Alteration of epigenetic program to recover memory and alleviate neurodegeneration: prospects of multi-target molecules
Ganesh N.
Pandian
a,
Rhys D.
Taylor
b,
Syed
Junetha
b,
Abhijit
Saha
b,
Chandran
Anandhakumar
b,
Thangavel
Vaijayanthi
b and
Hiroshi
Sugiyama
*ab
aInstitute for Integrated Cell-Material Sciences (iCeMS), Kyoto University, Sakyo, Kyoto 606-8502, Japan
bDepartment of Chemistry, Graduate School of Science, Kyoto University, Sakyo, Kyoto 606-8501, Japan. E-mail: hs@kuchem.kyoto-u.ac.jp; Fax: +81-75-753-3670; Tel: +81-75-753-4002
First published on 9th June 2014
Abstract
Epigenetic chromatin remodeling and signalling pathways play an integral role in transcription dependent neurodegeneration and long-term potentiation (LTP), a cellular model associated with learning and memory. Pathological epigenetic modifications associated with neurological disorders are inherently flexible and can be reversed through pharmacological intervention. Small molecules are the favored drugs for clinicians, and in neurological disorders associated with complex cellular mechanisms, epigenetic and/or signalling pathway enzymes inhibiting small molecules have shown clinical prospects. Recently, small molecules with two or more functionalities, such as sequence-specific recognition and signalling pathways and/or enzyme modulation, have shown capabilities as efficient transcriptional activators. Here, we give a balanced overview of the key factors associated with memory recovery and neurodegeneration, available chemical tools for modulation and the demand to develop next-generation small molecules with multi-functional activities to treat such intricate, multi-gene associated neurological disorders.
 (From left to right) – Abhijit Saha, Chandran Anandhakumar, Ganesh N. Pandian, Syed Junetha; seated (left to right) – Rhys D. Taylor, Hiroshi Sugiyama, Vaijayanthi Thangavel | Ganesh N. Pandian received his PhD in Biotechnology in 2009 from Niigata University with Monbukagakusho (Japanese Government) scholarship under the guidance of Prof. Hidetaka Hori. He continued his research with Prof. Hori as Assistant Professor (Research). Since 2010, he has been working at the Institute for Integrated Cell-Material Sciences (WPI-iCeMS), Kyoto University. Ganesh is now an Assistant Professor in the Sugiyama lab where he aims to develop artificial genetic switches for cell control. Rhys D. Taylor received his Masters in Chemistry from the University of Manchester, UK. He is currently a doctoral student in the Sugiyama Lab, Kyoto University. His research work focuses on the development of pyrrole–imidazole polyamide conjugates for selective gene suppression. Junetha Syed graduated with her Masters degree in Microbial Gene Technology from Madurai Kamaraj University, India, in 2009. Later, she joined as a Project Associate in the Department of Biotechnology, Indian Institute of Technology, Madras, and her research focus was on cancer biology. She is now a doctoral student under the guidance of Prof. Hiroshi Sugiyama in the Department of Chemistry, Kyoto University and her research interest is on epigenetics in cancer. Abhijit Saha received his Masters in organic chemistry from Cotton College, India, in 2008. He joined the Indian Institute of Technology, Guwahati, in 2009 as a Junior Research Fellow, working on peptide synthesis. Now he is a doctoral course student in Prof. Sugiyama’s Group. His research interests are the synthesis and biological evaluation of small molecules for epigenetic control, DNA photochemistry, and electron transfer. Chandran Anandhakumar received his Masters in Marine Genomics from Madurai Kamaraj University, India. Then, he joined Madras Veterinary College as a Senior Research Fellow. He has now joined Professor Sugiyama's Chemical Biology group at Kyoto University as a Doctoral student, studying global gene expression profiles in small molecule-mediated cellular reprogramming. Vaijayanthi Thangavel obtained her PhD in Chemistry at the Indian Institute of Technology-Madras, Chennai, India, in 2008 under the supervision of Prof. Anju Chadha. She then conducted post-doctoral research in the group of Prof. Andreas S. Bommarius at the Georgia Institute of Technology, Atlanta, USA. She continued her research as special assistant professor (Research) in the group of Prof. Hori Hidetaka, Niigata University, Japan. Since 2011 she has been doing her postdoctoral research with Prof. Hiroshi Sugiyama, Kyoto University, on the development and applications of pyrrole–imidazole polyamide–fluorophore conjugates. Hiroshi Sugiyama received his PhD in 1984, studying under Teruo Matuura at Kyoto University. After postdoctoral studies at the University of Virginia with Sidney M. Hecht, he returned to Kyoto University in 1986 as an Assistant Professor and became an Associate Professor in 1993. In 1996, he joined the Institute of Biomaterials and Bioengineering at Tokyo Medical and Dental University as a Professor. He has been Professor of chemical biology at Kyoto University since 2003. Among the honors he has received are the Nippon IBM Award and the Chemical Society of Japan Award for Creative Work. |
1. Introduction
More small molecules enter the clinic as new drugs than any other drugs such as vaccines, peptides, and siRNA.1 Modern biological and analytical techniques assist us to obtain a huge amount of biological information, which can be harnessed to devise novel therapeutic strategies for some complex disorders such as neurological diseases.2 Information is not only a powerful but also a critical resource that needs meticulous management and regulation. In living systems, DNA serves as the informational molecule, which can store and retrieve the genetic instructions governing the development and characteristics of all known living organisms.3,4 The term ‘memory’ could be defined as the process by which the information gets encrypted, accumulated, and recovered. From a functional point of view, memory is relevant to DNA because both encode, store and retrieve information and function as rewritable data storage in living cells.5 Depending on the mode, duration and capacity of storage, memory can be broadly classified into three types: (1) sensory memory, (2) short-term memory and (3) long-term memory.6 Long-term memory and its storage at the cellular level are associated with gene transcription/translation. Also, studies using protein synthesis inhibitors have further verified this phenomenon, as only the enduring memories were blocked.7 Cognitive processes like learning and memory are often associated with Long-term potentiation (LTP), a process that modulates the changes in neural synapses by enhancing the signal strength between neurons.8 Based on the distinctive molecular mechanisms, LTP gets categorized into two phases: the transcription independent early phase, which lasts for about 3 hours, and the transcription/translation-dependent late phase, which can last up to several weeks under in vivo conditions.9 Interestingly, this transcription dependency aspect of LTP is analogous to memory. N-Methyl-D-aspartate (NMDA) and its action on its receptors steer the influx of calcium and initiate phosphorylation of ERK (extracellular signal-regulated kinase), which in turn induces and maintains LTP.9 Studies with various histone deacetylase (HDAC) inhibitors and neural development have shown that in most cases, enrichment of the total acetylation rates in the brain can lead to an increased rate of transcription, which in turn can aid the memory process.10,11 CREB, a cAMP response element and transcriptional activator, binds with the transcriptional co-activator CBP (CREB-binding protein) to form a CREB:CBP complex for the activation of the target hippocampus-dependent, long-term memory associated genes.12 Haettig et al. implied the essential role of CBP through studies in mouse behavioural models, where the HDAC inhibition was shown to modulate long-term memory for object recognition.13 Their studies, with systemic delivery of a HDAC inhibitor, also suggested that different chromatin-modifying enzymes control distinctive memory-related genes in different brain regions. Although neurons are non-proliferating and terminally differentiated cells, the cellular proliferation- and differentiation-associated proteins and protein kinase cascades were shown to be vital for long-term memory formation.9,14–17 Recent developments in functional genomics reveal the essential role of noncoding RNAs (ncRNAs) in the regulation of chromatin remodelling, which controls the transcriptional programs conferring to LTP.18 The ncRNAs are generally accepted to contribute towards the intricacy and function of the mammalian brain.19 The ncRNAs play a vital role in LTP and are found more predominantly in the nervous system.20 For example, Scott et al. demonstrated through expression and functional deficit studies that miR-132 could co-ordinate the intricate transcriptional machinery associated with the synaptic mechanisms controlling memory formation.21 Thus, despite extensive available literature, it is hard to generalize cellular and molecular mechanisms governing the inception, consolidation, and retention of memory. The existing information regarding memory-associated mechanisms has been acquired through studies on neurological disorders.22 Several studies with HDAC-inhibiting small molecules have substantiated the essential role of epigenetics and transcriptional activation in memory formation and neurological disorders.23 Because alterations in epigenetic programming are merely a small part of a huge complicated interaction of signalling pathways controlling the mechanism of LTP expression, a comprehensive overview of the molecular mechanisms underlying LTP is beyond the scope of this review.In this review, we give a brief overview of the epigenetic aspects of transcription dependent LTP, an underlying cellular phenomenon associated with several neurological disorders. Also, by summarizing the potential of individual available bioactive small molecules, we suggest the importance of developing next generation small molecules with multiple activities such as (i) HDAC inhibition, (ii) signaling factor inhibition, (iii) sequence-specific recognition, and (iv) other such biologically significant properties. Development of such doubly active or multi-target small molecules can have a significant impact in elucidating the molecular mechanisms such as LTP, which in turn may lead to effective treatment for complex neurological disorders.
2. Long-term potentiation and memory
2.1. Long-term potentiation – a brief overview
Neurons are the fundamental building blocks of the nervous system, which manages the intricate organization of how humans sense, perceive, and act. Santiago Ramón y Cajal, an eminent neuroanatomist,24 first proposed that memories could be formed by improving the efficacy of neural communication through strengthening of the connections between existing neurons. In 1949, Donald Hebb reiterated this view and proposed a theory that cells develop fresh connections or undergo metabolic changes to enhance their communication capacity.25 In 1966, Terje Lømo discovered the process of long-term potentiation through a series of neurophysiological experiments on anesthetized rabbits and explored the relationship between short-term memory and the role of the hippocampus, a region long known to be essential for learning and memory.26 Subsequent reports characterized this phenomenon, and in 1975 Douglas and Goddard termed this phenomenon “long-term potentiation or LTP”.27 LTP occurs in other neural structures like the cerebral cortex, cerebellum and amygdala in the mammalian brain.28 Since different areas of the brain exhibit different forms of LTP, many factors such as age and anatomical location influence the specific type of LTP.29 For studies on mammalian LTP, the CA1 hippocampus gets preference, owing to its predictable organization and readily inducible LTP.28 LTP gains prominence over other kinds of synaptic plasticity, owing to its ability to persist and last from several minutes to months.30 Activation of NMDA receptors in hippocampal neurons trigger early phase LTP through the activation of calcium-dependent kinases, and involves an increase in the synaptic AMPA (α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid) receptor function.31 While early phase LTP is independent of protein synthesis, late phase LTP requires the persistent activity of atypical protein kinase C isoform (PKMζ) for active synaptic transmission in the central nervous system and to maintain an increased amount of AMPA receptors at potentiated synapses to consolidate memory.32 Transcription factors like Zif268 are suggested to trigger the signalling of PKMζ gene activation.33 Whitlock et al. demonstrated through inhibitory avoidance learning experiments in rats that learning induces similar changes in hippocampal glutamate receptors, like those observed in high frequency stimulation-induced LTP.342.2. LTP in neurological disorders
Impaired long-term potentiation plays an important role in the pathology of many progressive and immediate neurodegenerative disorders. Alzheimer's disease (AD) is an extensively studied progressive neurodegenerative disease characterized by the unnatural processing of amyloid precursor protein, hyperphosphorylation of tau protein, apoptotic-like cell death, cognitive decline and defective memory.35 Synaptic loss and dysfunction are the two main features associated with the cognitive decline in AD, which are attributed to soluble amyloid beta (Aβ) in the brain of AD patients. Aβ oligomers inhibit LTP and facilitate long term depression, a slow, weak, electrical stimulation of CA1 neurons, which reduces the number of AMPA receptors at the synapse.36 In addition, there are reports that AD impairs LTP by accumulation of the enzyme protein kinase M zeta (PKMζ) in neurofibrillary tangles, suggesting a link between PKMζ-mediated synaptic plasticity and memory impairment in AD.37 On account of the fact that AD is still incurable, a deeper understanding of the complex mechanisms, such as epigenetic dysfunction, associated with this neurodegenerative disease is of increasing importance.38 Gräff et al. showed that the hippocampus region in human AD brain has elevated levels of HDAC2, the enzyme known to modulate gene transcription associated with learning.39 Therefore, developing strategies to alter the epigenetic program could be useful in treating AD.Huntington's disease (HD) is an autosomal dominant neurodegenerative disorder that is characterized with progressive motor dysfunction, dementia, and psychiatric disturbance. Genetic and phenotypic studies of animal models of HD reveal certain LTP alterations. Crupi et al. discussed the possibility of damage to the LTP-like plasticity, due to alterations of the neuronal circuits in HD patients.40 Dallérac et al. showed that the D1 dopamine receptor activation rescued the impaired LTP in the mouse model of HD disease.41 Costa et al. showed that L-DOPA restores the hippocampal synaptic potentiation through D1/D5 receptors and improves the cognitive deficit in experimental models of Parkinson's disease.42 Transcriptional abnormalities leading to neuronal damage in HD are often associated with epigenetic alterations. The epigenetic enzymes known to control gene transcription such as DNMT and HDAC have been envisaged as potential therapeutic targets in HD treatment.43 Other major disorders include amnesia, where memory and learning are affected, among other cognitive functions, and agnosia, which is marked by the incapability to recognize certain objects, persons, or sounds. Because the loss of cognition is the common manifestation of these neurological disorders, strategies to regulate LTP could aid us to gain insights into general cellular mechanisms and facilitate strategies to treat cognitive impairment.
2.3. Transcription factor-mediated reprogramming for neurogenesis and cell replacement therapy
Neurogenesis, a dynamic cellular process that validates the precise functioning of learning and memory processes, could be achieved through the activation of cell fate specific transcription factors. Watanabe et al. showed the conversion of C2C12 myoblasts into neurons by overexpressing a recombinant form of REST (REST-VP16), which contains the transcriptional activation domain of the herpes simplex virus protein VP16.44 Vierbuchen et al. reprogrammed mouse embryonic and postnatal fibroblast cells into neurons by ectopic expression of three transcription factors Ascl1, Brn2, and Myt1 l.45 Neural stem cells (NSCs) that are capable of self-renewal or differentiation can generate new neurons and have a debatable role in alleviating memory loss after being transplanted into transgenic mouse AD models.46 An essential role of BDNF (brain derived neurotrophic factor) for the functional effect of transplanted NSCs was demonstrated in a study that clearly showed that only the rats that received stem cells and BDNF preserved learning and memory.47 Some of the LTP associated genes/factors, which are the potential targets of cellular reprogramming, and their characterized role in human and animal models are listed in Table 1.48–56| LTP-associated gene/factor | Function | Model | Reference |
|---|---|---|---|
| CREB | Positive regulation of memory consolidation by modulating BDNF expression by binding to CRE | Mice | 12 |
| BDNF | Positively modulates stable long-term potentiation (LTP) in hippocampus through TrkB receptor | Mice | 48 |
| Zif268 (EGR1) | Strengthening of memory trace by regulating proteins in the amygdala region | Mice | 49 |
| Arc | Govern the key translation factors during LTP consolidation | Rat | 50 |
| GAP43 | LTP persistence through release of glutamate by PRKCA mediated phosphorylation of GAP43 | Rat | 51 |
| Reelin | Signal transduction through Apoer2 for the induction of LTP | Mice | 52 |
| Synapsin I | Contributes to LTP by increasing the release of neurotransmitter and number of post synaptic neurons | Rat | 53 |
| Neurotensin | Regulation of working memory | Homo sapiens | 54 |
| DLG3 | Interaction with NMDA receptor and plays an important role in LTP and memory formation | Mice | 55 |
| Integrin associated protein | Memory formation | Rat | 56 |
Along with some of these key signal transduction factors, epigenetic modifications were also associated with cognitive recognition.57
3. Epigenetic enzyme inhibitors for transcriptional activation
3.1. HDAC and DNMT inhibiting small molecules for memory recovery
There are two major epigenetic mechanisms involved in learning and memory: (1) modification at cytosines in CpG dinucleotide sequences by methylation; and (2) chromatin remodelling. Studies were undertaken with animal models using different learning and memory methods,58 and with a post-mortem of the human brain59 to endorse this phenomenon. Methylation of both DNA and the histone proteins has been implicated in LTP and memory formation.60 DNA methylation induced DNA methyl transferases (DNMTs) such as DNMT1, DNMT3a, and DNMT3b are differentially expressed within the adult brain and suggest its critical role in cognition.61 Recent studies have shown that by blocking DNMT activity with inhibitors 5-AZA-dC and zebularine, contextual fear-conditioned memory formation could be disrupted.62 DNMT inhibition also resulted in increased expression level of the memory-stimulating gene Reelin through promoter demethylation, whereas a decreased expression level of memory-suppressing genes such as PPI1 was also observed.63 Taken together, it is evident that epigenetic regulation of memory promoting genes form a complex pattern as these processes cross-talk, interact with, and influence each other.HDAC enzymes govern gene transcription by conserving them in heterochromatin architecture and regulating the chromatin-remodelling event. Histone deacetylase inhibitors (HDACi), such as trichostatin A (TSA) and vorinostat [suberoylanilide hydroxamic acid (SAHA)], are capable of inhibiting HDACs with varying efficiency (at nanomolar to millimolar concentrations) and cause hyperacetylation of histones, which in turn triggers transcriptional activation of certain genes through relaxation of the DNA conformation.64 However, HDAC inhibition does not always restore the transcription of the gene under investigation. For instance, Lopez-Atalaya et al. showed through chromatin immune precipitation sequencing and gene expression profiling studies that TSA had only a moderate effect on hippocampal gene expression.65 Proteins other than histones can be deacetylated by HDACs, owing to the involvement of HDACs in many complex cellular processes.66,67 Prolonged treatment of adult rat neural progenitors with valproic acid (VPA), a known HDAC inhibitor, resulted in the reduced proliferation of adult neural progenitors, but observed an increase in neuronal differentiation.68 Further examination showed an increase in the Neurod1 mRNA level, a transcription factor required for neuronal differentiation after VPA treatment.69 Yu et al. showed that VPA intensely repressed the proliferation of progenitor cells and increased neuronal differentiation by inducing the expression of proneuronal transcription factors, such as Ngn1, Math1, and p15, which contribute to neuronal differentiation.70 VPA has also been shown to effectively block aberrant neurogenesis and prevent hippocampal seizure induced cognitive impairment.71 Kim et al. showed that neurogenesis is stimulated in the brain of ischemic rat models with NaB treatment.72 Overall, HDACi positively regulates differentiation of newborn neurons and has potential for the treatment of cognitive impairment.73 Also, a number of inhibitor-based experiments verified that HDACs can negatively regulate long-term memory formation.74,75 Recent studies on individual HDACs and their effects on LTP have revealed that HDAC2, and not HDAC1, is a key HDAC for regulating memory formation.76 HDAC3 is also known to negatively regulate memory formation.77Table 2 details the enzymes associated with specific neurological disorders.78–82
| Chromatin modifying enzymes | Highly expressed region | Inhibitor | Structure | Effect of inhibition | Disorder | Reference |
|---|---|---|---|---|---|---|
| DNMT1 | Cortical layers of brain interneurons | 5-AZA-dC |
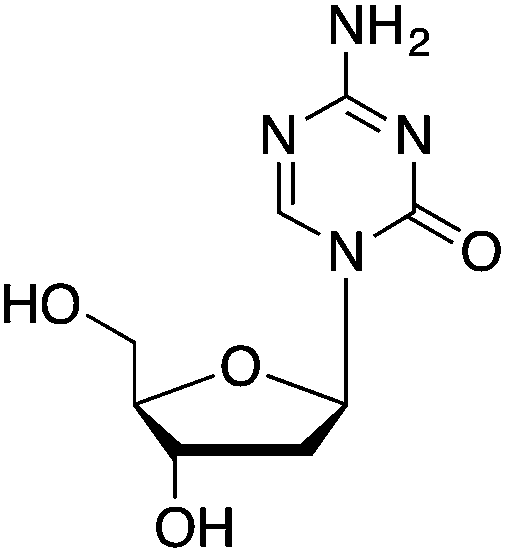
|
Synaptic plasticity and demethylation of Reelin gene | Schizophrenia | 35,36 |
| Embryogenesis of the brain | ||||||
| DNMT3a | Lateral nucleus of the amygdala (LA) neurons | Zebularine |
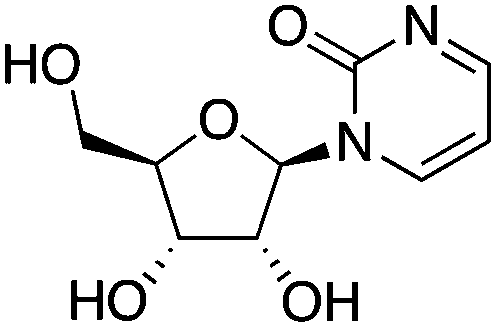
|
Contextual fear memory formation | 57 | |
| Embryogenesis of the brain | ||||||
| HDAC Class I/II | Hippocampus | Sodium butyrate |
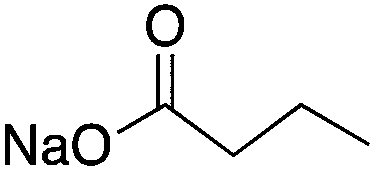
|
Modulated hippocampal LTP in a way that was entirely dependent on CBP | 13 | |
| HDAC Class I | Hippocampus | TSA |
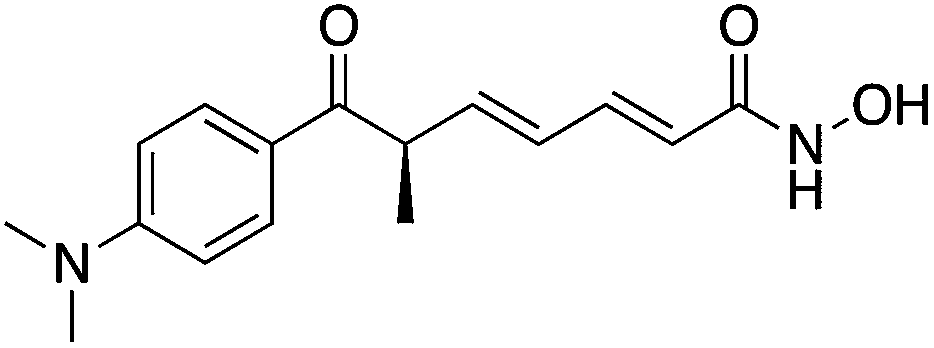
|
Enhancement of memory and synaptic plasticity | 11 | |
| HDAC2 HDACI | SAHA |
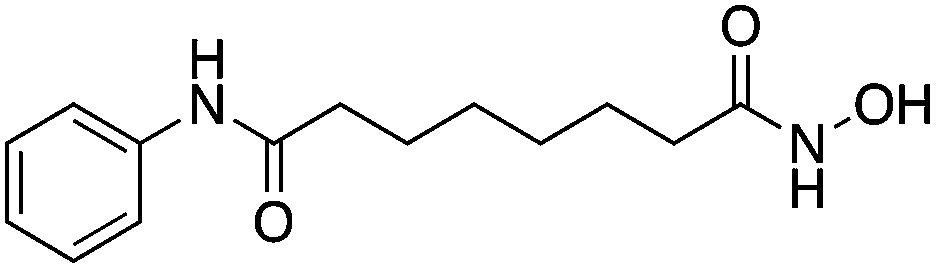
|
Prevent H3 deacetylation and promoted expression of neuroprotective proteins Bcl-2 and Hsp70 | Ischemic stroke | 78 | |
| HDAC3 (I) | Brain | RGFP966 |
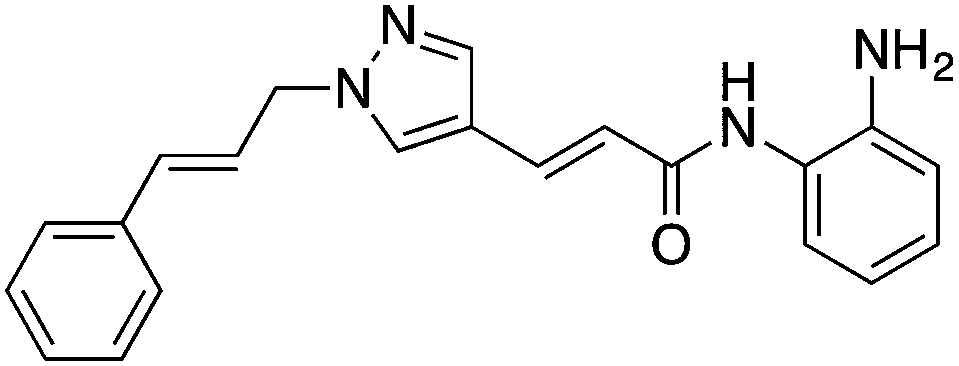
|
Reduced HDAC3 repression of c-fos and Nr4a2 and allowed increased acetylation of H4K8 and H3K14 | Cognitive disorders | 79 |
| HDAC Class I/II | Photoreceptor neuron | Phenyl butyrate |
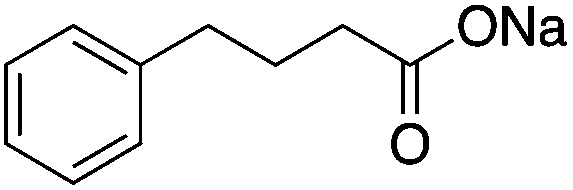
|
Improved histone acetylation and transcriptional regulation | Huntington's disease | 80 |
| HDAC Class I/II | Sodium butyrate/TSA + 5 aza-2′-deoxycytidine | Induced expression of FMRI 1 promoter region | Fragile X | 81 | ||
| DNMT3b | Embryogenesis of the brain | 5-AZA-dC, zebularine | ICF syndrome | 82 | ||
The process of aging results in reduced acetylation of histones H3 and H4 in the promoter regions of the BDNF gene, suppressing its expression and significantly contributing to the deficits in hippocampal synaptic structure and function.83 These changes can be repaired by HDAC inhibition or selective activation of trkB receptors. Taken together, a chromatin-modifying enzyme like HDAC is a potential therapeutic target that can generate a form of long-term memory, which persists beyond a point at which normal memory fails (Fig. 1).
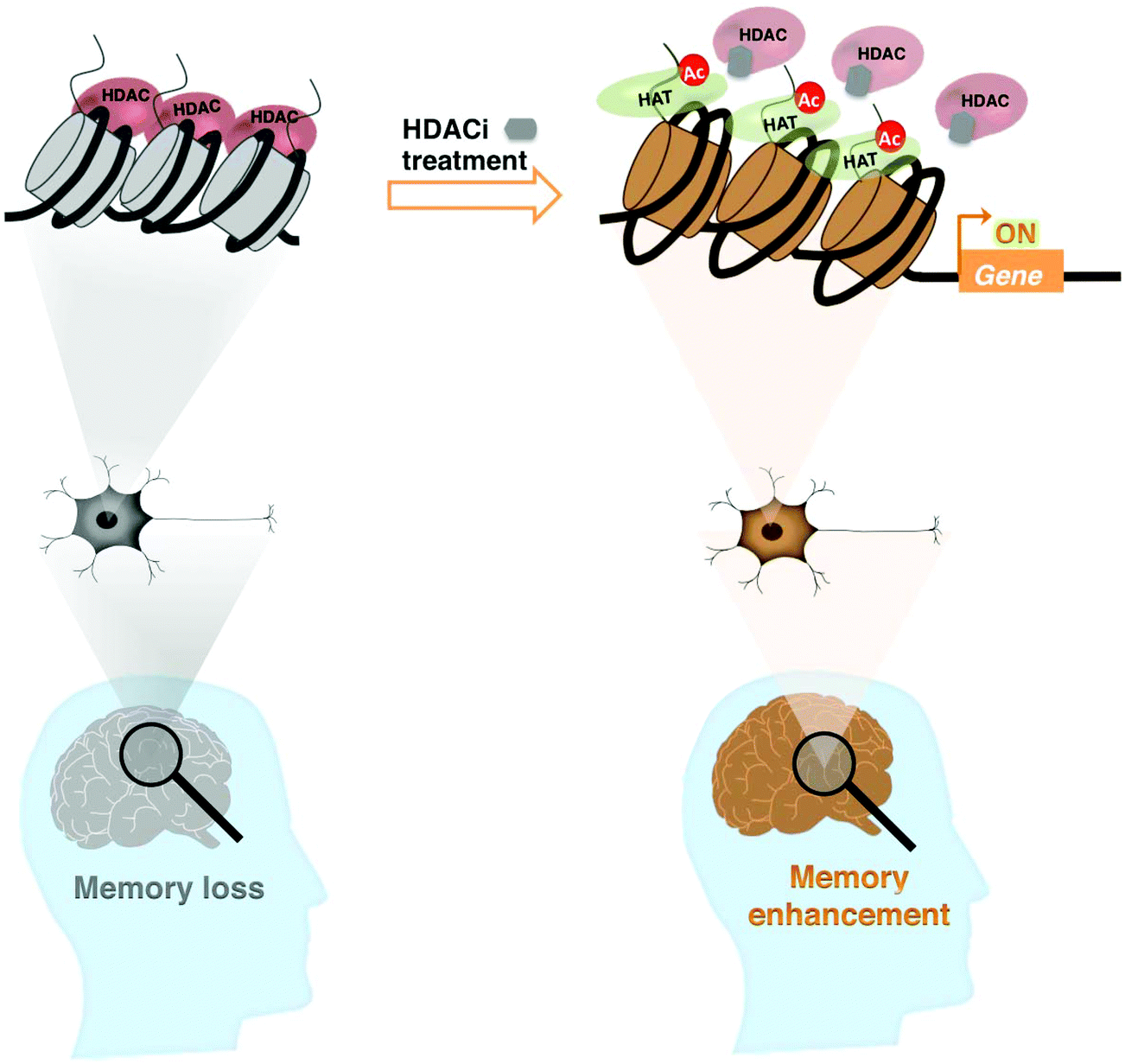 | ||
| Fig. 1 A person with memory loss (brain and neuron in grey) has a heterochromatin architecture protected by HDAC in which memory-associated gene(s) are silenced. HDACi treatment improves the memory (brain and neuron in orange) through chromatin remodeling (acetylation in histones (colored)). | ||
3.2. Therapeutic potential of HDACi in memory recovery
Pharmacological intervention of histone acetylation using HDACi has been useful to treat memory impairment in neurodegenerative disorders and to enhance cognition.84 However, the therapeutic use of HDACi needs careful validation, as studies in mice and some observations in patients have shown that memory-related processes could be impaired with chronic HDAC inhibition.85–87 By forming a complex with HDACs, methyl-CpG binding protein 2 (Mecp2) is believed to act as a transcriptional modulator in the basolateral amygdala (BLA) and is required for normal anxiety behaviour as well as certain types of learning and memory.88 Adachi et al. suggested the key role of Mecp2 as a transcriptional repressor in Rett Syndrome, an X-linked neurodevelopmental disorder.89 By infusing SAHA, they explored the effect of histone H3 acetylation on the anxiety phenotype of BLA-specific MECP2 knockdown mice. However, the SAHA infused mice demonstrated increased anxiety-like behaviour and impaired cue-dependent fear learning. This result is in contrast to that observed with a single drug dose, and the saturation of changes in neuronal plasticity due to the chronically elevated histone acetylation was hinted to be the reason.89 Since anxiety is a complex behaviour mediated by a combination of genes, SAHA may have targeted other genes not regulated by MeCP2. Hence, these molecules should be designed with the ability to recognize specific region(s) in the brain, as genes and systems required for LTP maintenance in one brain-region may not be required in the other. These findings demonstrate a potential shortcoming of employing the HDACis such as SAHA to recover memory, as they may produce undesirable psychiatric behavioural side effects, and gives rise to the need for customizing them to induce specific genes.3.3. Therapeutic potential of HDACi in neurological disorders
Memory loss and cognitive dysfunction are not typical characteristics of Friedrich's ataxia (FRDA), a neurodegenerative disease caused by the repression of FXN gene that codes for the essential mitochondrial protein, Frataxin.90 However, chemical approaches developed to treat this transcription dependent disorder could be mimicked for memory related disorders. For example, HDACis like SAHA, TSA, and sodium butyrate had no notable effect on FXN transcription in non-FRDA cells. However, the commercially available HDACi BML-210 and its derivative efficiently induced the silenced FXN gene in lymphoid cell lines derived from FRDA patients and increased the level of Frataxin in cells.91 In neurodegenerative HD, aberrant transcription caused by the mutant huntingtin involves functionally defective transcription factors and coactivators such as CBP, which impair CBP/CREB mediated gene expression and histone deacetylation.92,93 Steffan et al. suggested the therapeutic potential of a HDACi in HD, as the use of HDACi inhibited the polyglutamine-dependent neuronal degeneration and reduced lethality in a Drosophila model.94 Subsequently, Ferrante et al. showed that the HDACi sodium butyrate triggered specificity protein-1 acetylation, and ameliorated the neurodegenerative phenotype in R6/2 transgenic mouse model of HD.80 Administration of the HDACi phenylbutyrate at tolerable doses caused significant neuroprotective effects in the N171-82Q transgenic mouse model of HD.95 Preclinical trial experiments carried out in R6/2 HD mouse model showed that HDACi SAHA ameliorated motor deficits, by crossing the blood–brain barrier and increasing histone acetylation in the brain.96Genome-wide gene analysis through microarray studies showed that regular oral administration of HDACi 4b, an analog of BML-210, reversed histone H3 hypoacetylation and ameliorated the disease phenotype and transcriptional abnormalities in HD transgenic mice.97 Conversely, Beconi et al. demonstrated the shortcomings in the potential employment of pimelic diphenylamide HDACi to treat neurological disorders.98 Evaluation of HDAC isoform selectivity, cellular activity and in vitro and in vivo ADME (absorption, distribution, metabolism and excretion) properties of 4b have revealed that even when in vitro selectivity and binding mode were in agreement with previous reports,98 their physicochemical stability and metabolic characteristics cast serious doubts that CNS HDAC3 inhibition is a therapeutic target for HD.97 Hence, 4b was unsuitable as a molecular tool to examine class I HDAC inhibition in vivo, and the need for proper ADME assessment of compounds before in vivo target validation and therapeutic application was proposed. Specificity and off-target effects are the key issues that hamper the employment of small molecules in biological studies. Many studies tend to focus on target genes but ignore changes in the expression of other genes. The small molecules used (e.g. TSA, SAHA, VPA) in the experiments could also hit other targets not related to chromatin-remodeling or epigenetic regulation. This is often the case when unreasonably high concentrations of drugs are used. In this regard, there has been a rising demand for programmed small molecules targeting specific gene(s).
3.4. Programmable DNA binding small molecules in neurological disorders
DNA stores heritable information over a long period of time using only four nucleobases. Hence, small molecules interacting with these nucleobases can significantly alter the mutations that confer a specific neurological disorder. N-Methylpyrrole-N-methylimidazole polyamides (PIPs) are cell-permeable small molecules capable of binding to the minor groove of DNA with an affinity that is comparable to that of transcription factors.99 PIPs bind to DNA following a binding rule, where an antiparallel pairing of I opposite P (I/P) recognizes a G–C base pair, while a P/P pair recognizes A–T or T–A base pairs. Synthetic PIPs possessing an alkylating moiety have been successfully employed for selective gene silencing and to target cancer associated mutations.100,101 The interference of PIPs with transcription factors also has a positive induction effect, which suggests that PIPs can inhibit both basal and activated transcription. DNA-binding sequence-specific PIPs can potentially be used to reverse the effects and alter the FXN gene by targeting GAA TTC repeats.102 For a practical demonstration, Burnett et al. synthesized a PIP (P1) to target the 9 bp sequence, as in the GAA TTC repeating DNA sequence. These polyamides bound to the coding region of the gene without affecting transcription in mammalian cells. When polyamide P1-treated cell lines were taken from an FRDA patient who had a very low level (6%–13%) of Frataxin protein, and the results were compared with those of an unaffected sibling, it was found that the polyamide P1 increased the FXN transcription by about 2–3-fold. Similar treatment with a mismatched polyamide showed a modest result. A recent study by Dervan and colleagues has shown that these polyamides do not fit into single-stranded RNA or duplex regions of RNA, suggesting that they do not affect the translation of Frataxin mRNA.1034. Small molecules with dual characteristics: progress and prospects
4.1. Sequence-specific epigenetic activators
Recently, some novel strategies have emerged to obtain small molecules with versatile properties. One such novel small molecule, termed SAHA-PIP, encompasses both selective DNA-binding hairpin pyrrole–imidazole polyamides (PIPs) and the potent HDAC inhibitor, SAHA (Fig. 2A). The biological activity of SAHA-PIP was first demonstrated by synthesizing SAHA-PIPs that could sequence-specifically acetylate the promoter region of the tumour suppressing p16 in HeLa cells.104 When the effects of a library of distinct SAHA-PIPs were evaluated on the epigenetic-dependent and complicated pluripotency gene network in mouse embryonic fibroblasts (MEFs), certain SAHA-PIPs differentially induced pluripotency genes through the initiation of epigenetic marks that confer transcription accommodating chromatin, including histone H3 Lys9 and Lys14 acetylation105,106 (Fig. 2B). An advanced version of SAHA-PIP, termed ‘δ’, but not SAHA, rapidly induced multiple pluripotency genes.107 Interestingly, δ-OMe, the non-functional SAHA-PIP, did not activate any pluripotency genes to validate SAHA as the functional moiety in δ. Because SAHA-PIP encompasses both HDAC inhibitory activity and sequence-specific binding ability, interpretation of a definite mechanism is challenging. About 33% of the δ-induced genes belonged to the core pluripotency gene network that comprises 345 intertwined genes. Hence, it is reasonable to assume that the PIP in δ directs SAHA to the typically conserved core pluripotency gene network for site-specific epigenetic modifications (Fig. 2B). Analysis of the number of matching sites in the Nanog gene further substantiates this notion.105 However, more studies are needed to achieve a thorough understanding of the SAHA-PIP mediated epigenetic activation of certain genes. Recently, a SAHA-PIP termed ‘K’ was shown to be the first-ever small molecule capable of inducing the transcriptional activation of germ cell genes in a human somatic cell (Fig. 2C). This result is remarkable because meiotic process is specific to germ cells and could not occur in a somatic cell.108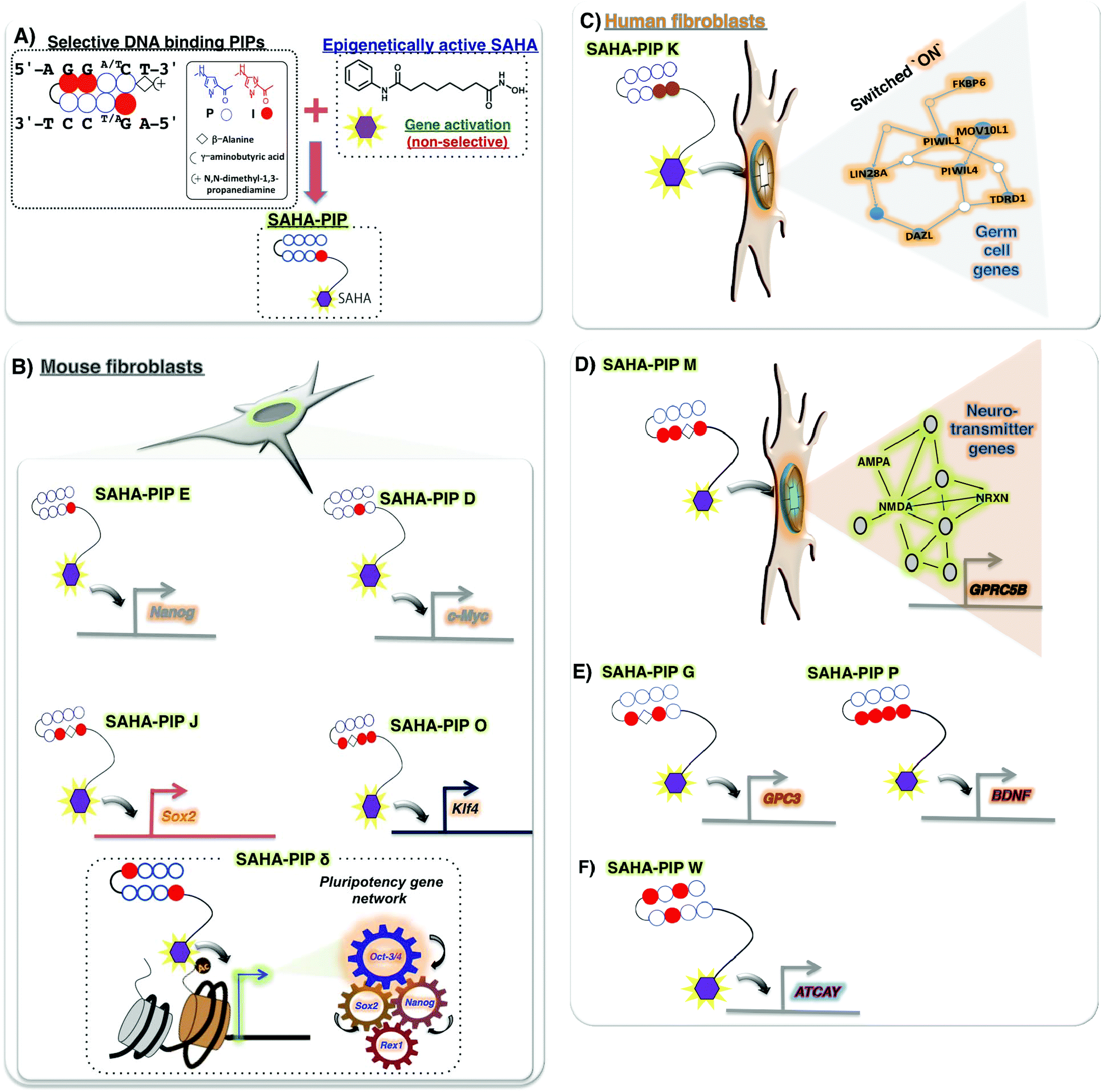 | ||
| Fig. 2 (A) Creation of a DNA-based epigenetic switch. An innovative small molecule called SAHA-PIP, capable of accessing both the genetic and epigenetic environments, was obtained by conjugating sequence-specific hairpin pyrrole imidazole polyamides (PIPs) with the chromatin modifying histone deacetylase inhibitor SAHA. (B) Biological activity of SAHA-PIP in mouse cells. SAHA-PIP E could cause site-specific acetylation by hindering the HDACs only in the promotion of Nanog in mouse fibroblasts. SAHA-PIPs D, J and O for c-Myc, Sox2 and Klf4, are also shown. An advanced version of SAHA-PIP δ induces site-specific chromatin remodelling of Oct-3/4 to trigger the core pluripotency gene network. (C) Biological activity of SAHA-PIP in human cells. SAHA-PIP K triggers unusual switching ON of the germ cell genes in a human somatic cell. (D) SAHA-PIP M trigger neurotransmitter related gene network. (E) SAHA-PIP G and P trigger neurogenesis related genes (GPC3 and BDNF) and (F) SAHA-PIP W triggers for ataxia related ATCAY gene. | ||
As shown before, both HDACi and PIPs were individually shown to be successful in treating neurodegenerative disorders but with certain disadvantages. Hence, conjugating them both to generate other HDACi-PIP could lead to site-specific activation of genes associated with memory formation and neurodegeneration. Recently, a SAHA-PIP called ‘M’ was shown to specifically activate a set of neurotransmitter genes, including NRXN and GPRC5B (Fig. 2D).109 SAHA-PIP ‘G’ activated the neurogenesis related GPC3, while SAHA-PIP ‘P’ activated BDNF (Fig. 2E). Interestingly, SAHA-PIP ‘W’ specifically activated the ataxia related ATCAY gene (Fig. 2F).109 Synthetic PIPs have a relative advantage to natural DNA-binding proteins with the following two properties: (i) presence of modifiable covalent sites and (ii) ability to alter the tightly packed chromatin structure by binding the methylated DNA sequences.110 Conjugation of a PIP with another HDACi conjugate called JAHA, which is HDAC8 specific, also elicited similar promoter specific functionality.111 Thus, it is possible to conjugate HDACis like BML-210, DNMTis like 5-azacytidine, and other such enzyme inhibitors to PIPs to have variable effects. Although it is difficult to predict the off-rate of PIPs inside the cells, the high binding affinity of SAHA-PIP to target DNA sequences, and their ability to induce pluripotency genes even at 72 h and 100 nM concentration, suggest it to be slow.112,113 Ligands that specifically recognize 15–16 base pairs like a PIP dimer was shown to target the regulatory region of the HIV-1 genome.114 Interestingly, a recent report suggests that partial cellular reprogramming promotes efficient neuroregeneration.115 In this regard, development of multi-gene targeting molecules like SAHA-PIPs could aid in the generation of clinically relevant neural cells.
Cell permeability is both an advantage and a bottleneck of the PIPs, as both the molecular size and pyrrole/imidazole content of the PIPs were shown to hamper their ability to permeate cells.100 But recent studies have shown that it is possible to increase the permeability of PIPs by engineering their chemical architecture.116 Although PIPs are well characterized to influence gene expression in cell culture, their clinical translation requires comprehensive characterization of their pharmacokinetic (PK) profiles and ADME properties. Fukasawa et al. have shown through intravenous administration of a set of PIPs in rat models that the area under the plasma concentration–time curve increases linearly as a function of dose, and the systemic clearance and volume distribution in the steady state remain unaltered.117 Synold et al. have suggested that size or minor structural modification of PIPs could affect their solubility and could lead to their accumulation in the lungs.118 Although SAHA does not hinder the binding properties of PIPs, it is difficult to generalize whether HDACi-PIPs will have the same properties as PIPs. Because the PK profiles and ADME properties of the PIPs showed that various results were obtained with minor changes to the structures, there is the need for a case-by-case study before employing PIPs and/or HDACi-PIPs.
4.2. Signalling pathway inhibitors
The design of innovative small molecules that could effectively modulate intricate signalling factors can be useful for restoring cellular homeostasis. Recently, a compound called Fisetin (Fig. 3A) was found to be an orally active, neuroprotective, cognition-enhancing small molecule. Fisetin possesses antioxidant activity and increases the intracellular level of glutathione and it has both neurotrophic and anti-inflammatory activity.119 Sivilia et al. recently demonstrated CHF5074, a multifunctional anti-inflammatory derivative, as a novel anti-Alzheimer compound in TG2576 mice.120 Several synthetic compounds are capable of a synergistic effect on multi-targets associated with a particular signalling pathway. For example, effective inhibitors of acetylcholinesterase (AChE) and Monoamine oxidase, such as imino 1,2,3,4-tetrahydrocyclopent[b]indole carbamates (Fig. 3B), Coumarin derivatives (Fig. 3C), chromone derivates (Fig. 3D) and Lipocrine (Fig. 3E), could be used to (i) target the catalytic activity of AChE, (ii) inhibit the AChE-induced Aβ aggregation and (iii) to protect against reactive oxygen species (ROS). Likewise, Caproctamine (Fig. 3F) prevented AChE-induced Aβ aggregation and stimulated the cholinergic activity by antagonizing the muscarinic M2 receptor.121 Bolognesi et al. showed the derivatives of bis-tacrine (Fig. 3G) as likely candidates for AD treatment, as they could reverse the AChE-induced amyloid fibrillogenesis by inhibiting AChE activity and chelating the metal ions.122 Rosini et al. developed a multi-target directed ligand carbacrine (Fig. 3H) to treat AD neurodegeneration by effectively blocking the in vitro AChE-induced Aβ aggregation, decreasing the oxidative stress and antagonizing the NMDA receptor activity.123 Memoquin (Fig. 3I) is another multi-target molecule capable of inhibiting acetylcholinesterase and β-secretase-1-activity. Capurro et al. showed that Memoquin could enhance cognition and prevent the Aβ-induced neurotoxicity in diseased mouse models.124 Youdim et al. prepared multi target drugs Ladostigil (Fig. 3J) and M30 (Fig. 3K), the derivatives of Rasagiline and demonstrated an improved effect on the depression, dementia and behavioral abnormalities related to AD in mouse models. M30 exhibited neurorestorative activity and induced the production of BDNF and glia-derived neurotrophic factor.125 Lu et al. have designed and synthesized the derivatives of resveratrol as a candidate for AD treatment that has the ability to inhibit Aβ aggregation. One of the derivatives 5d (Fig. 3L) showed no toxicity in a mouse model and in vitro studies have shown that the drug can enter the blood brain barrier.126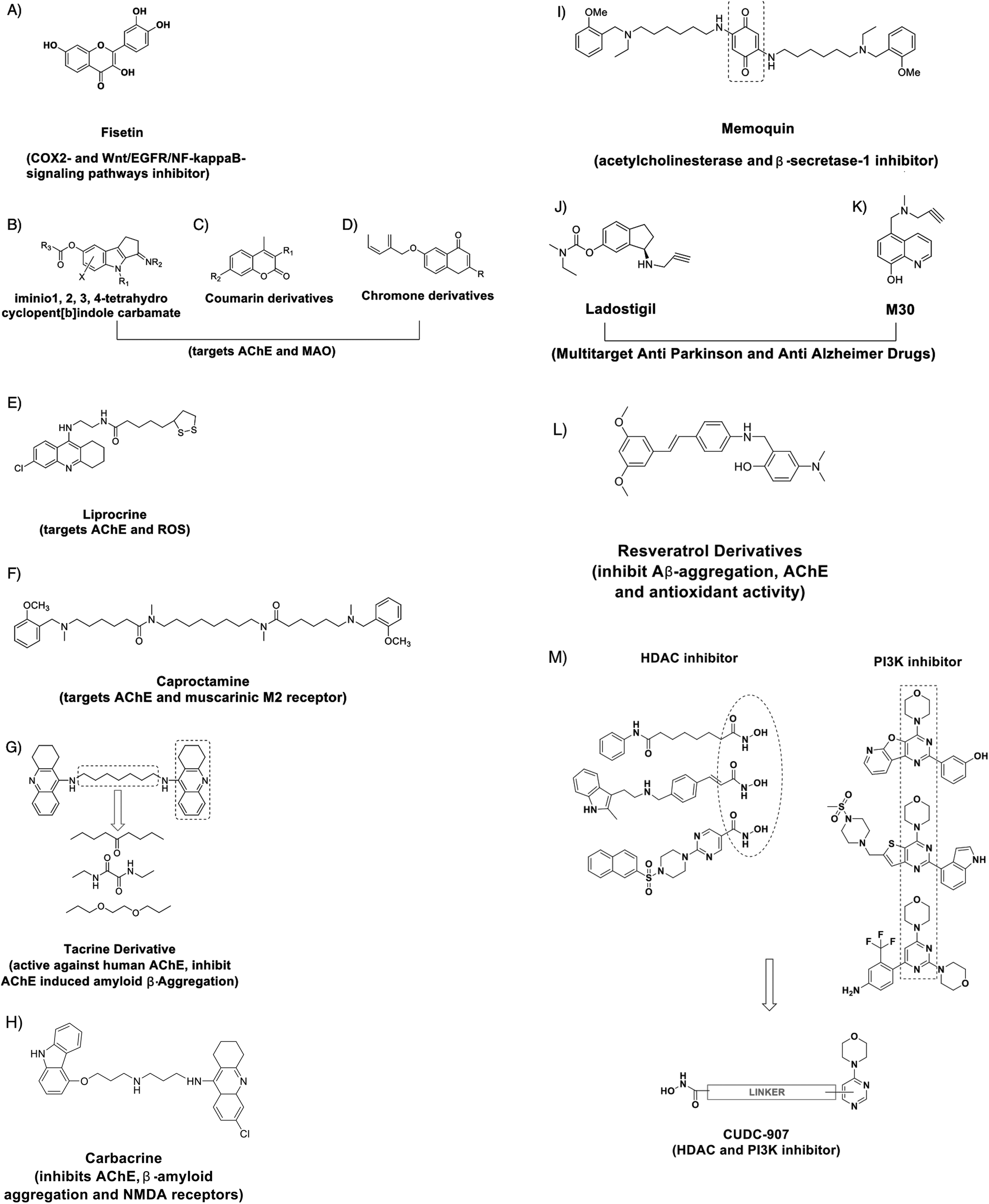 | ||
| Fig. 3 Chemical structure of (A) multi-target neuroprotective compound Fisetin, amyloid β-aggregation inhibiting multi-target compounds like (B) imino 1,2,3,4-tetrahydrocyclopent[b]indole carbamates, (C) coumarin derivatives, (D) chromone derivates, (E) Lipocrine, (F) Caproctamine, (G) bis-tacrine derivatives, (H) carbacrine, (I) Memoquin, (J) Ladostigil, (K) M30 and (L) resveratrol derivative. (M) A schematic representation of how the functional groups for inhibitory activity against both HDAC and PI3Ks (dotted box) are linked to design and generate CUDC-907. | ||
Small molecules capable of inhibiting epigenetic enzymes like HDAC and signalling pathway factors can have better efficacy. CUDC-907 is one such compound that has been designed with dual inhibitory activity against not only HDAC enzymes but also phosphatidylinositol 3-kinase (PI3Ks) (Fig. 3M).127 When compared with equivalent single target molecules, CUDC-907 displayed better growth inhibition in both cultured and implanted cancer cells by inhibiting class I and II HDAC enzymes as well as the PI3K-AKT-mTOR pathway and signaling molecules (including MEK, RAF (a protein oncogene protein kinase), STAT-3 (Signal transducer and activator of transcription), and MAPK (Mitogen activate protein kinase)) and the upstream receptor tyrosine kinases. Developing such dual inhibitory small molecules could be useful in recovering memory and alleviating neurodegeneration.
5. Conclusion and future perspective
Small molecules are often referred to as the missing link in the central dogma of biology.1 Recent developments in bioinformatics and techniques such as diversity-oriented synthesis suggest that it is possible to design small molecules to treat the diseases that were previously thought to be incurable. LTP governs a multitude of brain functions such as learning and memory and gets associated with incurable cognitive disorders. Small molecules that alter the chromatin architecture and signal transduction have shown clinical potential to modulate LTP mechanisms. However, to achieve effective regulation, artificial transcriptional activators capable of orchestrating a targeted gene network need to mimic its natural equivalents. By taking cues from nature, development of novel types of compounds like HDAC-PIPs, CUDC-907, CHF5074 and others (Fig. 3) could open up exciting opportunities to precisely orchestrate the intricate transcriptional machinery related to memory. However, precautions should be taken when developing such strategies, because chromatin remodelling and signalling pathways are not isolated events.128 Moreover, factors such as practicality and genome-wide specificity need to be focused to avoid side effects.129,130 Druggability is another major factor that needs consideration, as physicochemical characteristics, DMPA properties and safety profiles of these multi-target molecules could hamper their development. Strategies to make the sequence-specific epigenetic activators and/or signalling inhibitors penetrate CNS are other major issues that need profound study. Some factors, such as target–target interactions, and differing requirements for the percentage and duration of target engagement for different targets should also be considered, because the interaction of one target could affect the signaling of another target, while the activation/inhibition of multiple targets could result in synergic toxic effects. Innovative strategies to generate multi-target molecules with tunable activation potentials of differing strengths, by taking the above-mentioned factors into account, can lead to effective treatment of complex neurological disorders, as illustrated in Fig. 4.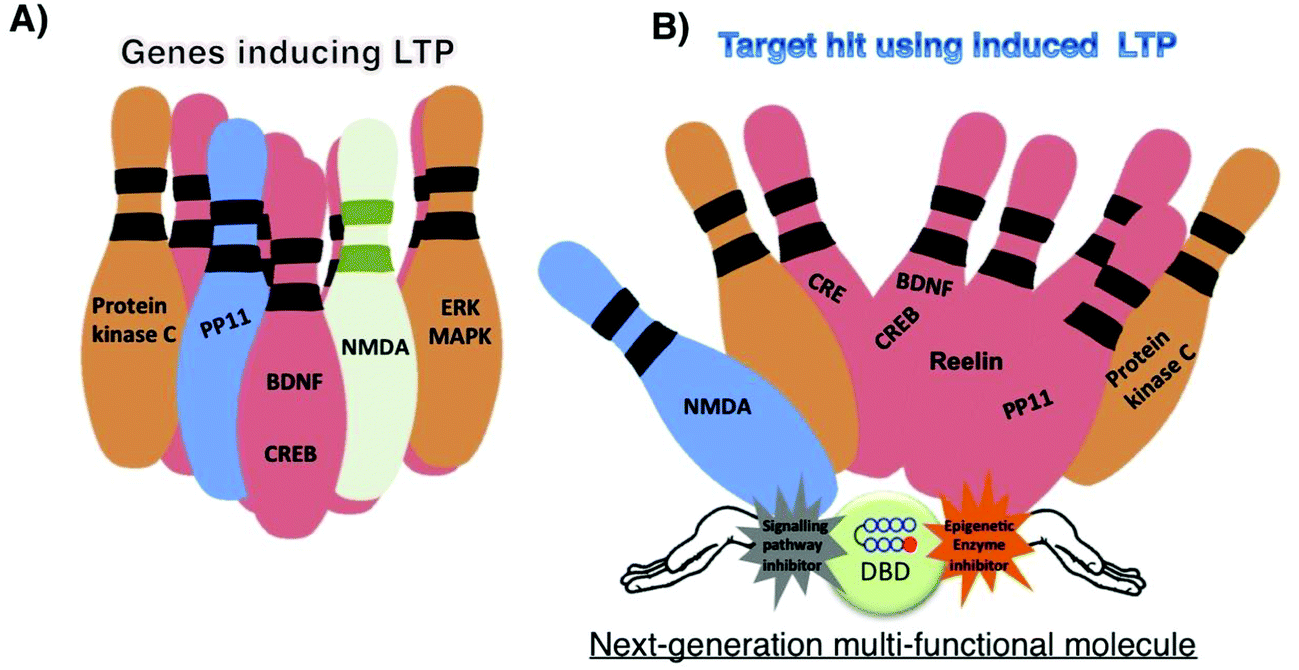 | ||
| Fig. 4 (A) Some major factors responsible for LTP induction are shown. (B) Next-generation small molecules encompassing multi-functional properties (DNA binding domain (DBD) and modulators of factors affecting the signalling pathway and epigenetic enzymes) may effectively target the set of genes associated with LTP. The potential gene targets for LTP are represented as bowling balls and the compound with multifunctional properties is shown as a ball with waving hands. | ||
Acknowledgements
This research was supported by the Ministry of Education, Culture, Sports, Science and Technology (MEXT) of Japan. The iCeMS is supported by World Premier International Research Center Initiative, MEXT, Japan. We thank Nagase Science and Technology foundation for their support. We thank Grants-in-aid for Young Scientists-B and challenging exploratory research from JSPS for support to G. N. P.References
- S. L. Schreiber, Nat. Chem. Biol., 2005, 1, 64–66 CrossRef CAS PubMed.
- A. L. Hopkins and C. R. Groom, Nat. Rev. Drug Discovery, 2002, 1, 727–730 CrossRef CAS PubMed.
- D. B. Matthew, C. Raymond and L. Andre, Science, 2012, 338, 334–335 CrossRef PubMed.
- D. Baltimore, Nature, 2001, 409, 814–816 CrossRef CAS PubMed.
- J. Cortés-Mendoza, S. Díaz de León-Guerrero, G. Pedraza-Alva and L. Pérez-Martínez, Int. J. Dev. Neurosci., 2013, 31, 359–369 CrossRef PubMed.
- I. Winkler and N. Cowan, Exp. Psychol., 2004, 51, 1–17 CrossRef PubMed.
- P. E. Gold, Neurobiol. Learn. Mem., 2008, 89, 201–211 CrossRef CAS PubMed.
- R. C. Malenka and R. A. Nicoll, Science, 1999, 285, 1870–1874 CrossRef CAS.
- M. A. Lynch, Physiol. Rev., 2004, 84, 87–136 CrossRef CAS PubMed.
- M. J. Morris and L. M. Monteggia, Int. J. Dev. Neurosci., 2013, 31, 370–381 CrossRef CAS PubMed.
- C. G. Vecsey, J. D. Hawk, K. M. Lattal, J. M. Stein, S. A. Fabian, M. A. Attner, S. M. Cabrera, C. B. McDonough, P. K. Brindle, T. Abel and M. A. Wood, J. Neurosci., 2007, 27, 6128–6140 CrossRef CAS PubMed.
- A. Suzuki, H. Fukushima, T. Mukawa, H. Toyoda, W. Long-Jun, Z. Ming-Gao, H. Xu, Y. Shang, K. Endoh, T. Iwamoto, N. Mamiya, E. Okano, S. Hasegawa, V. Mercaldo, Y. Zhang, R. Maeda, M. Ohta, S. A. Josselyn, M. Zhuo and S. Kida, Neuroscientist, 2011, 31, 8786–8802 CAS.
- J. Haettig, D. P. Stefanko, M. L. Multani, D. X. Figueroa, S. C. McQuown and M. A. Wood, Learn. Mem., 2011, 18, 71–79 CrossRef CAS PubMed.
- C. M. Alberini, Physiol. Rev., 2009, 89, 121–145 CrossRef CAS PubMed.
- M. A. Wood, J. D. Hawk and T. Abel, Learn. Mem., 2006, 13, 241–244 CrossRef CAS PubMed.
- Z. Guan, M. Giustetto, S. Lomvardas, J. H. Kim, M. C. Miniaci, J. H. Schwartz, D. Thanos and E. R. Kandel, Cell, 2002, 111, 483–493 CrossRef CAS.
- A. B. Danilova and L. N. Grinkevich, PLoS One, 2012, 7, e41828 CAS.
- T. R. Mercer, M. E. Dinger, J. Mariani, K. S. Kosik, M. F. Mehler and J. S. Mattick, Neuroscientist, 2008, 14, 434–445 CrossRef CAS PubMed.
- G. Barry, Mol. Psychiatry, 2014, 19, 410–416 CrossRef CAS PubMed.
- X. Cao, G. Yeo, A. R. Muotri, T. Kuwabara and F. H. Gage, Annu. Rev. Neurosci., 2006, 29, 77–103 CrossRef CAS PubMed.
- H. L. Scott, F. Tamagnini, K. E. Narduzzo, J. L. Howarth, Y. B. Lee, L. F. Wong, M. W. Brown, E. C. Warburton, Z. I. Bashir and J. B. Uney, Eur. J. Neurosci., 2012, 36, 2941–2948 CrossRef PubMed.
- D. H. Joshua, L. B. Angie, G. P. Shane, B. Morgan, J. R. Allison, E. S. Michael, T. K. Brian, J. M. David and T. Abel, J. Clin. Invest., 2012, 122, 3593–3602 Search PubMed.
- A. Fischer, F. Sananbenesi, X. Wang, M. Dobbin and L. H. Tsai, Nature, 2007, 447, 178–182 CrossRef CAS PubMed.
- C. S. Ramóny, Proc. R. Soc. London, 1894, 55, 444–468 CrossRef.
- D. O. Hebb, Organization of Behavior: a Neuropsychological Theory, John Wiley, New York, 1949 Search PubMed.
- T. Bliss and T. Lømo, J. Physiol., 1973, 232, 331–356 CAS.
- R. M. Douglas and G. V. Goddard, Brain Res., 1975, 86, 205–215 CrossRef CAS.
- R. Malenka and M. Bear, Neuron, 2004, 44, 5–21 CrossRef CAS PubMed.
- H. Yasuda, A. Barth, D. Stellwagen and R. Malenka, Nat. Neurosci., 2003, 6, 15–16 CrossRef CAS PubMed.
- W. C. Abraham, Philos. Trans. R. Soc. London, Ser. B, 2003, 358, 735–744 CrossRef PubMed.
- T. V. P. Bliss, G. L. Collingridge and S. Laroche, Science, 2006, 313, 1058–1059 CrossRef CAS PubMed.
- T. C. Sacktor, Mol. Brain, 2012, 5, 31 CrossRef CAS PubMed.
- M. W. Jones, M. L. Errington, P. J. French, A. Fine, T. V. P. Bliss, S. Garel, P. Charnay, B. Bozon, S. Laroche and S. Davis, Nat. Neurosci., 2001, 4, 289–296 CrossRef CAS PubMed.
- J. R. Whitlock, A. J. Heynen, M. G. Shuler and M. F. Bear, Science, 2006, 313, 1093–1097 CrossRef CAS PubMed.
- M. Goedert and M. G. Spillantini, Science, 2006, 314, 777–781 CrossRef CAS PubMed.
- G. M. Shankar, S. Li, T. H. Mehta, A. Garcia-Munoz, N. E. Shepardson, I. Smith, F. M. Brett, M. A. Farrell, M. J. Rowan and C. A. Lemere, Nat. Med., 2008, 14, 837–842 CrossRef CAS PubMed.
- J. F. Crary, C. Y. Shao, S. S. Mirra, A. I. Hernandez and T. C. Sacktor, J. Neuropathol. Exp. Neurol., 2006, 65, 319–326 CrossRef CAS PubMed.
- K. Lunnon and J. Mill, Am. J. Med. Genet. B. Neuropsychiatr. Genet., 2013, 162, 789–799 CrossRef CAS PubMed.
- J. Gräff, D. Rei, J. S. Guan, W. Y. Wang, J. Seo, K. M. Hennig, T. J. Nieland, D. M. Fass, P. F. Kao, M. Kahn, S. C. Su, A. Samiei, N. Joseph, S. J. Haggarty, I. Delalle and L. H. Tsai, Nature, 2012, 483, 222–226 CrossRef PubMed.
- D. Crupi, M. F. Ghilardi, C. Mosiello, A. D. Rocco, A. Quartarone and F. Battaglia, Brain Res. Bull., 2008, 75, 107–114 CrossRef CAS PubMed.
- G. M. Dallérac, S. C. Vatsavayai, D. M. Cummings, A. J. Milnerwood, C. Peddie, J. K. A. Evans, S. W. Walters, P. Rezaie, M. C. Hirst and K. P. Murphy, Neurodegener. Dis., 2011, 8, 230–239 CrossRef PubMed.
- C. Costa, C. Sgobio, S. Siliquini, A. Tozzi, M. Tantucci, V. Ghiglieri, M. Di Filippo, V. Pendolino, A. de Iure, M. Marti, M. Morari, M. G. Spillantini, E. C. Latagliata, T. Pascucci, S. Puglisi-Allegra, F. Gardoni, M. Di Luca, B. Picconi and P. Calabresi, Brain, 2012, 135, 1884–1899 CrossRef PubMed.
- J. M. Gil and A. C. Rego, Eur. J. Neurosci., 2008, 27, 2803–2820 CrossRef PubMed.
- Y. Watanabe, S. Kameoka, V. Gopalakrishnan, K. D. Aldape, Z. Z. Pan, F. F. Lang and S. Majumder, Genes Dev., 2004, 18, 889–900 CrossRef CAS PubMed.
- T. Vierbuchen, A. Ostermeier, Z. P. Pang, Y. Kokubu, T. C. Südhof and M. Wernig, Nature, 2010, 463, 1035–1041 CrossRef CAS PubMed.
- A. G. Xuan, M. Luo, W. D. Ji and D. H. Long, Neurosci. Lett., 2009, 450, 167–171 CrossRef CAS PubMed.
- M. Blurton-Jones, M. Kitazawa, H. Martinez-Coria, N. A. Castello, F. J. Muller, J. F. Loring, T. R. Yamasaki, W. W. Poon, K. N. Green and F. M. LaFerla, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 13594–13599 CrossRef CAS PubMed.
- L. Minichiello, Nat. Rev. Neurosci., 2009, 10, 850–860 CrossRef CAS PubMed.
- K. Baumgärtel, D. Genoux, H. Welzl, R. Y. Tweedie-Cullen, K. Koshibu, M. Livingstone-Zatchej, C. Mamie and I. M. Mansuy, Nat. Neurosci., 2008, 11, 572–578 CrossRef PubMed.
- C. R. Bramham, P. F. Worley, M. J. Moore and J. F. Guzowski, J. Neurosci., 2008, 28, 11760–11767 CrossRef CAS PubMed.
- M. M. Ryan, B. Ryan, M. Kyrke-Smith, B. Logan, W. P. Tate, W. C. Abraham and J. M. Williams, PLoS One, 2012, 7, e40538 CAS.
- U. Beffert, E. J. Weeber, A. Durudas, S. Qiu, I. Masiulis, J. D. Sweatt, L. Wei-Ping, G. Adelmann, M. Frotscher, R. E. Hammer and J. Herz, Neuron, 2005, 47, 567–579 CrossRef CAS PubMed.
- M. A. Lynch, K. L. Voss, J. Rodriguez and T. V. Bliss, Neuroscience, 1994, 60, 1–5 CrossRef CAS.
- J. Li, C. Chen, C. Chen, Q. He and H. Li, PLoS One, 2011, 6, e17365 CAS.
- P. Tarpey, J. Parnau, M. Blow, H. Woffenndin, G. Bignell, C. Cox, J. Coc, H. Davies, S. Edkins, S. Holden, A. Korny, U. Mallya, J. Moon, S. Omeara, A. Parker and P. Stephens, Am. J. Hum. Genet., 2004, 75, 318–324 CrossRef CAS PubMed.
- A. M. Huang, H. L. Wang, Y. P. Tang and E. H. Lee, J. Neurosci., 1998, 18, 4305–4313 CAS.
- J. Gräff and I. M. Mansuy, Behav. Brain Res., 2008, 192, 70–87 CrossRef PubMed.
- J. J. Day and J. D. Sweatt, Neurobiol. Learn. Mem., 2011, 96, 2–12 CrossRef CAS PubMed.
- V. Yuferov, D. A. Nielsen, O. Levran, M. Randesi, S. Hamon, A. Ho, S. Morgello and M. J. Kreek, Pharmacogenet. Genomics, 2011, 21, 185–196 CAS.
- F. D. Lubin, T. L. Roth and J. D. Sweatt, J. Neurosci., 2008, 28, 10576–10586 CrossRef CAS PubMed.
- J. Feng, Y. Zhou, S. L. Campbell, T. Le, E. Li, J. D. Sweatt, A. J. Silva and G. Fan, Nat. Neurosci., 2010, 13, 423–430 CrossRef CAS PubMed.
- J. J. Day and J. D. Sweatt, Neuropsychopharmacology, 2012, 37, 247–260 CrossRef CAS PubMed.
- C. A. Miller and J. D. Sweatt, Neuron, 2007, 15, 857–869 CrossRef PubMed.
- P. A. Jones and S. B. Baylin, Nat. Rev. Genet., 2002, 3, 415–428 CrossRef CAS.
- J. P. Lopez-Atalaya, S. Ito, L. M. Valor, E. Benito and A. Barco, Nucleic Acids Res., 2013, 41, 8072–8084 CrossRef CAS PubMed.
- L. J. Juan, W. J. Shia, M. H. Chen, W. M. Yang, E. Seto, Y. S. Lin and C. W. Wu, J. Biol. Chem., 2000, 275, 20436–20443 CrossRef CAS PubMed.
- W. G. Zhu, R. R. Lakshmanan, M. D. Beal and G. A. Otterson, Cancer Res., 2001, 61, 1327–1333 CAS.
- J. Hsieh, K. Nakashima and T. Kuwabara, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 16659–16664 CrossRef CAS PubMed.
- Z. Gao, K. Ure and J. L. Ables, Nat. Neurosci., 2009, 12, 1090–1092 CrossRef CAS PubMed.
- I. T. Yu, J. Y. Park and S. H. Kim, Neuropharmacology, 2009, 56, 473–480 CrossRef CAS PubMed.
- S. Jessberger, K. Nakashima and G. D. Clemenson, J. Neurosci., 2007, 27, 5967–5975 CrossRef CAS PubMed.
- H. J. Kim, P. Leeds and D. M. Chuang, J. Neurochem., 2009, 110, 1226–1240 CrossRef CAS PubMed.
- A. Contestabile, S. Sintoni and B. Monti, Curr. Psychopharmacol., 2012, 1, 14–28 CAS.
- C. M. Alberini, Physiol. Rev., 2009, 89, 121–145 CrossRef CAS PubMed.
- D. P. Stefanko, R. M. Barrett, A. R. Ly, G. K. Reolon and M. A. Wood, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 9447–9452 CrossRef CAS PubMed.
- J. S. Guan, S. J. Haggarty, E. Giacometti, J. H. Dannenberg, N. Joseph, J. Gao, T. J. Nieland, Y. Zhou, X. Wang and R. Mazitschek, Nature, 2009, 459, 55–60 CrossRef CAS PubMed.
- S. C. McQuown, R. M. Barrett, D. P. Matheos, R. J. Post, G. A. Rogge, T. Alenghat, S. E. Mullican, S. Jones, J. R. Rusche and M. A. Lazar, J. Neurosci., 2011, 31, 764–774 CrossRef CAS PubMed.
- G. Faraco, T. Pancani, L. Formentini, P. Mascagni, G. Fossati, F. Leoni, F. Moroni and A. Chiarugi, Mol. Pharmacol., 2006, 70, 1876–1884 CrossRef CAS PubMed.
- M. Malvaez, S. C. McQuowna, G. A. Roggea, M. Astarabadia, V. Jacquesc, S. Carreiroc, J. R. Ruschec and M. A. Wood, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 2647–2652 CrossRef CAS PubMed.
- R. J. Ferrante, J. K. Kubilus, J. Lee, H. Ryu, A. Beesen, B. Zucker, K. Smith, N. W. Kowall, R. R. Ratan, R. Luthi-Carter and S. M. Hersch, J. Neurosci., 2003, 23, 9418–9427 CAS.
- P. Chiurazzi, M. G. Pomponi, R. Pietrobono, C. E. Bakker, G. Neri and B. A. Oostra, Hum. Mol. Genet., 1999, 8, 2317–2323 CrossRef CAS PubMed.
- H. M. Amir and C. Frédéric, J. Mol. Biol., 2011, 409, 758–772 CrossRef PubMed.
- G. Lynch, C. S. Rex and C. M. Gall, Ageing Res. Rev., 2006, 5, 255–280 CrossRef CAS PubMed.
- J. Gräff and L.-H. Tsai, Annu. Rev. Pharmacol., 2013, 53, 331–330 CrossRef PubMed.
- S. Sintoni, E. Kurtys, M. Scandaglia, A. Contestabile and B. Monti, Pharmacol., Biochem. Behav., 2013, 106, 8–15 CrossRef CAS PubMed.
- C. T. Gualtieri, L. G. Johnson and K. B. Benedict, Journal of Neuropsychiatry, 2006, 18, 217–225 CrossRef PubMed.
- C. Hommet, K. Mondon, B. de Toffol and T. Constans, J. Am. Geriatr. Soc., 2007, 55, 628 CrossRef PubMed.
- E. T. Kavalali, E. D. Nelson and L. M. Monteggia, J. Neurodev. Disord., 2011, 3, 250–256 CrossRef PubMed.
- M. Adachi, A. E. Autry, H. E. Covington and L. M. Monteggia, J. Neurosci., 2009, 29, 4218–4227 CrossRef CAS PubMed.
- V. Campuzano, L. Montermini, M. D. Molto, L. Pianese, M. Cossee, F. Cavalcanti, E. Monros, F. Rodius, F. Duclos, A. Monticelli, F. Zara, J. Canizares, H. Koutnikova, S. I. Bidichandani, C. Gellera, A. Brice, P. Trouillas, G. D. Michele, A. Fila, R. D. Frutos, F. Palau, P. I. Patel, S. D. Donato, J. L. Mandel, S. Cocozza, M. Koenig and M. Pandolfo, Science, 1996, 271, 1423–1427 CAS.
- M. Rai, E. Soragni, C. J. Chou, G. Barnes, S. Jones, J. R. Rusche, J. M. Gottesfeld and M. Pandolfo, PLoS One, 2010, 5, e8825 Search PubMed.
- T. Abel and R. S. Zukin, Curr. Opin. Pharmacol., 2008, 8, 57–64 CrossRef CAS PubMed.
- K. L. Sugars and D. C. Rubinsztein, Trends Genet., 2003, 19, 233–238 CrossRef CAS.
- J. S. Steffan, L. Bodai, J. Pallos, M. Poelman, A. McCampbell, B. L. Apostol, A. Kazantsev, E. Schmidt, Y. Z. Zhu, M. Greenwald, R. Kurokawa, D. E. Housman, G. R. Jackson, J. L. Marsh and L. M. Thompson, Nature, 2001, 413, 739–743 CrossRef CAS PubMed.
- G. Gardian, S. E. Browne, D. K. Choi, P. Klivenyi, J. Gregorio, J. K. Kubilus, H. Ryu, B. Langley, R. R. Ratan, R. J. Ferrante and M. F. Beal, J. Biol. Chem., 2005, 280, 556–563 CAS.
- E. Hockly, V. M. Richon, B. Woodman, D. L. Smith, X. Zhou, E. Rosa, K. Sathasivam, S. Ghazi-Noori, A. Mahal, P. A. Lowden, J. S. Steffan, J. L. Marsh, L. M. Thompson, C. M. Lewis, P. A. Marks and G. P. Bates, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 2041–2046 CrossRef CAS PubMed.
- E. A. Thomas, G. Coppola, P. A. Desplats, B. Tang, E. Soragni, R. Burnett, F. Gao, K. M. Fitzgerald, J. F. Borok, D. Herman, D. H. Geschwind and J. M. Gottesfeld, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 15564–15569 CrossRef CAS PubMed.
- M. Beconi, O. Aziz, K. Matthews, L. Moumné, C. O'Connell, D. Yates, S. Clifton, H. Pett, J. Vann, L. Crowley, A. F. Haughan, D. L. Smith, B. Woodman, G. P. Bates, F. Brookfield, R. W. Bürli, G. McAllister, C. Dominguez, I. Munoz-Sanjuan and V. Beaumont, PLoS One, 2012, 7, e44498 CAS.
- G. Kashiwazaki, T. Bando, T. Yoshidome, S. Masui, T. Takagaki, K. Hashiya, G. N. Pandian, J. Yasuoka, K. Akiyoshi and H. Sugiyama, J. Med. Chem., 2012, 55, 2057–2066 CrossRef CAS PubMed.
- G. N. Pandian and H. Sugiyama, Biotechnol. J., 2012, 7, 798–809 CAS.
- R. D. Taylor, S. Asamitsu, T. Takenaka, M. Yamamoto, K. Hashiya, Y. Kawamoto, T. Bando, H. Nagase and H. Sugiyama, Chem. - Eur. J., 2013, 19, 1–9 CrossRef.
- R. Burnett, C. Melander, J. W. Puckett, L. S. Son, R. D. Wells, P. B. Dervan and J. M. Gottesfeld, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 11497–11502 CrossRef CAS PubMed.
- D. M. Chenoweth, J. L. Meier and P. B. Dervan, Angew. Chem., Int. Ed., 2013, 52, 415–418 CrossRef CAS PubMed.
- A. Ohtsuki, M. T. Kimura, M. Minoshima, T. Suzuki, M. Ikeda, T. Bando, H. Nagase, K. ichi Shinohara and H. Sugiyama, Tetrahedron Lett., 2009, 50, 7288–7292 CrossRef CAS PubMed.
- G. N. Pandian, K. Shinohara, A. Ohtsuki, Y. Nakano, M. Masafumi, T. Bando, H. Nagase, Y. Yamada, A. Watanabe, N. Terada, S. Sato, H. Morinaga and H. Sugiyama, ChemBioChem, 2011, 12, 2822–2828 CrossRef CAS PubMed.
- G. N. Pandian, A. Ohtsuki, T. Bando, S. Sato, K. Hashiya and H. Sugiyama, Bioorg. Med. Chem., 2012, 20, 2656–2660 CrossRef CAS PubMed.
- G. N. Pandian, Y. Nakano, S. Sato, H. Morinaga, T. Bando, H. Nagase and H. Sugiyama, Sci. Rep., 2012, 2, e544 Search PubMed.
- L. Han, G. N. Pandian, S. Junetha, S. Sato, C. Anandhakumar, J. Taniguchi, A. Saha, T. Bando, H. Nagase and H. Sugiyama, Angew. Chem., Int. Ed., 2013, 52, 13410–13413 CrossRef CAS PubMed.
- G. N. Pandian, J. Taniguchi, S. Junetha, S. Sato, L. Han, A. Saha, C. AnandhaKumar, T. Bando, H. Nagase, T. Vaijayanthi, R. D. Taylor and H. Sugiyama, Sci. Rep., 2014, 4, e3843 Search PubMed.
- G. N. Pandian and H. Sugiyama, Pharmaceuticals, 2012, 6, 1–24 CrossRef PubMed.
- A. Saha, G. N. Pandian, S. Sato, J. Taniguchi, K. Hashiya, T. Bando and H. Sugiyama, Bioorg. Med. Chem., 2013, 21, 4201–4209 CrossRef CAS PubMed.
- T. Vaijayanthi, T. Bando, G. N. Pandian and H. Sugiyama, ChemBioChem, 2012, 13, 2170–2185 CrossRef CAS PubMed.
- T. Vaijayanthi, T. Bando, K. Hashiya, G. N. Pandian and H. Sugiyama, Bioorg. Med. Chem., 2013, 21, 852–855 CrossRef CAS PubMed.
- J. W. Trauger, E. E. Baird and P. B. Dervan, J. Am. Chem. Soc., 1998, 120, 3534–3535 CrossRef CAS.
- T. Matsui, M. Takano, K. Yoshida, S. Ono, C. Fujisaki, Y. Matsuzaki, Y. Toyama, M. Nakamura, H. Okano and W. Akamatsu, Stem Cells, 2012, 30, 1109–1119 CrossRef CAS PubMed.
- J. L. Meier, D. C. Montgomery and P. B. Dervan, Nucleic Acids Res., 2012, 40, 2345–2356 CrossRef CAS PubMed.
- A. Fukasawa, T. Aoyama, T. Nagashima, N. Fukuda, T. Ueno, H. Sugiyama, H. Nagase and Y. Matsumoto, Biopharm. Drug Dispos., 2009, 30, 81–89 CrossRef CAS PubMed.
- T. W. Synold, B. Xi, J. Wu, Y. Yen, B. C. Li, F. Yang, J. W. Phillips, N. G. Nickols and P. B. Dervan, Cancer Chemother. Pharmacol., 2012, 70, 617–625 CrossRef CAS PubMed.
- C. Chiruta, D. Schubert, R. Dargusch and P. Maher, J. Med. Chem., 2012, 55, 378–389 CrossRef CAS PubMed.
- S. Sivilia, L. Lorenzini, A. Giuliani, M. Gusciglio, M. Fernandez, V. A. Baldassarro, C. Mangano, L. Ferraro, V. Pietrini, M. F. Baroc, A. R. Viscomi, S. Ottonello, G. Villetti, B. P. Imbimbo, L. Calzà and L. Giardino, BMC Neurosci., 2013, 14, 44 CrossRef CAS PubMed.
- H. Y. Zhang, FEBS Lett., 2005, 579, 5260–5264 CrossRef CAS PubMed.
- M. L. Bolognesi, A. Cavalli, L. Valgimigli, M. Bartolini, M. Rosini, V. Andrisano, M. Recanatini and C. Melchiorre, J. Med. Chem., 2007, 50, 6446–6449 CrossRef CAS PubMed.
- M. Rosini, E. Simoni, M. Bartolini, A. Cavalli, L. Ceccarini, N. Pascu, D. W. McClymont, A. Tarozzi, M. L. Bolognesi, A. Minarini, V. Tumiatti, V. Andrisano, I. R. Mellor and C. Melchiorre, J. Med. Chem., 2008, 51, 4381–4384 CrossRef CAS PubMed.
- V. Capurro, P. Busquet, J. P. Lopes, R. Bertorelli, G. Tarozzo, M. L. Bolognesi, D. Piomelli, A. Reggiani and A. Cavalli, PLoS One, 2013, 8, e56870 CAS.
- M. B. H. Youdim, Exp. Neurobiol., 2013, 22, 1–10 CrossRef PubMed.
- C. Lu, Y. Guo, J. Yan, Z. Luo, H.-B. Luo, M. Yan, L. Huang and X. Li, J. Med. Chem., 2013, 56, 5843–5859 CrossRef CAS PubMed.
- C. Qian, C.-J. Lai, R. Bao, D.-G. Wang, J. Wang, G.-X. Xu, R. Atoyan, H. Qu, L. Yin, M. Samson, B. Zifcak, A. W. S. Ma, S. DellaRocca, M. Borek, H.-X. Zhai, X. Cai and M. Voi, Clin. Cancer Res., 2012, 18, 4104–4113 CrossRef CAS PubMed.
- Y. L. Wu, G. N. Pandian, Y. P. Ding, W. Zhang, Y. Tanaka and H. Sugiyama, Chem. Biol., 2013, 20, 1311–1322 CrossRef CAS PubMed.
- G. N. Pandian, J. Taniguchi and H. Sugiyama, Clin. Transl. Med., 2014, 3, 6 CrossRef PubMed.
- S. Masuda, J. Wu, T. Hishida, G. N. Pandian, H. Sugiyama and J. C. I. Belmonte, J. Mol. Cell. Biol., 2013, 5, 354–355 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2014 |