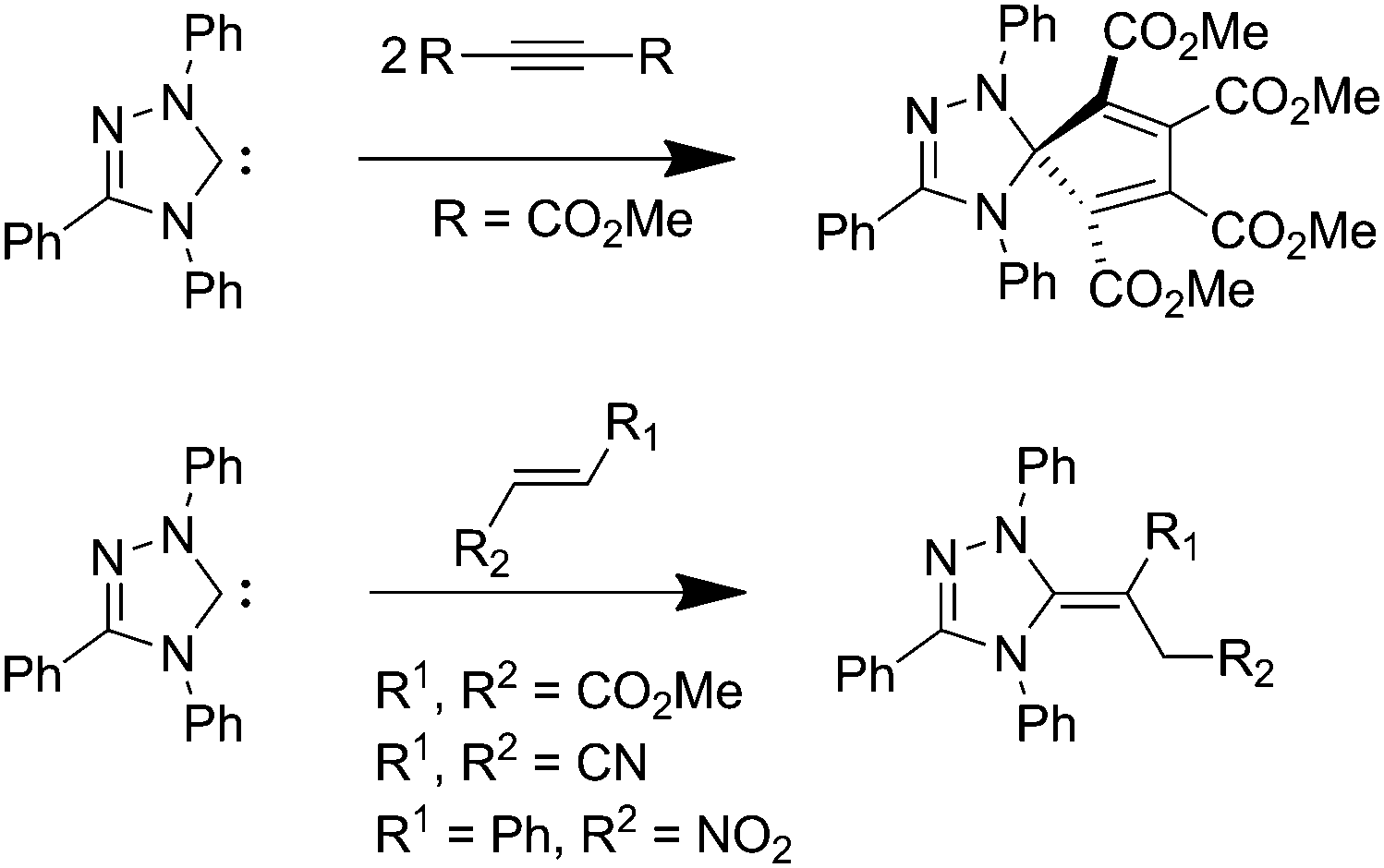 | ||
| Scheme 1 Reactivity of a triazolylidene towards electron-poor double and triple bonds. | ||
Nucleophilic attack by NHCs at C–C triple bonds is extremely rare, being limited to very electron-poor alkynes. Our synthesis of ethynyldimesitylborane (reported herein), an electron-poor alkyne with a number of possible sites for nucleophilic attack, prompted us to probe its reactivity towards various NHCs. Herein, we present the diverse reactivity of this electron-poor alkyne with NHCs of different sizes, leading to attack at either the boron atom or the β-alkynyl carbon atom. The latter leads either to C–H activation and boraindane formation, or alternatively, to isolation of a reactive zwitterionic borataallene compound. This formation of a borataallene is analogous to the zwitterionic intermediate proposed by Enders to occur prior to hydrogen migration. However in the presence of a boryl substituent, the unfavourable negative charge on the carbon atom can be transferred onto the boron atom, partially stabilising the molecule.
Reaction of ethynylmagnesium bromide with Mes2BF in THF at 0 °C affords ethynyldimesitylborane (1), which is isolated as a colourless solid in moderate yield after workup. Reaction of 1 with 1,3-bis-methylimidazol-2-ylidene (IMe) yields adduct 2, which was isolated as a colourless solid showing two 1H NMR signals (δ 6.89 and 6.74) for the carbene backbone protons and a broad signal for the ortho methyl groups (δ 2.06), suggesting rotational hindrance. The signal of the alkynyl proton (δ 2.17) was found upfield of that of 1. The 13C{1H} NMR spectrum exhibits broad signals at δ 168, 146, and 105, with partially resolved JBC coupling constants of 63, 48, and 73 Hz respectively. These signals represent the carbon atoms attached to the boron center. Partially resolved 1JBC couplings have previously been reported for borirene–IMe adducts.11 The resonance of the terminal alkynyl carbon atom is shifted upfield to δ 83.8 and the 11B{1H} NMR signal was observed at δ −18.5. In contrast to the respective 1H and 13C NMR shifts, the ![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C–H vibrational band (3238 cm−1) is little affected by quaternisation of the boron center.
C–H vibrational band (3238 cm−1) is little affected by quaternisation of the boron center.
In contrast, addition of larger NHCs results in nucleophilic attack at the H–C moiety of 1. Reaction of 1 with 1,3-bis-(2,6-diisopropylphenyl)imidazol-2-ylidene (IDipp) in C6D6 resulted in a colour change from colourless to red-violet and finally to red-orange. After workup, the new species 3 was isolated in high yield as a thermally stable red solid sensitive to air, moisture and protic substances (mp 150.4 °C; decomp. ∼206.3 °C). Spectroscopic and structural data revealed 3 to be an unusual 1-boraindane. These species are commonly synthesised by pyrolysis12,13 of alkyl- or arylboranes, a route which is rather harsh and has limited scope. Moreover, the alternative synthesis of 2-dialkylamino-boraindanes by double salt elimination14 results in yields of only ca. 50%. The 1H NMR spectrum of 3 shows doublets at δ 3.07 (1H, 2JHH = 9.0 Hz) and at δ 2.76 (2H, 2JHH = 5.8 Hz). An associated doublet of triplets is detected at δ 2.41 and has an integral of unity. The signals are assigned to the vinylic proton and the protons of the five-membered boron heterocycle. The 11B{1H} NMR spectrum displays a signal at δ 81.4. The newly formed C1–C5 bond has a length of 1.558(3) Å and is longer than the sum of the covalent radii (ΣC(sp2)C(sp3) = 1.49 Å)15 as well as the relevant bond in the known 1-boraindane (1.50(3) Å) (Fig. 1, left).16 In contrast, the respective bond in another 1-boraindane (1.587(4) Å)17 is marginally longer. The C1–C2 bond (1.502(3) Å) is 26% longer than in the alkyne 1 (1.194(5) Å). The new C2–C6 bond (1.346(3) Å) is a typical C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond.
C bond.
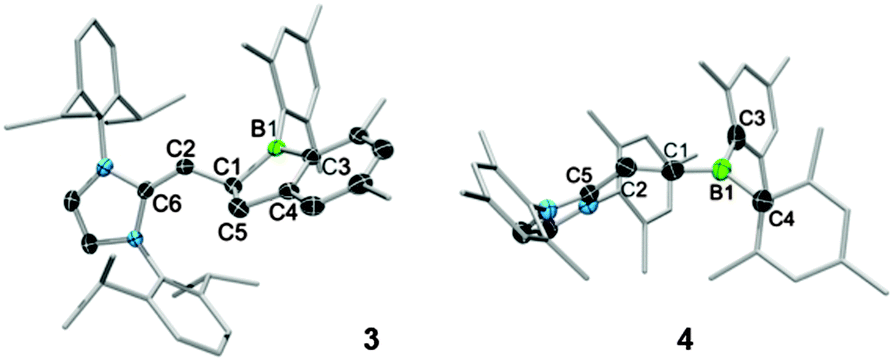 | ||
| Fig. 1 Molecular structures of 3 and 4 in the solid state. Thermal ellipsoids displayed at the 50% probability level. Hydrogen atoms and some ellipsoids have been omitted. Relevant bond lengths [Å] and angles [°] for 3: C1–C2: 1.502(3), C1–C5: 1.558(3), C2–C6: 1.346(3), C4–C5: 1.502(3), B1–C1: 1.583(3), B1–C3: 1.546(3), C3–C4: 1.403(3); C6–C2–C1: 131.0(2), C5–C1–B1: 103.9(2), C1–B1–C3: 106.7(2), B1–C3–C4: 108.3(2), C3–C4–C5: 112.8(2), C4–C5–C1: 106.9(2). For 4: C1–C2: 1.329(6), C2–C5: 1.441(6), B1–C1: 1.408(6), B1–C3: 1.605(7), B1–C4: 1.625(5); C1–B1–C3: 117.7(4), C3–B1–C4: 118.7(3), C4–B1–C1: 123.6(4), B1–C1–C2: 175.4(2), C1–C2–C5: 124.9(4). | ||
We initially assumed the intermediacy of a boracarbene, which is then inserted into a methyl C–H bond to form 3. However, reports of boracarbenes are rare and they are yet to be isolated.18,19 In order to isolate or identify an intermediate in this process, we performed the reaction of 1 with IDipp in hexane, observing the formation of a violet solid. This solid was unstable and turned red immediately after being dissolved in C6H5F. The 11B NMR spectrum of this solution showed the presence of 3 (ca. δ 80, major) and signals at ca. δ 27 (broad) and a number of sharp unidentified signals at ca. δ 3. However, on the basis of the analogous reaction of 1 with a different NHC (vide infra), we propose that the borataallene structure 3′ corresponds to the violet solid and the 11B NMR signal at δ 27.
In order to slightly alter the reaction conditions, we reacted 1 with IMes in C6D6 and again a colour change from colourless to red-violet and finally to red is observed, with the red-violet colour lasting significantly longer than when IDipp was used. Directly after mixing the reagents the 11B NMR spectrum of the mixture showed a major signal at ca. δ 22 (4, Scheme 2) along with a minor signal at ca. δ 73 which we assumed to be that of the boraindane compound analogous to 3. Over 3 d at rt, the 11B NMR signal for 4 disappeared, replaced by signals at ca. δ 74, 3 and −6; compounds corresponding to these signals could not be isolated from the mixture. To isolate the intermediate we carried out the reaction in hexane, resulting in the immediate formation of a red-violet (in one case orange) solid (4, Scheme 2) which shows no further reaction in the solid state at ambient temperature. The 1H NMR spectrum of 4 shows singlets at δ 6.83 and 6.53 for the aryl protons of the mesityl groups and the methyl protons of the mesityl groups appear at δ 2.31, 2.27, 2.12, and 1.76. Interestingly, the mesityl groups of the BMes2 group as well as those of the NHC fragment showed no rotational hindrance. The 13C{1H} NMR spectrum exhibited a signal at δ 286.8 for the cationic carbon atom of the imidazole ring. The boron-bound carbon atom of the borataallene moiety was detected at δ 137 and the terminal carbon nucleus signal at δ 87.2. The 11B NMR signal was detected at δ 22.3. The solid-state 13C variable-amplitude cross-polarization/magic-angle spinning (VACP/MAS) NMR spectrum exhibited a signal for the cationic carbon atom at δ 276.1 and two signals at δ 121.9 and δ 119.9 for the backbone of the imidazolium ring. The terminal carbon atom of the borataallene moiety is detected at δ 88.0, whereas the signal for the boron-bound carbon atom is obscured by the signals of the aromatic carbon atoms. For the aromatic mesityl carbon atoms, nine broad signals between δ 146 and δ 126 were found. In addition, eight signals for the mesityl methyl carbon atoms were found between δ 26 and δ 15. In the 11B VACP/MAS NMR one signal at δ 20.5 showing a η(quad) value of 0.3 was found.
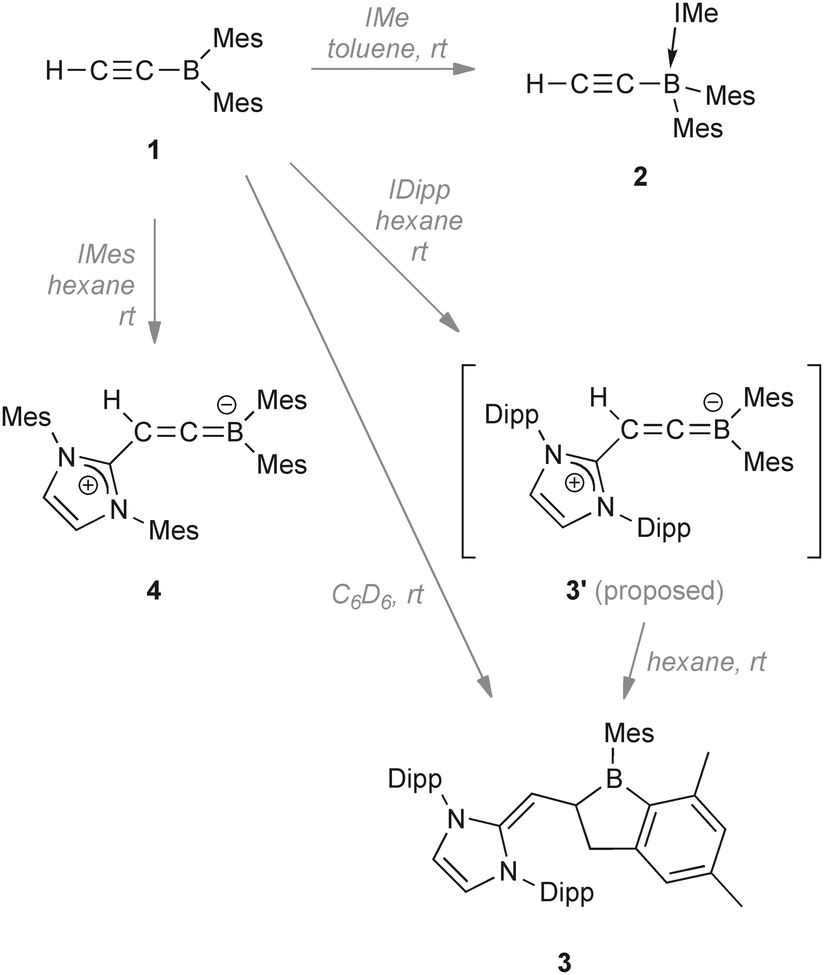 | ||
| Scheme 2 Reactivity of ethynyldimesitylborane (1) towards different N-heterocyclic carbenes (NHCs). | ||
The C1–C2 bond of 4 (1.329(6) Å) is longer than that of 1 (1.194(5) Å) and represents a conventional CC double bond (Fig. 1, right). In contrast, the B1–C1 bond of 4 (1.408(6) Å) is shorter than that of 1 (1.527(6) Å), thus indicating the significant BC double bond character. BC double bond systems are rare, however they exist in aminoalkylideneboranes,20–24 alkylalkylideneboranes,25 a 1,3-diborataallene,26,27 and the boron methylidenide ion Li[Mes2BCH2].28 The B1–C1 distance of 4 (1.408(6) Å) is slightly shorter than in the ionic Li[Mes2BCH2] (1.444(8) Å),28 longer than BC double bonds in alkylalkylideneboranes (1.36(1) Å),25 and comparable to those of aminoalkylideneboranes20–24 and a 1,3-diborataallene26,27 (∼1.40 Å). The newly formed C2–C5 bond (1.441(6) Å) has a typical length of a C(sp2)–C(sp3) bond. The B1–C1–C2 angle is 175.2(4)°, in comparison to 168.4(4)° in a 1,3-diborataallene,26,27 thus suggesting a more linear coordination of C1.
Attempts to convert a pure sample of 4 into a boraindane analogous to 3 by heating the compound to 140 °C led to a mixture, the 11B NMR spectrum of which contained signals for 4, the presumed boraindane (ca. δ 75), and a sharper signal at ca. δ 3. The extreme broadness of the signals precluded the estimation of the relative amounts of the compounds.
We have reported the synthesis of dimesitylborylacetylene (1), the IMe adduct 2, the 1-boraindane 3, and the first ever structurally characterised borataallene 4. The borataallene structure is presumably an intermediate in the boraindane formation process, however we were unable to definitively confirm this. Depending on the size of the NHC, either quaternisation of the boron center or attack at the terminal acetylenic carbon atom is observed. The nucleophilic attack is noteworthy as alkyne deprotonation by the NHC is another possibility given the high acidity of the acetylenic proton. Boron–carbon double bond systems remain rare and only a few have been reported in the literature. Our facile synthesis of the first borataallene 4 opens possibilities for exploring the properties of this rare and reactive family of molecules. The convenient formation of 1-boraindanes (3) in a controlled and mild manner is also unprecedented.
Financial support from the Deutsche Forschungsgemeinschaft is gratefully acknowledged.
Notes and references
- G. A. Grasa, R. M. Kissling and S. P. Nolan, Org. Lett., 2002, 4, 3583–3586 CrossRef CAS PubMed.
- D. Enders, O. Niemeier and A. Henseler, Chem. Rev., 2007, 107, 5606–5655 CrossRef CAS PubMed.
- A. Grossmann and D. Enders, Angew. Chem., Int. Ed., 2012, 51, 314–325 CrossRef CAS PubMed.
- J. Izquierdo, G. E. Hutson, D. T. Cohen and K. A. Scheidt, Angew. Chem., Int. Ed., 2012, 51, 11686–11698 CrossRef CAS PubMed.
- X. Bugaut and F. Glorius, Chem. Soc. Rev., 2012, 41, 3511–3522 RSC.
- D. T. Cohen and K. A. Scheidt, Chem. Sci., 2012, 3, 53–57 RSC.
- N-Heterocyclic Carbenes in Synthesis, ed. S. P. Nolan, Wiley-VCH, Weinheim, 2006 Search PubMed.
- D. Enders, K. Breuer, G. Raabe, J. Runsink, J. H. Teles, J. P. Melder, K. Ebel and S. Brode, Angew. Chem., Int. Ed. Engl., 1995, 34, 1021–1023 CrossRef CAS.
- D. Enders, K. Breuer, J. Runsink and J. H. Teles, Liebigs Ann., 1996, 2019–2028 CrossRef CAS.
- C. E. I. Knappke, A. J. Arduengo, H. J. Jiao, J. M. Neudorfl and A. Jacobi von Wangelin, Synthesis, 2011, 3784–3795 CAS.
- H. Braunschweig, A. Damme, R. D. Dewhurst, S. Ghosh, T. Kramer, B. Pfaffinger, K. Radacki and A. Vargas, J. Am. Chem. Soc., 2013, 135, 1903–1911 CrossRef CAS PubMed.
- R. Koster, G. Benedikt, W. Fenzl and K. Reinert, Liebigs Ann. Chem., 1967, 702, 197–223 CrossRef.
- W. Schacht and D. Kaufmann, Chem. Ber., 1987, 120, 1331–1338 CrossRef CAS.
- G. E. Herberich, U. Eigendorf and U. Englert, Chem. Ber., 1993, 126, 1397–1402 CrossRef CAS.
- B. Cordero, V. Gomez, A. E. Platero-Prats, M. Reves, J. Echeverria, E. Cremades, F. Barragan and S. Alvarez, Dalton Trans., 2008, 2832–2838 RSC.
- M. T. Reetz, H. Brummer, M. Kessler and J. Kuhnigk, Chimia, 1995, 49, 501–503 CAS.
- H. Michel, D. Steiner, S. Wocadlo, J. Allwohn, N. Stamatis, W. Massa and A. Berndt, Angew. Chem., Int. Ed. Engl., 1992, 31, 607–610 CrossRef.
- M. Suginome, T. Fukuda and Y. Ito, J. Organomet. Chem., 2002, 643–644, 508–511 CrossRef CAS.
- D. Bourissou, O. Guerret, F. P. Gabbaï and G. Bertrand, Chem. Rev., 2000, 100, 39–92 CrossRef CAS PubMed.
- B. Glaser and H. Nöth, Angew. Chem., Int. Ed. Engl., 1985, 24, 416–417 CrossRef.
- H. Nöth, W. Rattay and U. Wietelmann, Chem. Ber., 1987, 120, 859–861 CrossRef.
- B. Glaser, E. P. Mayer, H. Nöth, W. Rattay and U. Wietelmann, Z. Naturforsch., B, 1988, 43, 449–456 CAS.
- R. Boese, P. Paetzold and A. Tapper, Chem. Ber., 1987, 120, 1069–1071 CrossRef CAS.
- A. Tapper, T. Schmitz and P. Paetzold, Chem. Ber., 1989, 122, 595–601 CrossRef CAS.
- R. Boese, P. Paetzold, A. Tapper and R. Ziembinski, Chem. Ber., 1989, 122, 1057–1060 CrossRef CAS.
- R. Hunold, J. Allwohn, G. Baum, W. Massa and A. Berndt, Angew. Chem., Int. Ed. Engl., 1988, 27, 961–963 CrossRef.
- M. Pilz, J. Allwohn, P. Willershausen, W. Massa and A. Berndt, Angew. Chem., Int. Ed. Engl., 1990, 29, 1030–1032 CrossRef.
- M. M. Olmstead, P. P. Power, K. J. Weese and R. J. Doedens, J. Am. Chem. Soc., 1987, 109, 2541–2542 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic, electrochemical and crystallographic details. CCDC 948685–948688. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c3cc46947f |
| This journal is © The Royal Society of Chemistry 2014 |