Shining light on integrity of a tetracobalt-polyoxometalate water oxidation catalyst by X-ray spectroscopy before and after catalysis†‡
Rafael
Schiwon
a,
Katharina
Klingan
b,
Holger
Dau
*b and
Christian
Limberg
*a
aHumboldt-Universität zu Berlin, Institut für Chemie, Brook-Taylor-Str. 2, 12489 Berlin, Germany. E-mail: christian.limberg@chemie.hu-berlin.de
bFreie Universität Berlin, FB Physik, Arnimallee 14, 14195 Berlin, Germany. E-mail: holger.dau@fu-berlin.de
First published on 11th November 2013
Abstract
Modification of the Co-oxo cores of cobalt-polyoxometalate water oxidation catalysts is detectable by X-ray absorption spectroscopy (XAS) as demonstrated by comparison of Na10[Co4(H2O)2(PW9O34)2] (1) and Na17[((Co(H2O))Co2PW9O34)2(PW6O26)] (2). XAS reveals the integrity of 1 uncompromised by oxidant-driven water oxidation, which proceeds without formation of catalytic cobalt oxide.
Striving for new solar fuels, the water oxidation reaction is currently considered to be a bottleneck, hampering progress in the development of applicable technologies for the conversion of light into storable fuels.1 Intense research efforts are thus being pursued in order to develop both heterogeneous and molecular catalysts, which enable water oxidation at potentials close to the thermodynamic limit. A breakthrough has recently been achieved in the area of heterogeneous catalysis. Nocera and coworkers reported an in situ formed, self-healing Co-based catalyst (CoCat), which operates at moderate overpotentials as a water oxidation electrocatalyst.2,3 The CoCat is readily formed by electrodeposition from a buffered Co2+ solution as an amorphous film on the electrode surface; its atomic structure is characterized by cobalt oxide units composed of edge-sharing CoO6 octahedra.4 Also homogeneous WOCs are developing rapidly.1,5–9 The majority of the latter are based on (tailor-made) organic ligands which naturally represent the Achilles' heel of such systems. In this context the use of defective polyoxometalate (POM) derivatives as ligands is an interesting approach for the design of WOCs with enhanced stability, because oxidative ligand degradation is not possible. Therefore, the recent report10 that the purely inorganic POM Na10[Co4(H2O)2(PW9O34)2], 1, based on earth-abundant cobalt represents an active WOC with [Ru(bpy)3](ClO4)3 (bpy = tris(2,2′-bipyridyl)) as the oxidant has received significant attention.11–13 To us this system seemed to be an interesting subject for X-ray absorption spectroscopy (XAS) studies as it may be viewed as a molecular and perhaps more defined version of Nocera's heterogeneous CoCat, which some of us have investigated structurally and electronically with the aid of X-ray absorption near-edge structure (XANES) and extended X-ray absorption fine-structure (EXAFS) spectroscopy.4,14 Recently it has been put into question that 1 is stable as such under turnover conditions.11,15 Therefore we decided to investigate the hydrolytic stability and possible catalyst degradation during catalysis by XAS.19
The synthesis of 1 has been performed according to a procedure established previously16 and its identity was confirmed by single-crystal X-ray diffraction (XRD, Fig. S1 and Table S2, ESI‡) and mass spectroscopy (Table S3 and Fig. S2–S8, ESI‡) as well as by XANES and EXAFS. These structural methods were complemented by UV/Vis spectroscopy (Fig. S9–S19, ESI‡).
As there were conflicting reports on the solution stability of 1 even before catalysis,10,11,15 we have monitored its UV/Vis spectrum under various conditions. While 1 (1 mM) is indeed stable in pure water, addition of sodium phosphate (NaPi), used in the previous investigations10 to ensure a pH of 8 (30 mM for solutions being 3.2 μM 1), triggers a slow decrease of its main absorbance band at 580 nm, thus indicating decomposition to some extent. Interestingly, the rate depends on the NaPi concentration: for a 30 mM NaPi solution a band decrease of 2% was observed within 1.5 h while it amounted to 3.5% in the case of 400 mM NaPi. However, very recent results20 suggest that such aging does not play a role in catalytic activity. Stability of the oxidant [Ru(bpy)3](ClO4)3 used has been investigated before, too.17 Albeit not the focus of our investigation, related results obtained by UV/Vis spectroscopy may be of interest and thus are reported herein. In contrast to the rather slow corrosion of 1 in aqueous solution, decomposition of [Ru(bpy)3]3+ in a 30 mM buffer of NaPi is more significant (Fig. S9, ESI‡). Monitoring the UV/Vis absorption maximum of [Ru(bpy)3]3+ at 670 nm with time (Fig. S10, ESI‡) suggests redox-degradation with a t1/2 of ca. 75 s. The product(s) showed a new band at 800 nm.
The addition of a 3.2 μM solution of 1 to a solution being 1.5 mM in [Ru(bpy)3](ClO4)3 and 30 mM in NaPi significantly accelerates the bleaching of the 670 nm band belonging to [Ru(bpy)3]3+ (Fig. S11 and S12, ESI‡), as expected for water oxidation catalyzed by 1, and indeed O2 evolution with a yield of 66.7% has been shown.10 Hence, under these conditions reduction of [Ru(bpy)3]3+ to [Ru(bpy)3]2+ is much faster than degradation.
For investigation of the atomic structure of the WOCs, Co K-edge XAS spectra of the following samples were collected: (a) microcrystalline powder of 1, (b) 3.2 μM of 1, (c) 3.2 μM of 1 with 1.5 mM [Ru(bpy)3]3+, (d) 1 mM of 1, (e) 1 mM of 1 with 8 mM [Ru(bpy)3]3+ (b–e, 1 in aqueous solutions being 30 mM in NaPi, pH 8). All samples were frozen in liquid nitrogen and spectra were collected at 20 K. The oxidant-containing samples were not immediately frozen but after water oxidation catalysis.
XANES analysis was employed to investigate 1 in aqueous solution at the same low concentration (3.2 μM) used previously for assessment of its catalytic performance.10 A clearly higher concentration of 1 (1 mM) facilitated collection of high-quality EXAFS spectra. To verify the ability of XAS to detect structural changes in the cobalt oxo core of 1, in addition the known Co-POM Na17[((Co(H2O))Co2PW9O34)2(PW6O26)], 2, with a similar but somewhat different metal oxo core (ref. S5, ESI†) was synthesized and, after verification of its structure by XRD, investigated by XAS in form of a microcrystalline powder.
The XANES spectra are shown in Fig. 1. Visual inspection immediately reveals that for 1 dissolved at low and high concentration (3.2 μm and 1 mM), the spectra recorded before and after the RuIII-driven oxygen-evolving reaction (OER) are virtually identical. Moreover, these four solution spectra are highly similar to the spectrum of the microcrystalline powder of 1, but differ slightly from the spectrum of 2 (absence of a shoulder at 7730 eV in 2). We also compared 1 to two conceivable products of hydrolytic decomposition or oxidative transformation: (I) [Co(H2O)6]2+ and (II) the CoCat. The XANES spectra of I and II differ pronouncedly from the spectra of 1. In conclusion, already the visual comparison of the XANES suggests that hydrolytic or oxidative transformation of 1 does not take place to an extent detectable by XANES.
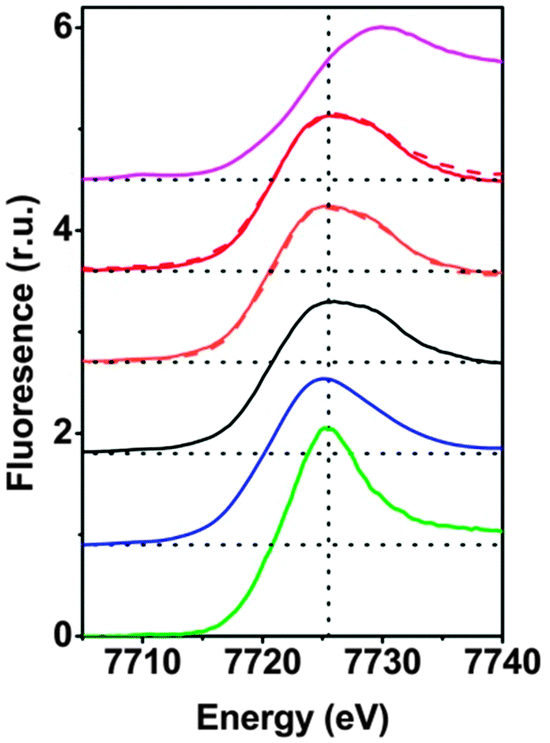 | ||
| Fig. 1 Cobalt K-edge XANES spectra. (i) Heterogeneous CoIII-oxide catalyst (CoCat, magenta), (ii) 3.2 μM of 1 with 1.5 mM [Ru(bpy)3]3+ (red dashed line), (iii) 1 mM of 1 with 8 mM [Ru(bpy)3]3+ (red), (iv) 3.2 μM of 1 (orange dashed line), (v) 1 mM of 1 (orange); (vi) microcrystalline powder of 1 (black), (vii) microcrystalline powder of 2 (blue), (viii) [CoII(H2O)6](NO3)2 in aqueous solution (green). In (ii–v), 1 and [Ru(bpy)3]3+ were mixed in an aqueous phosphate buffer (30 mM NaPi). All samples were frozen in liquid nitrogen before XAS data collection (at 20 K). We note that the spectra (ii)–(v) are virtually identical and highly similar to (vi). | ||
The K-edge positions on the energy axis are influenced by the oxidation state of the respective transition metal. An empirical relationship between the integral edge position18 and the oxidation state facilitates estimation of cobalt ion oxidation states of cobalt oxides and related Co-compounds.14 The edge positions of 1 before dissolution, after dissolution and after catalysis are, within the error limit, identical (Table S5, ESI‡) and confirm a Co oxidation state of +2, as in the crystallographically characterized form of 1. The estimated edge position of CoCat is close to +3, as reported before;4 the oxidation state of the Co2+ hexaaquo complex was estimated to be 2.0 ± 0.1 as expected. Thus we conclude that the oxidation state of 1 is conserved upon dissolution of the microcrystalline powder and upon water oxidation using [Ru(bpy)3]3+. We note that the latter finding does not exclude that higher cobalt oxidation states are formed during water oxidation because the herein investigated samples were frozen at a point where water oxidation had come to an end because of exhaustion of the [Ru(bpy)3]3+ oxidant.
Whereas the XANES region is sensitive mostly to oxidation states and the coordination geometry in the first coordination sphere, the EXAFS region of the spectrum can provide specific structural information on the first, second, third and higher coordination spheres. Fig. 2 compares Fourier-transformed (FT) EXAFS spectra; the x-axis indicates the reduced distance between the X-ray-absorbing Co ion and ‘backscattering atoms’ of the first and higher coordination spheres. The indicated reduced distances are by about 0.4 Å shorter than the real distances obtained by EXAFS simulations and presented in Table 1 as well as Table S4 (ESI‡). The five prevalent FT-peaks (a–e) of the POM samples are assigned to atom pairs as shown in Fig. 3.
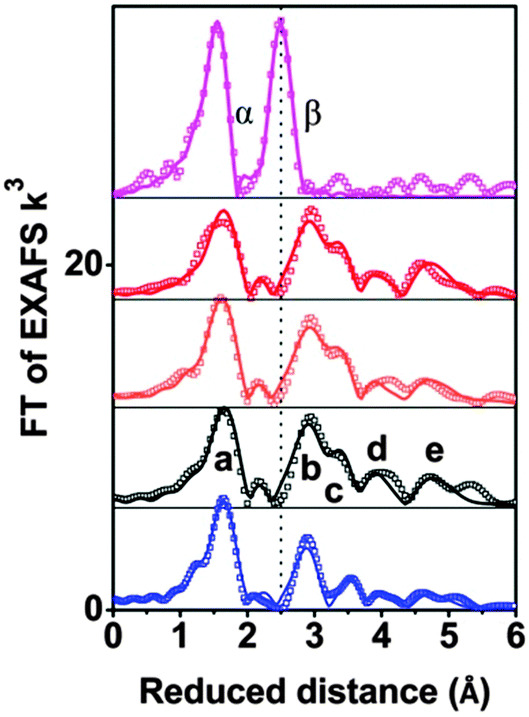 | ||
| Fig. 2 Fourier-transformed EXAFS spectra of CoCat (deposited in KPi, magenta), 2 powder (blue), 1 powder (black), 1 mM 1 in 30 mM NaPi (orange); and 1 mM 1 and 8 mM [Ru(bpy)3]3+ in 30 mM NaPi (red) are shown. Experimental data are indicated by symbols and EXAFS simulations (parameters given in Table 1) are shown as lines. Note that the distance on the x-axis is reduced by 0.3–0.4 Å relative to the real distance. Peaks (a–e) as well as α and β correspond to distance vectors shown in Fig. 2. | ||
| Sample | Co–O | |||
|---|---|---|---|---|
| N O | R O (Å) | σ O (Å) | R f | |
| CoCat | 6.0 ± 0.3 | 1.89 ± 0.01 | 0.064 ± 0.002 | 17 |
| 1, [Ru(bpy)3]3+ | 5.9 ± 0.8 | 2.05 ± 0.01 | 0.081* | 20 |
| 1 (l) | 6.7 ± 0.8 | 2.04 ± 0.01 | 0.081* | 20 |
| 1 (s) | 6.0 ± 0.8 | 2.05 ± 0.01 | 0.081* | 20 |
| 2 (s) | 6.5 ± 0.4 | 2.05 ± 0.01 | 0.081* | 22 |
| Sample | Co–Co | |||
|---|---|---|---|---|
| N Co | RCo (Å) | σ Co (Å) | R f | |
| CoCat | 4.3 ± 0.3 | 2.81 ± 0.01 | 0.068 ± 0.002 | 17 |
| 1, [Ru(bpy)3]3+ | 2.4 ± 0.4 | 3.19 ± 0.01 | 0.044* | 20 |
| 1 (l) | 2.5 ± 0.4 | 3.19 ± 0.01 | 0.044* | 20 |
| 1 (s) | 2.5 ± 0.4 | 3.19 ± 0.01 | 0.044* | 20 |
| 2 (s) | 1.5 ± 0.2 | 3.18 ± 0.01 | 0.044* | 22 |
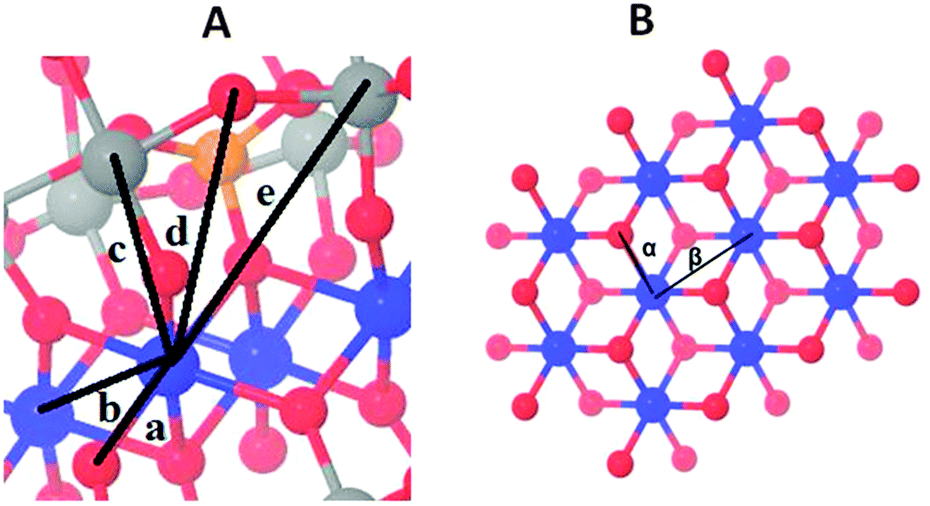 | ||
| Fig. 3 Co oxo core and distance vectors in 1 (A) and the Co-based heterogenous catalysts (CoCat, B). The distances resolved by EXAFS analysis between the X-ray absorbing Co ion and neighbouring atoms are shown for 1 (a–e in panel A) and the CoCat (α and β in panel B) (Co blue, O red, P orange, and W grey). | ||
Visual comparison of the EXAFS spectra (Fig. 2) as well as the structural parameters determined by EXAFS simulation (Table 1) reveal that neither dissolving 1 in aqueous buffer nor oxidant exposure and water oxidation catalysis causes structural modification. The comparison with CoCat reveals that the Co–Co peak distance of 2.81 Å typical of edge-sharing octahedra in CoCat (FT peak labeled as β) is fully absent for 1, before and after catalysis of water oxidation.
Comparison of 1 and 2 illustrates the high sensitivity of the EXAFS spectra to structural changes within Co oxo cores: the two POMs only differ in the number of Co atoms interconnected by oxo-bridges and sandwiched between oxo tungstates (four Co in 1versus three Co in 2, see Fig. S20, ESI‡). Consequently the number of Co–Co vectors per X-ray absorbing Co ion is 2.5 in 1 and 1.5 in 2. The EXAFS simulations reproduce the predicted difference in the coordination number perfectly well (NCo–Co of 1.5 in 2vs. 2.5 in 1, Table 1). We conclude that for Co4-POM 1 even the loss of a single Co ion would be detectable by qualitative comparison of EXAFS spectra (i.e., visual comparison of Fourier-transformed spectra) and quantitative analysis (that is, determination of structural parameters by curve-fitting).
In conclusion, the EXAFS analysis corroborates the absence of hydrolytic or oxidative transformation of 1. Recently it was shown that electrocatalytic water oxidation in the presence of 1 involves its partial decomposition and release of Co2+ followed by deposition of a thin CoCat oxide film on the working electrode; not 1 but the amorphous Co oxide was suggested to be the WOC.11 Avoiding relatively harsh electrocatalytic conditions and using a chemical oxidant at neutral pH, there are no indications of formation of a catalytically active Co oxide. We cannot exclude that, e.g., 5% of the Co ions are transformed into CoCat-type oxides. If these 5% of the Co ions were responsible for water oxidation catalysis, their TOF would exceed 2 s−1, though, whereas the TOF for the CoCat is clearly below 0.1 s−1.1 Thus it is highly unlikely that a minority fraction of Co ions present in oxide form could be responsible for WOC. A further recent proposal has been hydrolytic fragmentation of 1 into soluble species,15 which then perform the catalysis. If so, these fragments reassemble to give back 1 after catalysis, according to our results or would exhibit an extreme activity.
The herein presented evidence suggests that Co-POMs, which are composed exclusively of chemical elements of relatively high abundance, can be stable under catalytic conditions and thus are indeed promising WOCs.
This work has been pursued within the framework of the Berlin cluster of excellence on ‘Unifying Concepts in Catalysis’ (UniCat).
Notes and references
- H. Dau, C. Limberg, T. Reier, M. Risch, S. Roggan and P. Strasser, ChemCatChem, 2010, 2, 724–761 CrossRef CAS.
- M. W. Kanan and D. G. Nocera, Science, 2008, 321, 1072–1075 CrossRef CAS PubMed.
- S. Y. Reece, J. A. Hamel, K. Sung, T. D. Jarvi, A. J. Esswein, J. J. H. Pijpers and D. G. Nocera, Science, 2011, 334, 645–648 CrossRef CAS PubMed.
- M. Risch, V. Khare, I. Zaharieva, L. Gerencser, P. Chernev and H. Dau, J. Am. Chem. Soc., 2009, 131, 6936–6937 CrossRef CAS PubMed.
- X. Sala, I. Romero, M. Rodríguez, L. Escriche and A. Llobet, Angew. Chem., Int. Ed., 2009, 48, 2842–2852 CrossRef CAS PubMed.
- R. Brimblecombe, G. C. Dismukes, G. F. Swiegers and L. Spiccia, Dalton Trans., 2009, 9374–9384 RSC.
- M. Yagi, A. Syouji, S. Yamada, M. Komi, H. Yamazaki and S. Tajima, Photochem. Photobiol. Sci., 2009, 8, 139–147 CAS.
- C. W. Cady, R. H. Crabtree and G. W. Brudvig, Coord. Chem. Rev., 2008, 252, 444–455 CrossRef CAS PubMed.
- M. R. g. I. Romero, C. Sens, J. Mola, M. R. Kollipara, L. Francás, E. Mas-Marza, L. Escriche and A. Llobet, Inorg. Chem., 2008, 47, 1824 CrossRef PubMed.
- Q. Yin, J. M. Tan, C. Besson, Y. V. Geletii, D. G. Musaev, A. E. Kuznetsov, Z. Luo, K. I. Hardcastle and C. L. Hill, Science, 2010, 328, 342–345 CrossRef CAS PubMed.
- J. J. Stracke and R. G. Finke, J. Am. Chem. Soc., 2011, 133, 14872 CrossRef CAS PubMed.
- D. Lieb, A. Zahl, E. F. Wilson, C. Streb, L. C. Nye, K. Meyer and I. Ivanović-Burmazović, Inorg. Chem., 2011, 50, 9053–9058 CrossRef CAS PubMed.
- C. A. Ohlin, S. J. Harley, J. G. McAlpin, R. K. Hocking, B. Q. Mercado, R. L. Johnson, E. M. Villa, M. K. Fidler, M. M. Olmstead, L. Spiccia, R. D. Britt and W. H. Casey, Chem.–Eur. J., 2011, 17, 4408 CrossRef CAS PubMed.
- M. Risch, K. Klingan, F. Ringleb, P. Chernev, I. Zaharieva, A. Fischer and H. Dau, ChemSusChem, 2012, 5, 542–549 CrossRef CAS PubMed.
- M. Natali, S. Berardi, A. Sartorel, M. Bonchio, S. Campagna and F. Scandola, Chem. Commun., 2012, 48, 8808–8810 RSC.
- R. G. Finke, M. W. Droege and P. J. Domaille, Inorg. Chem., 1987, 26, 3886–3896 CrossRef CAS.
- C. Creutz, H. A. Schwarz and N. Sutin, J. Am. Chem. Soc., 1984, 106, 3036–3037 CrossRef CAS.
- H. Dau, P. Liebisch and M. Haumann, Anal. Bioanal. Chem., 2003, 376, 562–583 CrossRef CAS PubMed.
- For XAS on POM WOC also see: S. Piccinin, A. Sartorel, G. Aquilanti, A. Goldoni, M. Bonchio and S. Fabris, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 4917 CrossRef CAS PubMed.
- J. W. Vickers, H. Lv, J. M. Sumliner, G. Zhu, Z. Luo, D. G. Musaev, Y. V. Geletii and C. L. Hill, J. Am. Chem. Soc., 2013, 135, 14110–14118 CrossRef CAS PubMed.
Footnotes |
| † Dedicated to Prof. Dr Werner Uhl on the occasion of his 60th birthday. |
| ‡ Electronic supplementary information (ESI) available: Experimental section and details of the synthesis of 1, 2, [Ru(bpy)3](ClO4)3 and the spectroscopic details of the UV/Vis, EXAFS and XANES experiments. CCDC 943020. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c3cc46629a |
| This journal is © The Royal Society of Chemistry 2014 |