
Graphene oxide modified with aptamer-conjugated gold nanoparticles and heparin: a potent targeted anticoagulant†
Yi-Heng
So
a,
Huan-Tsung
Chang
b,
Wei-Jane
Chiu
a and
Chih-Ching
Huang
*acd
aInstitute of Bioscience and Biotechnology, National Taiwan Ocean University, Keelung, 20224, Taiwan. E-mail: huanging@ntou.edu.tw; Fax: +011-886-2-2462-2320; Tel: +011-886-2-2462-2192 ext. 5517
bDepartment of Chemistry, National Taiwan University, Taipei, 10617, Taiwan
cCenter of Excellence for the Oceans, National Taiwan Ocean University, Keelung, 20224, Taiwan
dSchool of Pharmacy, College of Pharmacy, Kaohsiung Medical University, Kaohsiung, 80708, Taiwan
First published on 23rd July 2014
Abstract
Graphene oxide (GO) modified with 29-mer thrombin-binding-aptamer-conjugated gold nanoparticles (TBA29–Au NPs/GO) can synergistically inhibit the thrombin-mediated cleavage of fibrinogen to fibrin. To further improve anticoagulation efficiency in human plasma, TBA29–Au NPs/heparin/GO has been prepared from TBA29–Au NPs/GO and heparin that can also bind thrombin. The dose-dependence of thrombin clotting time (TCT) delay caused by TBA29–Au NPs/heparin/GO is 21.4, 17.0 and >100 times higher than that caused by the TBA29–Au NPs, TBA29–Au NPs/GO and commercially available drugs (heparin, argatroban, hirudin or warfarin), respectively.
Introduction
Blood coagulation involves a series of enzymatic reactions, largely localized on vascular and cellular surfaces, and results in the formation of a fibrin clot.1 Thrombin (activated factor II) is a multifunctional serine protease produced by the cleavage of prothrombin at two positions by the factor Xa-involved prothrombinase complex (factor Va, negatively charged phospholipid vesicles, and calcium).2 Thrombin plays many vital roles in the coagulation cascade, converting soluble fibrinogen into insoluble strands of fibrin, catalyzing many other coagulation-related reactions, including directly such as activating protein C and platelets, and providing feedback amplification of factors V, VIII, XI, and XII.3 Therefore, thrombin is a very attractive target for anticoagulation factors.Many specific inhibitors have been used for regulating thrombin activity during surgical procedures and for treating patients with arterial or venous thromboembolisms.4 Thrombin (which has a molecular weight of 37 kDa) is a heterodimer consisting of two polypeptide chains, A and B. The catalytic domain in thrombin has a deep cleft around its catalytic site and two large electropositive surfaces, called exosite I (the fibrinogen-binding site) and exosite II (the heparin-binding site), on opposite sides of the active site cleft.5 The biological activity of thrombin is strongly affected by substrates, cofactors, and inhibitors that bind to exosites I and II.6,7 Many direct thrombin inhibitors, which block the enzymatic active site, are currently available for clinical use, but most of them cause serious side effects and suffer from narrow therapeutic windows due to their low specificity and indirect action.8,9 Thus, more efficient and specific anticoagulation agents are required for treating coagulation-related cardiovascular diseases. A 15-mer thrombin-binding-aptamer (TBA15) has been shown to be a potential anticoagulation drug because it can inhibit the activity of thrombin through its interaction with fibrinogen-binding exosite I.10 However, TBA15 at a high concentration is required in order to give an appropriate anticoagulant response because of its low binding affinity. Moreover, oligonucleotide aptamers are easily degraded by blood nucleases and have a short lifetime (∼2 min) in blood, limiting their potential use.11 Conjugation of aptamers through phosphorothioate linkages and by the use of locked-nucleic acids has been shown to be efficient in improving the half-life and binding affinity of aptamers.12 However, toxicity of the expensive non-natural modified aptamers is a concern.
Recent studies have demonstrated that linearly assembled 15-mer thrombin-binding-aptamer (TBA15) and TBA29 (using a poly-dA linker or a polyethylene glycol linker), synthetic dendritic TBAs, and nanostructured TBAs can inhibit thrombin activity efficiently through their multivalent interactions.13–18 More recently, we found that TBA29-conjugated gold nanoparticles (TBA29–Au NPs) have a strong anticoagulant potency (82 times stronger than TBA29) because they cause steric blocking and have high binding affinity for thrombin in a biological plasma mimic.19 In this study, we prepared a TBA29–Au NP functionalized graphene oxide (GO) nanocomposite (TBA29–Au NPs/GO) with the aim of further improving the anticoagulant potency. TBA29 is easily folded to a G-quadruplex structure.20 The TBA29–Au NPs functionalized on GO mainly through the π–π stacking interactions between the square planar guanine tetrad and GO.21–24 The multivalent TBA29 units on Au NPs result in strong cooperative π–π stacking interaction between TBA29–Au NPs and GO. The hydrophobic and the hydrogen bonding interactions between TBA29 and GO could not be excluded.25 TBA29–Au NPs/GO can bind to thrombin effectively to inhibit the activity of thrombin to catalyze the cleavage of fibrinogen to form fibrin (Scheme 1a). The TBA29–Au NPs/GO in comparison with TBA29–Au NPs provided higher inhibitory potency toward thrombin, mainly because of the synergistic effects of TBA29–Au NPs and GO. The anticoagulant efficiency of TBA29–Au NPs/GO was 3.7 times stronger than that of TBA29–Au NPs in a mimic physiological solution. We found that heparin-modified TBA29–Au NPs/GO (TBA29–Au NPs/heparin/GO; Scheme 1b) had an ultrahigh anticoagulation potency toward thrombin in plasma. We measured the dose-dependence of the thrombin clotting time (TCT) delay, showing that the as-prepared TBA29–Au NPs/heparin/GO had a much stronger anticoagulation potency than that of TBA29–Au NPs/GO and commercially available drugs (heparin, argatroban, hirudin, and warfarin).
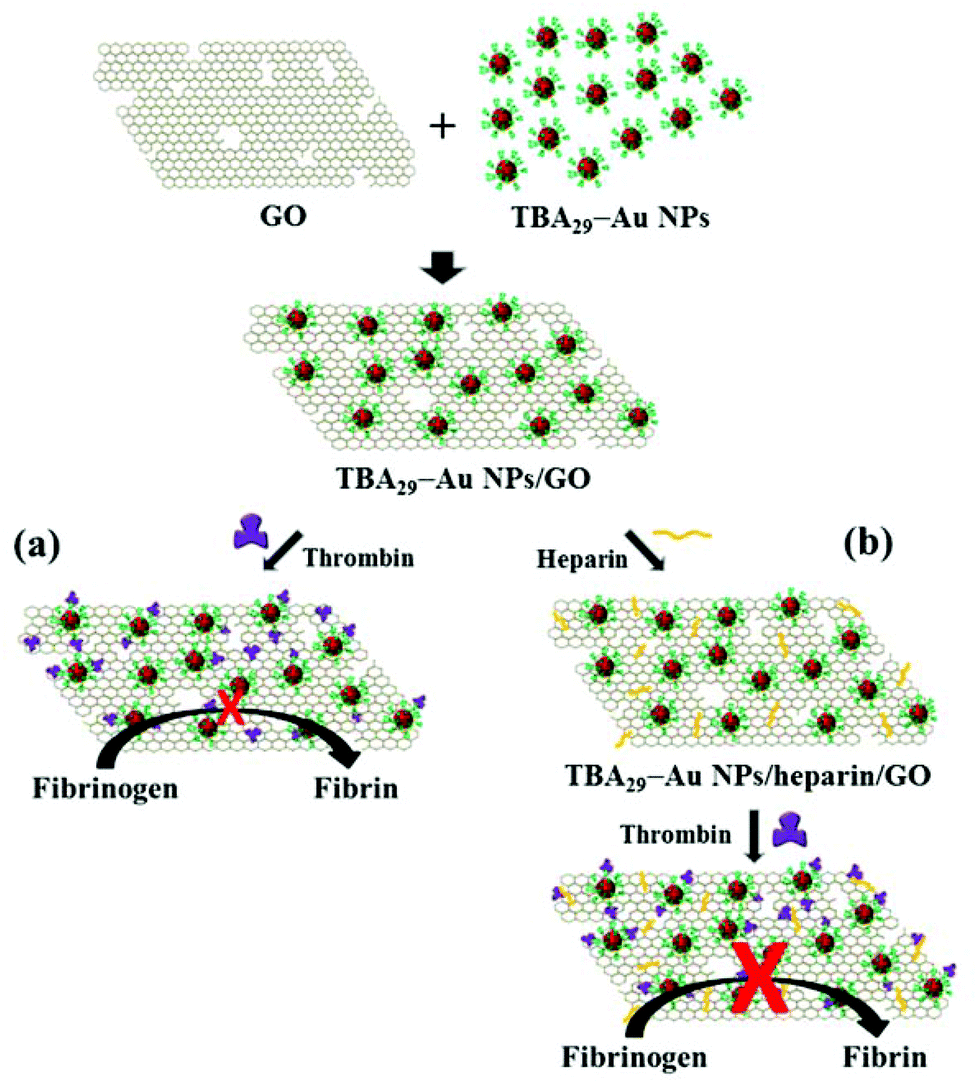 | ||
| Scheme 1 Schematic of the preparation of (a) graphene oxide (GO) modified with 29-mer thrombin-binding aptamer-conjugated gold nanoparticles (TBA29–Au NPs) and (b) GO modified with TBA29–Au NPs and heparin, showing enhanced inhibitory efficiency toward thrombin. | ||
Results and discussion
Preparation and characterization of TBA29–Au NPs/GO nanomaterials
GO was synthesized using an improved Hummers’ method from graphite with a particle size of 7–11 μm.26,27 Transmission electron microscopy (TEM) and atomic force microscopy (AFM) images show that the average size of the single-layer GO is about 400 nm, and the monolayer thickness is about 1.8 nm (Fig. 1a and 1c). From dynamic light scattering (DLS) measurements (Fig. S1†), we estimated the hydrodynamic diameters of GO to be ∼300 nm with a narrow size distribution, indicating that the GO was well dispersed in the solution. As-prepared GO (24 mg L−1) and TBA29–Au NPs (0.5 nM; 13 nm) in a sodium phosphate solution (5 mM; pH 7.0) were used to prepare TBA29–Au NPs/GO. About 90% of the TBA29–Au NPs was determined to be adsorbed onto the GO by comparing the absorbance values at 520 nm of the TBA29–Au NPs in standard solution and in the supernatant after centrifugation (at a relative centrifugation force (RCF) of 1500g for 10 min) of the TBA29–Au NPs/GO mixture. Furthermore, the results of agarose gel separation of the mixtures of TBA29–Au NPs (10 nM) and GO (0–600 mg L−1) indicated that >90% TBA29–Au NPs were loaded on GO when the concentration of GO was higher than 120 mg L−1 (Fig. S2, ESI†). A TEM image of TBA29–Au NPs/GO (Fig. 1b) reveals that the TBA29–Au NPs were assembled homogeneously on the surfaces of GOs. The AFM (Fig. 1d) and UV-vis absorption spectra of TBA29–Au NPs/GO (Fig. S3, ESI†) further confirm that the Au NPs were dispersed (no aggregation) and well-distributed on the surface of each GO. | ||
| Fig. 1 (a and b) TEM images and (c and d) tapping mode AFM images of (a and c) GO and (b and d) TBA29–Au NPs/GO. (e) TEM image of TBA29–Au NPs/heparin/GO. | ||
Effect of TBA29–Au NPs/GO on the inhibition of thrombin activity
Two well-known TBAs, a 15-mer (TBA15) and a 29-mer (TBA29), were used to prepare TBA-modified Au NPs (TBA–Au NPs) previously and employed to detect and control the activity of thrombin.28–30 We demonstrated that TBA29–P8T15–Au NPs (Au NPs conjugated with TBA29 with a linker of 15 thymine (T) and 8 stem pairs (P) at the termini of the TBA29) interact strongly with thrombin (Kd < 0.01 nM) due to its high local concentration of TBA ligands on the particles’ surfaces.28 In addition, we also demonstrated that 15 thymine units in the linker and 8 stem pairs at the termini of the TBA are important to provide stability and flexibility for strong binding with thrombin.28 Here, the descriptor “TBA29–Au NPs” was simply used to represent Au NPs conjugated with approximately 70 TBA29 units presenting T15 and P8. We tested the inhibitory potencies of TBA29, GO, TBA29–Au NPs, and TBA29–Au NPs/GO by performing typical clotting tests using fibrinogen (1.14 μM), bovine serum albumin (BSA; 100 μM) and thrombin (15 nM) under physiological conditions. The sequences of TBA29 and other DNA fragments used in this study are listed in Table S1 (ESI†). The real-time coagulation kinetics of thrombin-induced fibrin formation in mimic physiological solution (25 mM tris(hydroxymethyl)aminomethane (Tris)-HCl at pH 7.4, 150 mM NaCl, 5.0 mM KCl, 1.0 mM MgCl2, and 1.0 mM CaCl2) containing BSA (100 μM) was investigated by measuring the scattered-light intensity caused by fibrin as a function of time (Fig. S4, ESI†). The amount of each oligonucleotide present was kept constant (35 nM). Because of the formation of fibrin-gel, the turbidity and light scattering of the reaction mixtures increased with increasing thrombin activity.29 Through comparing the inhibition potencies of these inhibitors toward thrombin (based on the initial reaction rates, i.e., within 800 s; a high rate reveals a weak inhibitory potency), we found that the potencies decreased in the order: TBA29–Au NPs/GO > TBA29–Au NPs > GO > TBA29 (Fig. S5, ESI†). As controls, the 25-mer random-sequence DNA with a T15 linker (rDNA, 35 nM), rDNA-modified Au NPs (rDNA–Au NPs), rDNA–Au NPs/GO, and BSA-modified Au NPs (BSA–Au NPs) all provide negligible inhibition toward thrombin (data not shown). We also found that TBA15–Au NPs than TBA29–Au NPs and TBA15–Au NPs/GO than TBA29–Au NPs/GO provided much lower inhibitory abilities, mainly because TBA15 than TBA29 has a weaker binding affinity for thrombin.20 Notably, the TBA29–Au NPs/GO inhibition potency (128 cps s−1) was much stronger than that (476 cps s−1) of the TBA29–Au NPs, mainly because of ultrahigh binding ligands present on the surface of GO (Fig. S5, ESI†) and/or synergistic effects of TBA29–Au NPs and GO. Compared with free TBA29 (Kd ∼ 0.5 nM),20 TBA29–Au NPs/GO had a much higher binding affinity for thrombin (Kd = 7.0 × 10−12 M; Fig. S6, ESI†). The binding affinity of TBA29–Au NPs/GO for thrombin was comparable to our previous bivalent TBA15/TBA29–Au NPs (Kd = 8.86 × 10−12).28 The ultrahigh TBA density on the surface of the Au NPs and TBA29–Au NPs on the surface of GO provided high local concentrations of TBA ligands, enhancing the binding affinity for thrombin.19,31 In addition, steric blocking and electrostatic interactions between thrombin and GO (zeta potential ∼ −45 mV; specific surface area ∼2600 m2 g−1) could not be excluded for such a strong inhibition. Thrombin activity was strongly inhibited by unmodified 60 mg L−1 GO (Fig. S7, ESI†) in the absence of background protein (BSA, 100 μM). We believe that these synergistic effects (interactions of thrombin with TBA29 and with GO, and steric effects) are the main contributors to the ultrahigh inhibitory function of TBA29–Au NPs/GO. Compared to GO covalently modified with oligonucleotides,32 the preparation of TBA29–Au NPs/GO is relatively very simple. Highly dense aptamer molecules could be easily loaded on the GO through multivalent base-graphene pi stacking. When the aptamers were covalently bonded to GO, the aptamer molecules most likely existed as flattened structures, leading to weak affinity toward its targeted molecules. However, the TBA29 on the surfaces of the Au NPs/GO provided high flexibility and an appropriate orientation, allowing stronger interaction with thrombin.Thrombin clotting time
We showed that TBA29–Au NPs/GO had an ultrahigh capacity for inhibiting the activity of thrombin in a mimic biological solution. We further tested its capacity in human plasma samples by performing TCT tests. TCT tests are commonly performed on patients suspected of suffering from coagulopathy and are used to screen for factors I, IIa, and XIII.33 We first compared the inhibitory potencies of TBA29–Au NPs/GO, TBA29–Au NPs, and commercial anticoagulant drugs, including three direct thrombin inhibitors (heparin, argatroban, and hirudin) and one indirect thrombin inhibitor (warfarin) by performing TCT assays (Fig. 2 and S8, ESI†). The dose-dependence of the TCT delay indicated that the anticoagulation potency of TBA29–Au NPs/GO in plasma was stronger than that of the four commercial drugs, but only slightly stronger than that of the TBA29–Au NPs. The TCT using TBA29–Au NPs/GO ([TBA29] = 8.75 nM) was 87 ± 12 s, which is longer than the TCT in the absence of the inhibitor (20 ± 3 s), whereas the TCTs were 69 ± 8 s, 23 ± 3 s, 21 ± 2 s, 22 ± 4 s, and 21 ± 5 s for TBA29–Au NPs ([TBA29] = 8.75 nM), heparin (8.75 nM), argatroban (8.75 nM), hirudin (8.75 nM), and warfarin (8.75 nM), respectively. We suggest that the nonspecific binding of the plasma proteins to GO weakened the binding strength between TBA29–Au NPs/GO and thrombin.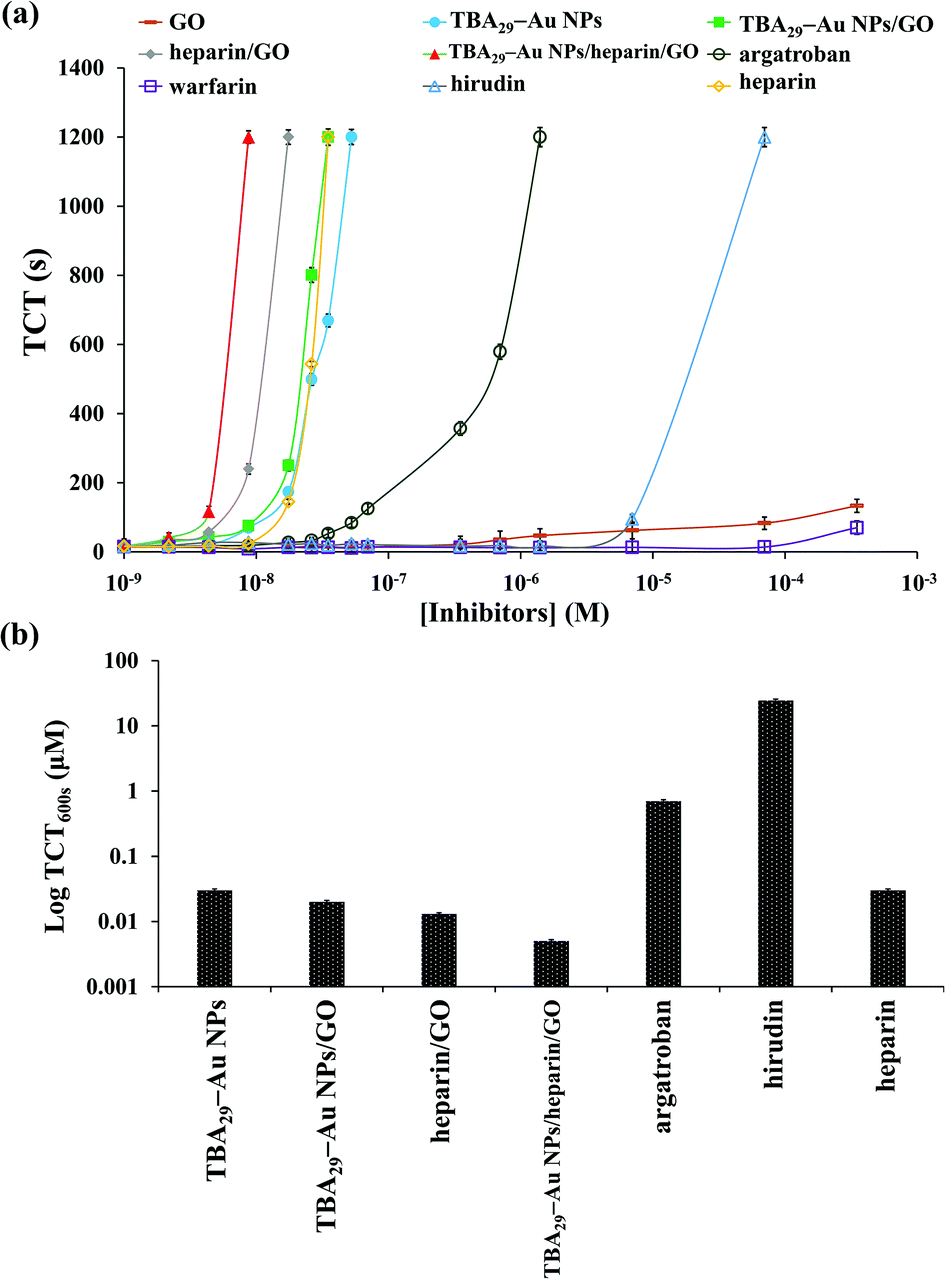 | ||
| Fig. 2 (a) Dose-dependence of the TCTs using commercial drugs (heparin, argatroban, hirudin, and warfarin) and nanomaterials prepared in this study (GO, TBA29–Au NPs, TBA29–Au NPs/GO, heparin/GO, TBA29–Au NPs/heparin/GO) in human plasma samples. The TCT was taken as the point at which the scattering signal was halfway between the lowest and highest points. The longest time of clotting assays monitored was set at 1200 s. (b) TCT delay to 600 s (TCT600 s) of TBA29–Au NPs, TBA29–Au NPs/GO, heparin/GO, TBA29–Au NPs/heparin/GO, argatroban, hirudin, and heparin. The error bars represent the standard deviations of three repeated measurements. Other conditions were the same as those described in Fig. S4.† | ||
Heparin-modified TBA29–Au NPs/GO (TBA29–Au NPs/heparin/GO) was prepared (see the Experimental section for detailed preparation of TBA29–Au NPs/heparin/GO) to try to decrease nonspecific binding and improve the binding affinity. The heparin molecules conjugated with GO probably through multivalent hydrogen bonding and hydrophobic interactions.34,35 Analysis by light scattering experiments revealed that about 95% of the heparin was adsorbed onto the GO, by comparing the concentration of heparin in the supernatant after centrifugation of the TBA29–Au NPs/heparin/GO mixture to that of control solutions. The heparin did not cause aggregation of TBA29–Au NPs/GO after heparin adsorbed on GO (Fig. 1e and S3†). We noted that the TBA29–Au NPs were not desorbed from the GO after heparin adsorbed on GO from the comparison of absorbance values at 520 nm of the TBA29–Au NPs in standard solution and in the supernatant after centrifugation (at an RCF of 1500g for 10 min) of the TBA29–Au NPs/heparin/GO mixture. The inactivation of thrombin and activated factor X (factor Xa) induced by heparin (a sulfated polysaccharide) is through an antithrombin-dependent mechanism.36 The inactivation of thrombin by antithrombin III can increase by nearly three orders of magnitude in the presence of heparin. The binding of thrombin to the highly negatively charged heparin appears to be a predominantly electrostatic interaction at a site proximal to the pentasaccharide of the heparin, with an intrinsic Kd of 5–10 μM.37 The dose-dependence of the TCT delay and TCT delay to 600 s (TCT600 s) (Fig. 2) shows that the anticoagulation ability of TBA29–Au NPs/heparin/GO was not only much higher than that of the four commercial drugs but also much higher than that of TBA29–Au NPs/GO, TBA29–Au NPs, and heparin-modified GO (heparin/GO). The TCTs of TBA29–Au NPs/heparin/GO ([TBA29] = 8.75 nM), heparin/GO ([heparin] = 8.75 nM), TBA29–Au NPs/GO ([TBA29] = 8.75 nM), and TBA29–Au NPs ([TBA29] = 8.75 nM) were approximately 75.0, 15.0, 4.4, and 3.5 times longer (t/t0; t0 is the TCT in the absence of the inhibitor, and t is the TCT in the presence of the inhibitor), respectively, than that of the TCTs in the absence of any inhibitor (Fig. S9, ESI†). The TCT delay caused by TBA29–Au NPs/heparin/GO was 21.4 times higher than that caused by the TBA29–Au NPs and 17.0 times higher than that caused by TBA29–Au NPs/GO. The results clearly show the advantages of using TBA29–Au NPs/heparin/GO over TBA29–Au NPs/GO and TBA29–Au NPs with respect to blood coagulation time. The ultrahigh anticoagulant potency of TBA29–Au NPs/heparin/GO was mainly caused by heparin-passivated GO, decreasing the nonspecific binding between plasma background proteins and GO. In addition, multivalent binding of thrombin with TBA29 and heparin molecules also accounts for enhanced potency. Steric blocking of fibrinogen provided by heparin and TBA29–Au NPs on the GO surfaces cannot be excluded for the obtained stronger inhibitions.
We have further evaluated the anticoagulant capability of TBA29–Au NPs/heparin/GO by the measurement of prothrombin time (PT) and activated partial thromboplastin time (aPTT) in plasma samples. aPTT measurement is a screening test for coagulant factors II, V, VIII, IX, X, XI, and XII of the intrinsic and common pathways, while PT measurement is the test for factors II, V, VII, and X of the extrinsic and common pathways.38,39Fig. 3 shows the comparison of elongation of PT and aPPT by TBA29–Au NPs/heparin/GO and four commercial drugs (heparin, argatroban, hirudin, and warfarin). Prolonging the PT and aPTT (t/t0; t0 and t are the PT or aPPT in the absence and presence of the inhibitor, respectively) in plasma samples by TBA29–Au NPs/heparin/GO, heparin, argatroban, hirudin, and warfarin led to values of 5.9/3.9, 1.5/1.2, 1.0/1.1, 1.1/1.0, and 1.0/1.0 times, respectively. The results further clearly demonstrate the advantages of our TBA29–Au NPs/heparin/GO over commercial drugs with respect to blood coagulation time.
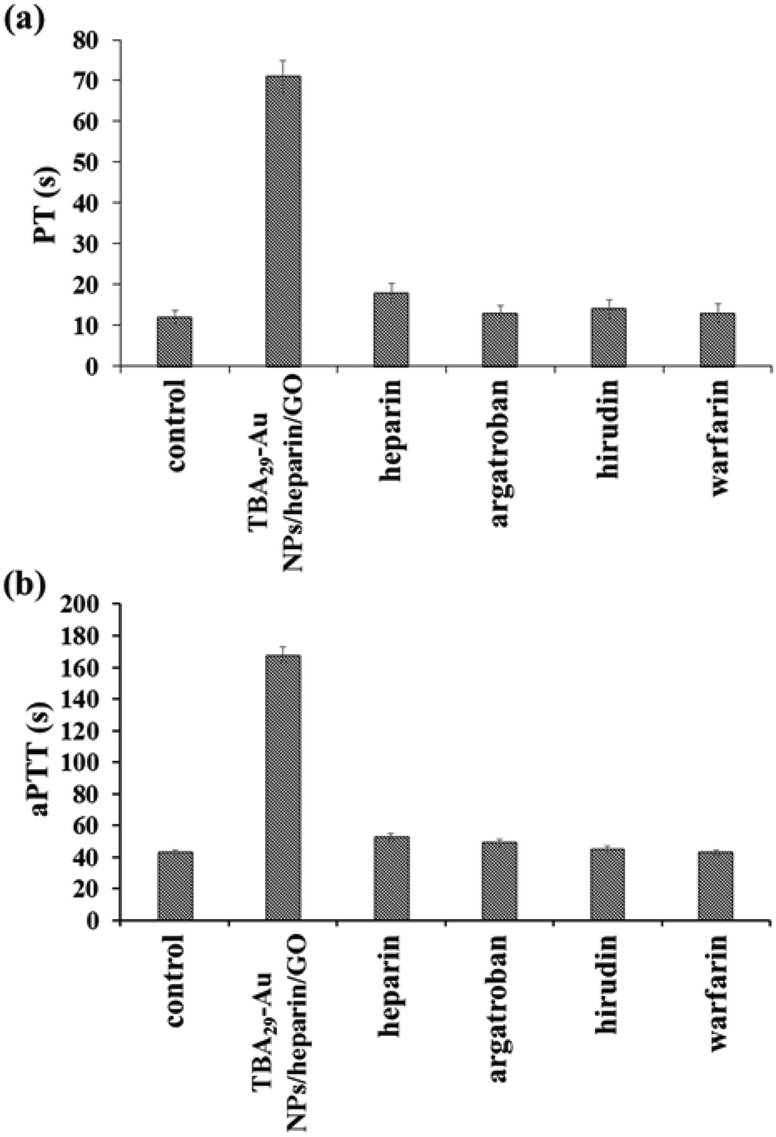 | ||
| Fig. 3 (a) PT and (b) aPTT measurements, comparison of the anticoagulant potency of TBA29–Au NPs/heparin/GO and four commercial drugs (heparin, argatroban, hirudin, and warfarin) in human plasma. The concentrations of TBA29 or commercial drugs in PT and aPPT assays were 105 nM and 35 nM, respectively. To calculate PT and aPTT, the end time was chosen to be the point where the scattering signal reached halfway between the lowest and maximum points. Other conditions were the same as those described in Fig. 2. | ||
Stability of TBA29–Au NPs/heparin/GO
First, we study the long-term stability of TBA29–Au NPs/heparin/GO in physiological buffer by DLS measurements. The DLS results indicated that the TBA29–Au NPs/heparin/GO solution was stable (no aggregation) for at least two months when stored in physiological buffer (Fig. S1d, ESI†). The stability of nucleic acids toward nucleases is also an important factor that affects their therapeutic and diagnostic applications.12,40,41 A major problem with therapeutic oligonucleotide aptamers is their short plasma half-lives, typically of only a few minutes, which usually do not allow time for a sufficient amount of oligonucleotide drug to be delivered to the target site. Prolonging the plasma half-life is a prerequisite for the potential clinical use of a number of drug candidates. We found that the TCT delay for TBA29–Au NPs/heparin/GO was almost constant after 72 h of incubation in the presence of human plasma (Fig. S10, ESI†), revealing that the TBA29–Au NPs and heparin did not release from GO in plasma. Therefore, we suggest that the bleeding risk from the releasing heparin is quite low because the anticoagulation inhibitory potency of TBA29–Au NPs/heparin/GO is >100-fold higher than free heparin (Fig. 2). It has been suggested that high local salt concentrations in the highly negatively charged TBA29–Au NPs/heparin/GO cause increased resistance to nuclease digestion in plasma.30,42 In addition, the steric blocking of nuclease from accessing TBA molecules, caused by heparin molecules, also contributed to the ultrahigh stability of TBA29–Au NPs/heparin/GO in the plasma samples. Nanoparticles smaller than 10 nm will be rapidly cleared by the kidneys; hence, TBA29–Au NPs/heparin/GO particles larger than 10 nm shall have lower renal clearance rates.43 In addition it has been demonstrated that GO nanosheets exhibited long blood circulation time and low uptake in the reticuloendothelial system even if their sizes are larger than 100 nm when compared with other carbon nanomaterials.44 The extraordinary stability of TBA29–Au NPs/heparin/GO suggests that our newly developed anticoagulant may have a long plasma half-life. The small sized GO may be employed for loading of TBA29–Au NPs and heparin if the TBA29–Au NPs/heparin/GO are easy to uptake in the reticuloendothelial system in animal studies in future.45,46Biocompatibility of TBA29–Au NPs/heparin/GO
Recent reports suggest that aptamer-modified Au NPs have good biocompatibility for mammalian cells.47 Although GO could cause some rupture to cell membrane, most studies report that mammalian cell viabilities decrease by less than 20% after exposure to GO concentrations and lower than 10 mg L−1 during 24 h or longer.48 The cytotoxicity of TBA29–Au NPs/heparin/GO towards mammalian cells was evaluated using an Alamar Blue assay. After 24 h of incubation of human embryonic kidney cells (293 T cell line) and breast adenocarcinoma cells (MDA-MB-231 and MCF-7 cell lines) with the TBA29–Au NPs/heparin/GO ([GO] = 0–24 mg L−1), we found that the TBA29–Au NPs/heparin/GO had little influence on cell viability (Fig. S11, ESI†). Therefore, we suggest that the TBA29–Au NPs/heparin/GO have good biocompatibility toward mammalian cells, considering that the TBA29–Au NPs/heparin/GO at the concentration of 24 mg L−1 (GO) show ultrahigh inhibitory ability to thrombin in plasma (Fig. 2). Similarly to TBA29–Au NPs/heparin/GO, TBA29–Au NP/GO had little influence on cell viability when the concentration was lower than 24 mg L−1 (data not shown). The TBA29–Au NPs/heparin/GO and TBA29–Au NPs/GO show very low cytotoxicity in selected human cell lines, probably due to the GO surfaces passivated by highly biocompatible aptamer molecules.We further studied the in vitro hemolysis of TBA29–Au NPs/heparin/GO to verify the satisfactory biocompatibility of TBA29–Au NPs/heparin/GO. The hemolysis assay was performed in defibrinated blood (human) to test the rupture of red blood cells (RBCs) to withstand swelling in contact with the TBA29–Au NPs/heparin/GO solution. The hemolysis of RBCs was not observed after incubation with the TBA29–Au NPs/heparin/GO solution at 37 °C for 4 h (Fig. S12, ESI†), revealing that the TBA29–Au NPs/heparin/GO could serve as potentially safe anticoagulant nanomaterials. The TBA29–Au NPs/heparin/GO exhibit insignificant cytotoxicity to the mammalian cells and hemolysis insinuates the emergence of this nanocomposite as a future putative anticoagulation drug. However, acute and chronic studies using animal models in the future should be conducted to confirm the potential of TBA29–Au NPs/heparin/GO as a viable drug.
Conclusions
We synthesized a nanocomposite TBA29–Au NPs/heparin/GO which acts as a highly effective anticoagulant by controlling the thrombin activity towards fibrinogen. Such a high anticoagulant activity could be due to: (i) the high selectivity of TBA29 towards thrombin that helps heparin to easily access the target and the anticoagulant activity of heparin that leads to the inhibition of thrombin activity, and (ii) the steric hindrance caused by the TBA29–Au NPs to thrombin, minimizing the access of fibrinogen to the active sites of thrombin. More importantly, the easily prepared and highly active TBA29–Au NPs/heparin/GO is an efficient anticoagulant in human plasma, showing its potential for treating coagulation-related cardiovascular diseases. Furthermore, the in vitro cytotoxicity and hemolysis experiment results show that TBA29–Au NPs/heparin/GO has low cytotoxicity and hemolysis effect, revealing that TBA29–Au NPs/heparin/GO could serve as a potentially safe anticoagulant. In addition, this study shows that GO modified with nanomaterials may be useful in regulating molecular function, controlling protein and DNA binding, and manipulating enzyme activities.Acknowledgements
This study was supported by the Ministry of Science and Technology of Taiwan under contracts NSC 101-2628-M-019-001-MY3, 102-2113-M-019-001-MY3, and 102-2627-M-019-001-MY3.Notes and references
- K. A. Tanaka, N. S. Key and J. H. Levy, Anesth. Analg., 2009, 108, 1433 CAS.
- M. S. Chatterjee, W. S. Denney, H. Jing and S. L. Diamond, PLoS Comput. Biol., 2010, 6, e1000950 Search PubMed.
- A. S. Wolberg and R. A. Campbell, Transfus. Apher. Sci., 2008, 38, 15 CrossRef PubMed.
- S. Yates and R. Sarode, Curr. Opin. Hematol., 2013, 20, 552 CrossRef CAS PubMed.
- R. C. Becker and F. A. Spencer, J. Thromb. Thrombolysis, 1998, 5, 215 CrossRef CAS.
- H. Nar, Trends Pharmacol. Sci., 2012, 33, 279 CrossRef CAS PubMed.
- A. Y. Mehta, Y. Jin and U. R. Desai, Expert Opin. Ther. Pat., 2014, 24, 47 CrossRef CAS PubMed.
- E. Schaden and S. A. Kozek-Langenecker, Intensive Care Med., 2010, 36, 1127 CrossRef CAS PubMed.
- D. A. Garcia, T. P. Baglin, J. I. Weitz and M. M. Samama, Chest, 2012, 141, 24 CrossRef PubMed.
- K. Padmanabhan, K. P. Padmanabhan, J. D. Ferrara, J. E. Sadler and A. Tulinsky, J. Biol. Chem., 1993, 268, 17651 CAS.
- I. V. Gribkova, V. A. Spiridonov, A. S. Gorbatenko, S. S. Denisov, F. I. Ataullakhanov and E. I. Sinauridze, Blood Coagul. Fibrinolysis, 2014, 25, 39 CrossRef CAS PubMed.
- A. Avino, C. Fabrega, M. Tintore and R. Eritja, Curr. Pharm. Des., 2012, 18, 2036 CrossRef CAS.
- F. Rohrbach, M. I. Fatthalla, T. Kupper, B. Pötzsch, J. Müller, M. Petersen, E. B. Pedersen and G. Mayer, ChemBioChem, 2012, 13, 631 CrossRef CAS PubMed.
- Y. Kim, D. M. Dennis, T. Morey, L. Yang and W. Tan, Chem. – Asian J., 2010, 5, 56 CrossRef CAS PubMed.
- A. Rangnekar, A. M. Zhang, S. S. Li, K. M. Bompiani, M. N. Hansen, K. V. Gothelf, B. A. Sullenger and T. H. LaBean, Nanomedicine, 2012, 8, 673 CrossRef CAS PubMed.
- N. S. Petrera, A. R. Stafford, B. A. Leslie, C. A. Kretz, J. C. Fredenburgh and J. I. Weitz, J. Biol. Chem., 2009, 284, 20620 CrossRef PubMed.
- Y. Kim, Z. Cao and W. Tan, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 5664 CrossRef CAS PubMed.
- D. Musumeci and D. Montesarchio, Pharmacol. Ther., 2012, 136, 202 CrossRef CAS PubMed.
- Y.-C. Shiang, C.-C. Huang, T.-H. Wang, C.-W. Chien and H.-T. Chang, Adv. Funct. Mater., 2010, 20, 3175 CrossRef CAS.
- D. M. Tasset, M. F. Kubik and W. Steiner, J. Mol. Biol., 1997, 272, 688 CrossRef CAS PubMed.
- J. Liu, Phys. Chem. Chem. Phys., 2012, 14, 10485 RSC.
- L. Tang, Y. Wang, Y. Liu and J. Li, ACS Nano, 2011, 5, 3817 CrossRef CAS PubMed.
- F. Li, H. Pei, L. Wang, J. Lu, J. Gao, B. Jiang, X. Zhao and C. Fan, Adv. Funct. Mater., 2013, 23, 4140 CrossRef CAS.
- P. J. Huang and J. Liu, Small, 2012, 8, 977 CrossRef CAS PubMed.
- S. Akca, A. Foroughi, D. Frochtzwajg and H. W. Ch. Postma, PLoS One, 2011, 6, e18442 CAS.
- D. C. Marcano, D. V. Kosynkin, J. M. Berlin, A. Sinitskii, Z. Sun, A. Slesarev, L. B. Alemany, W. Lu and J. M. Tour, ACS Nano, 2010, 4, 4806 CrossRef CAS PubMed.
- W. S. Hummers Jr. and R. E. Offeman, J. Am. Chem. Soc., 1958, 80, 1339 CrossRef.
- C.-L. Hsu, S.-C. Wei, J.-W. Jian, H.-T. Chang, W.-H. Chen and C.-C. Huang, RSC Adv., 2012, 2, 1577 RSC.
- C.-L. Hsu, H.-T. Chang, C.-T. Chen, S.-C. Wei, Y.-C. Shiang and C.-C. Huang, Chem. – Eur. J., 2011, 17, 10994 CrossRef CAS PubMed.
- Y.-C. Shiang, C.-L. Hsu, C.-C. Huang and H.-T. Chang, Angew. Chem., Int. Ed., 2011, 123, 7802 CrossRef.
- C. Zhang, J. Ma, J. Yang, S. Liu and J. Xu, Anal. Chem., 2013, 85, 11973 CrossRef CAS PubMed.
- V. Georgakilas, M. Otyepka, A. B. Bourlinos, V. Chandra, N. Kim, K. C. Kemp, P. Hobza, R. Zboril and K. S. Kim, Chem. Rev., 2012, 112, 6156 CrossRef CAS PubMed.
- V. Ignjatovic, Methods Mol. Biol., 2013, 992, 131 Search PubMed.
- D. Y. Lee, Z. Khatun, J. H. Lee, Y. K. Lee and I. In, Biomacromolecules, 2011, 12, 336 CrossRef CAS PubMed.
- Y. Wang, P. Zhang, C. F. Liu, L. Zhan, Y. F. Lia and C. Z. Huang, RSC Adv., 2012, 2, 2322 RSC.
- R. D. Rosenberg, Am. J. Med., 1989, 87, 2 CrossRef.
- J. P. Sheehan and J. E. Sadler, Proc. Natl. Acad. Sci. U. S. A., 1994, 91, 5518 CrossRef CAS.
- E. Ejlersen, T. Melsen, J. Ingerslev, R. B. Andreasen and H. Vilstrup, Scand. J. Gastroenterol., 2001, 36, 1081 CrossRef CAS.
- R. D. Langdell, R. H. Wagner and K. M. Brinkhous, J. Lab. Clin. Med., 1953, 41, 637 CAS.
- R. E. Wang, H. Wu, Y. Niu and J. Cai, Curr. Med. Chem., 2011, 18, 4126 CrossRef CAS.
- M. A. Campbell and J. Wengel, Chem. Soc. Rev., 2011, 40, 5680 RSC.
- D. S. Seferos, A. E. Prigodich, D. A. Giljohann, P. C. Patel and C. A. Mirkin, Nano Lett., 2009, 9, 308 CrossRef CAS PubMed.
- B. Wang, X. He, Z. Zhang, Y. Zhao and W. Feng, Acc. Chem. Res., 2013, 46, 761 CrossRef CAS PubMed.
- X. Zhang, J. Yin, C. Peng, W. Hu, Z. Zhu, W. Li, Q. Fan and Q. Huang, Carbon, 2011, 49, 986 CrossRef CAS PubMed.
- J. T. Robinson, S. M. Tabakman, Y. Liang, H. Wang, H. S. Casalongue, D. Vinh and H. Dai, J. Am. Chem. Soc., 2011, 133, 6825 CrossRef CAS PubMed.
- H. Zhang, C. Peng, J. Yang, M. Lv, R. Liu, D. He, C. Fan and Q. Huang, ACS Appl. Mater. Interfaces, 2013, 5, 1761 CAS.
- L. Yang, X. Zhang, M. Ye, J. Jiang, R. Yang, T. Fu, Y. Chen, K. Wang, C. Liu and W. Tan, Adv. Drug Delivery Rev., 2011, 63, 1361 CrossRef CAS PubMed.
- A. M. Pinto, I. C. Gonçalves and F. D. Magalhães, Colloids Surf., B, 2013, 111, 188 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: DNA sequences, size measurements by DLS, gel separations, UV-vis absorption of TBA29–Au NPs/GO, initial coagulation reaction rates, plots for calculating the dissociation constant Kd, inhibition of thrombin by unmodified graphene oxide, representative scattering intensity as a function of time, dose-dependent delay in the TCT (t/t0), stability of TBA29–Au NPs/GO/heparin in plasma, cytotoxicity and hemolysis assays. See DOI: 10.1039/c4bm00156g |
| This journal is © The Royal Society of Chemistry 2014 |