Immobilization of bioactive factor-loaded liposomes on the surface of electrospun nanofibers targeting tissue engineering
Nelson
Monteiro
ab,
Albino
Martins
ab,
Ricardo
Pires
ab,
Susana
Faria
c,
Nuno A.
Fonseca
d,
João N.
Moreira
d,
Rui L.
Reis
ab and
Nuno M.
Neves
*ab
a3B's Research Group – Biomaterials, Biodegradables and Biomimetics, Department of Polymer Engineering, University of Minho, Headquarters of the European Institute of Excellence on Tissue Engineering and Regenerative Medicine, AvePark, Zona Industrial da Gandra S. Cláudio do Barco, 4806-909 Caldas das Taipas, Guimarães, Portugal. E-mail: nelson.monteiro@dep.uminho.pt; amartins@dep.uminho.pt; rpires@dep.uminho.pt; rgreis@dep.uminho.pt; nuno@dep.uminho.pt
bICVS/3B's, PT Government Associate Laboratory, Braga/Guimarães, Portugal
cDepartment of Mathematics for Science and Technology, Research CMAT, University of Minho, Campus de Azurém, 4800-058 Guimarães, Portugal. E-mail: sfaria@math.uminho.pt
dCNC – Center for Neurosciences and Cell Biology, FFUC – Faculty of Pharmacy of the University of Coimbra, 3000 Coimbra, Portugal. E-mail: nuno.a.c.fonseca@gmail.com; jmoreira@ff.uc.pt
First published on 19th May 2014
Abstract
Electrospun nanofiber meshes (NFM), due to their morphology and fibrous structure, are extensively proposed as biomedical devices, for tissue engineering on scaffolds and also as drug delivery systems. Liposomes are nanoparticles prepared from a biologically derived material (phospholipid), which are already in clinical use as a drug release device. Liposomes may be combined with biomaterial scaffolds to promote a local and sustained delivery of loaded bioactive agents. The main objective of the present study is to evaluate the efficacy of dexamethasone (Dex)-loaded liposomes immobilized on the surface of electrospun polycaprolactone (PCL) NFM for promoting the osteogenic differentiation of human bone marrow-derived mesenchymal stem cells (hBMSCs). The in vitro release profile demonstrates a sustained release of Dex over 21 days, after an initial burst release over 12 h. Biological assays show that Dex-loaded liposomes immobilized on the surface of electrospun PCL NFMs do not exhibit any cytotoxic effect, being able to successfully promote the osteogenic differentiation of hBMSCs. We herein validate the concept of using liposomes immobilized on the surface of a nanostructured fibrous system to be used as an advanced cell carrier device with autonomous release of growth/differentiation factors relevant for tissue engineering and regenerative medicine strategies.
1. Introduction
There is a huge need for more effective treatment modalities capable of regenerating damaged or diseased tissues. This important clinical need stimulates the development of new technologies in many fields, but particularly in the interdisciplinary areas encompassing tissue engineering and regenerative medicine (TERM). Therefore, by applying combinations of biomaterial scaffolds, cells and bioactive molecules the tissue regeneration can be guided and stimulated.1 Biomaterial scaffolds were already previously developed to deliver cell regulatory signals such as growth differentiation factors and cytokines. Those scaffolds provide powerful microenvironments to positively influence and guide the stem cell fate, both in ex vivo culture and post-implantation in vivo.2 Biomaterial scaffolds for TERM can also be used as reservoirs or release systems for bioactive molecules.3–5 They can be functionalized to serve as local release systems modulating the activities of communities of cells in direct contact with the device including their coordinated differentiation.6–8 Specifically, the drug delivery kinetics is particularly relevant when the therapeutic agent is a growth/differentiation factor aimed to achieve effective dose and spatiotemporal release kinetics at the intended site of injury.9,10 Strategies that combine scaffolds and drug delivery systems have the potential to provide more effective tissue regeneration when compared with currently available therapies.10Electrospinning has attracted huge interest from the biomaterials research community as a simple and versatile technique enabling production of polymeric ultrafine fibers with diameters ranging from a few micrometers down to tens of nanometers.11,12 Electrospun fiber meshes exhibit a high specific surface area, excellent mechanical properties and flexibility in surface functionalities.11,13 Moreover, electrospun nanofiber meshes (NFM) enable mimicking of the morphology of the extracellular matrix of native tissues.14 Electrospun NFM surfaces have been chemically functionalized to achieve sustained delivery of various bioactive molecules, just by physical adsorption.12,15 Optimized surfaces may be achieved in the nanofibers by gas plasma treatments,16 UV–ozone treatments,17 wet chemical methods,15 surface graft polymerization,18 and co-electrospinning of bioactive agents and polymers.19 Several bioactive molecules including antibiotics,20 siRNA,21 growth or differentiation factors,4,22 anti-cancer drugs, enzymes and cytokines have been entrapped within electrospun NFMs for controlled drug delivery.15,20 Electrospun NFMs have also been chemically modified to allow immobilization of specific cell ligands to enhance cell adhesion, proliferation and differentiation by mimicking the morphology and biological functions of the native natural ECM.15,23–25
The most important advantages of combining TE scaffolds with growth factor-loaded nanoparticles are the possibility and flexibility of obtaining a well-controlled, sustained and local release. Examples of release systems are liposomes, dendrimers and polymeric nanoparticles.1,26–28 Immobilized nanoparticles interact not only with the target cells, but also with the substrates on which the cells are cultured. These combined release systems may be used to improve the therapeutic efficacy and safety of growth factors, by delivering them at the site of action and at a rate dictated by the need of the physiological environment.29,30 Indeed, it is well described that nanoparticle immobilization is thought to increase the growth factor efficiency by increasing its local concentration closer to the cell surfaces and also by increasing its bioavailability and half-life.31,32
Liposomes have specific advantages in controlling the delivery of growth factors, when immobilized/incorporated onto biomaterial scaffolds. Both the structure and the surface properties can be optimized for that specific application. An example is the incorporation of polyethylene glycol (PEG) molecules bearing terminal amine and thiol groups onto the liposome surfaces. These modified liposomes were already covalently bound onto stainless steel disks.33 Other studies described the sustained release systems that use liposomes loaded with proteins or drugs and that are further incorporated into fibrin sealants.34–36 Using these carriers it is possible to maintain therapeutic drug levels at the defect site, minimizing the total amount of drug required when compared with the systemic administration dose and its side effects.3,37 Three different model drugs (carboxyfluorescein, doxorubicin, and lysozyme) were previously encapsulated into liposomes and immobilized onto collagen/hydroxyapatite composite scaffolds.37 The drugs entrapped into the liposomes showed a slow release from scaffolds. Mickova et al. demonstrated that electrospinning does not allow processing intact liposomes.38 Alternatively, coaxial electrospinning allowed the successful incorporation of liposomes into the nanofibers.38
Cell culture devices using this new approach may be efficiently used to control the behavior of stem cells. MSCs hold great potential in several therapies due to their unique biological characteristics, such as self-renewal and capability to differentiate into specific cells. These attributes make them excellent candidates for cell and tissue regeneration therapies.4,39–41 Herein, we developed multi-functionalized electrospun polycaprolactone (PCL) NFMs with the potential to be used as stem cell differentiation inductive structures, through the covalent immobilization of drug-loaded liposomes carrying Dex. For that, electrospun PCL NFMs were functionalized using UV–ozone irradiation and aminolysis, followed by the insertion of thiol groups using 2-iminothiolane (2IT). PCL is hydrophobic and lacks the functional groups intended for liposome immobilization. The UV–ozone irradiation increases the hydrophilicity of the polymer surface by generating polar groups such as –OH and –COOH.42 The polar groups can be advantageous for the insertion of NH2 groups by aminolysis and further insertion of the thiol groups, when used for liposome immobilization. Furthermore, we intended to analyze the effect of Dex-loaded liposomes immobilized on the surface of electrospun PCL NFMs on the viability, proliferation, protein synthesis and differentiation of human bone marrow-derived mesenchymal stem cells (hBMSCs).
2. Results
2.1. Development and characterization
One aim of this study was to develop a method to generate functional groups on the surface of electrospun PCL NFMs, which provide binding sites suitable for immobilizing drug-loaded liposomes. Previously, we optimized the encapsulation of Dex into the liposomes (formulation F1).49 In this work, we used the optimized liposome formulation, and we added DSPE-PEG-Mal to the liposome formulations (F2 and F3) to bind to the generated functional groups. In Table 1, various liposome formulations used in this study are presented (see the description in the Experimental section). The formulation F1 was used to perform the biological assays from liposomes in suspension. The formulations F2 (with cholesterol – Chol) and F3 (without Chol) were used for the analysis of the presence of liposomes immobilized on the surface of the PCL NFM by florescence microscopy and also for the biological assays. Fig. 1 shows a scheme of the liposome formulations and the chemical pathway followed for the covalent immobilization of the Dex-loaded liposomes on the surface of PCL NFM (NFM4_F2 and NFM4_F3).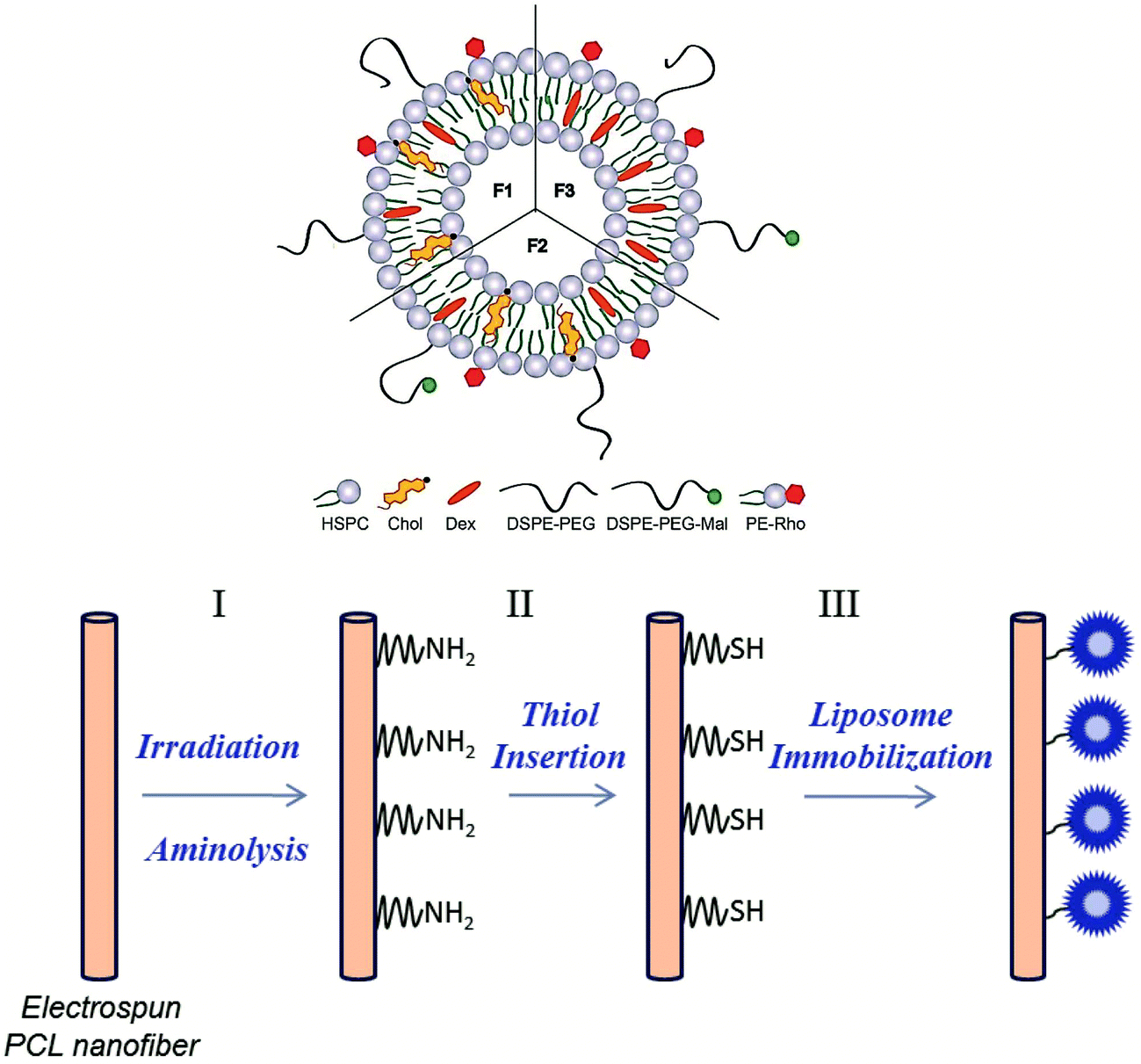 | ||
| Fig. 1 Liposome formulations and the chemical pathway used for the immobilization of liposomes on the surface of modified PCL NFM (NFM_F2 and NFM_F3). (I) UV–ozone irradiation and aminolysis treatment, (II) primary amine group insertion and 2-iminothiolane reaction (at pH 8), (III) thiol group insertion and maleimide reaction (at pH 7). | ||
| Dex | HSPC | Chol | DSPE-PEG | DSPE-PEG-Mal | PE-Rho | |
|---|---|---|---|---|---|---|
| F1 | 0.25 | 2 | 0.1 | 0.1 | — | 0.02 |
| F2 | 0.25 | 2 | 0.1 | 0.04 | 0.06 | 0.02 |
| F3 | 0.25 | 2 | — | 0.04 | 0.06 | 0.02 |
| UV–ozone time (min) | SH/NH2 (nmol cm−2) |
|---|---|
| 0 | 0 |
| 1 | 0.60 ± 0.09 |
| 2 | 1.17 ± 0.09 |
| 3 | 1.87 ± 0.37 |
| 4 | 2.83 ± 0.11 |
| 5 | 2.94 ± 0.11 |
The UV–ozone irradiation time was selected taking into account the morphology and the concentration of SH groups. If the selection is only based on the quantification of SH groups on the surface our choice would be 5 min UV–ozone treatment time. However, this treatment has an impact on the morphology of the meshes, thus the UV–ozone treatment time was set at 4 min. Other parameters may affect the efficiency of grafting the SH groups on the surface of the NFMs, namely the reaction time of the aminated surface with 2IT (used to generate the free SH groups) and of the SH-functionalized surface with DTNB (used to quantify the SH groups). The effect of both parameters is shown in Table 3. The effect of the DTNB seems to be higher than the effect of 2IT. Given those results the condition selected was 3 h 2IT and 3 h DTNB.
| 2IT (h) | DTNB (h) | SH/NH2 (nmol cm−2) |
|---|---|---|
| 1 | 1 | 1.62 ± 0.17 |
| 2 | 2 | 3.15 ± 0.40 |
| 3 | 1 | 2.11 ± 0.17 |
| 1 | 3 | 5.01 ± 0.42 |
| 3 | 3 | 5.52 ± 0.53 |
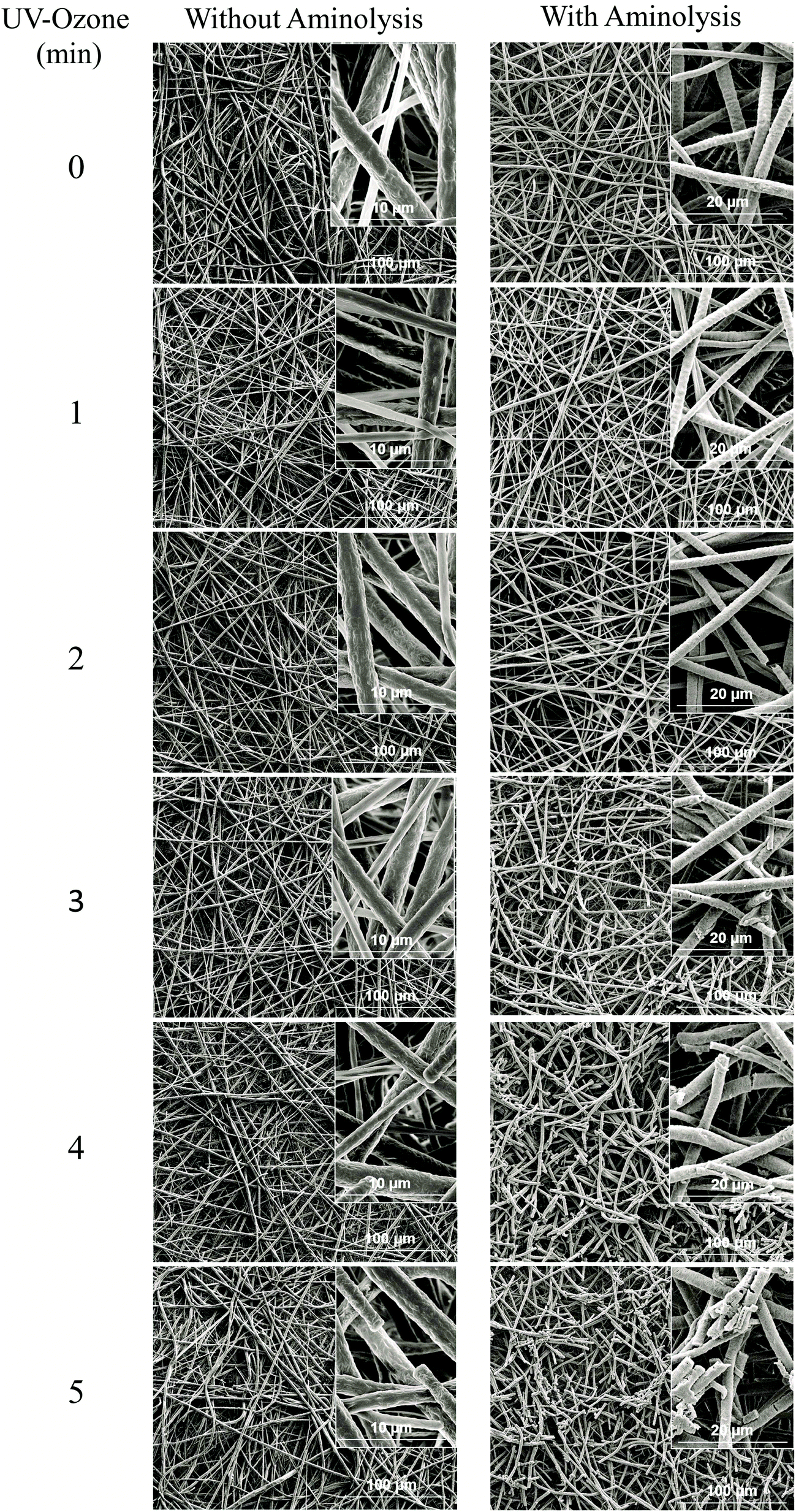 | ||
| Fig. 2 SEM micrographs of UV–ozone irradiated NFMs (exposure time ranging from 0 to 5 min) without and with aminolysis (t = 1 h). | ||
 | ||
| Fig. 3 Fluorescence micrograph of the electrospun PCL NFM (A); negative control (NFM) exposed to BODIPY but without SH groups (B) and SH-functionalized NFM reacted with BODIPY (C). | ||
| Formulation | ζ-Potential | Size (nm) |
|---|---|---|
| F1 | −23.10 ± 1.68 | 103.70 ± 15.74 |
| F2 | −22.50 ± 3.40 | 119.10 ± 6.57 |
| F3 | −19.96 ± 2.60 | 113.20 ± 6.11 |
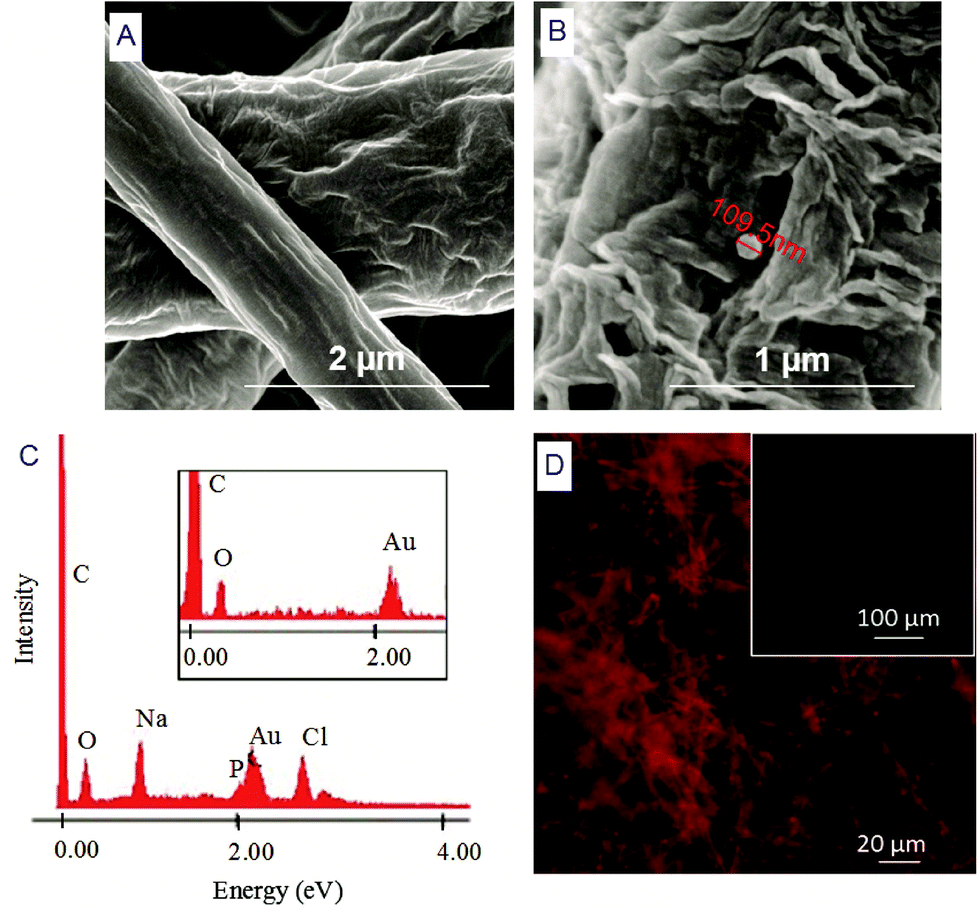 | ||
| Fig. 4 SEM, EDS and fluorescence micrographs. (A) NFM without liposomes; (B) details of a liposome (F3) immobilized on the surface of functionalized NFMs (t = 4 min); (C) EDS of the surface of electrospun NFM showing the presence of phosphorus (P) (absent in the inset represents the EDS of NFM without liposome immobilization); (D) fluorescence micrograph of PE-Rho marked liposomes immobilized on the surface of SH functionalized NFM (the inset shows negative controls without PE-Rho liposomes). | ||
| Sample | Cholesterol (ratio) | Nominal size | Volume of liposomes | Washing | Dex (μM) |
|---|---|---|---|---|---|
| NFM_F2 | 0.1 | 100 nm | 1.5 mL | 1 | 2.91 ± 0.75 |
| 100 nm | 1.5 mL | No | 15.98 ± 2.25 | ||
| 1 | 11.00 ± 0.92 | ||||
| 2 | 11.05 ± 0.34 | ||||
| 3 | 11.38 ± 0.65 | ||||
| NFM_F3 | 0 | 400 nm | 1.5 mL | No | 20.95 ± 0.73 |
| 1 | 18.76 ± 0.92 | ||||
| 2 | 17.77 ± 1.32 | ||||
| 3 | 17.70 ± 0.65 | ||||
| 100 nm | 4.5 mL | No | 31.46 ± 4.12 | ||
| 1 | 19.65 ± 1.54 |
These results show that the SH groups were saturated, and some of the liposomes were not covalently linked to the nanofiber mesh surface. Thus, the amount of liposomes immobilized is specifically controlled by the amount of SH groups available on the surface, the remaining liposomes being removed with the first washing. The conditions selected for further Dex release and biological tests were liposomes immobilized on the surface of the activated and functionalized NFMs with cholesterol (NFM4_F2) and without cholesterol, with an average size of 100 nm, 4.5 mL and first washing (NFM4_F3).
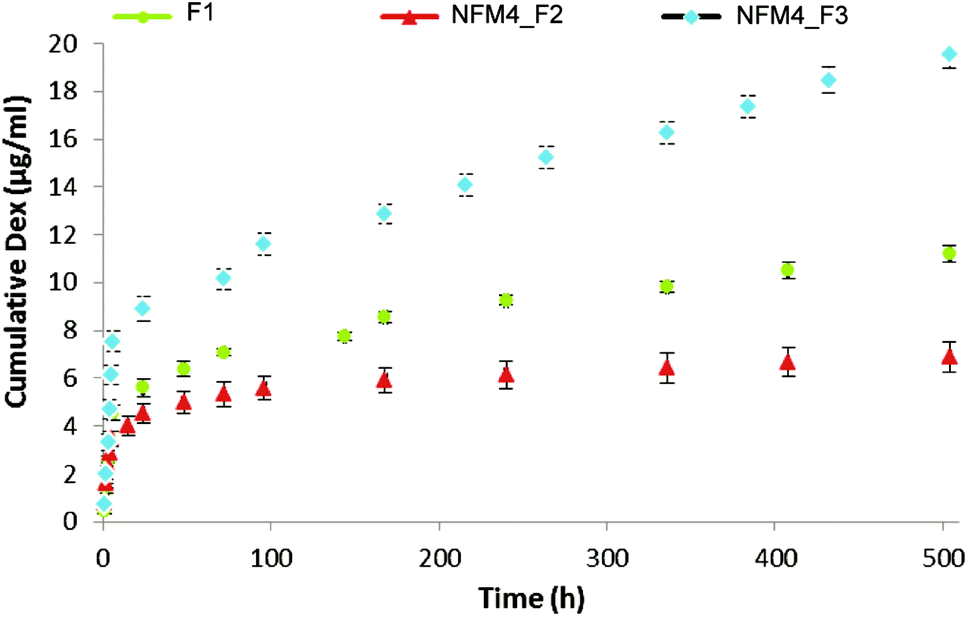 | ||
| Fig. 5 In vitro cumulative Dex release from liposomes in suspension (F1) and immobilized on the surface of activated and functionalized NFMs with and without cholesterol (NFM4_F2 and NFM4_F3). | ||
2.2. Biological activity
The effectiveness of these multifunctionalized systems was assessed by culturing hBMSCs in osteogenic differentiation medium. Table 6 describes the experimental conditions used in the biological assays (see the description in the Experimental section).| Condition | Description |
|---|---|
| NFM0_Ost | Non-functionalized PCL NFMs in standard osteogenic medium |
| NFM0_F1 | Non-functionalized PCL NFMs with Dex-loaded liposomes (F1) in suspension in Dex-absent osteogenic differentiation medium |
| NFM4_Ost | SH-functionalized PCL NFMs in standard osteogenic medium |
| NFM4_F2 | SH-functionalized PCL NFMs with Dex-loaded liposomes (F2 with Chol) immobilized on the surface in Dex-absent osteogenic differentiation medium |
| NFM4_F3 | SH-functionalized PCL NFMs with Dex-loaded liposomes (F3 without Chol) immobilized on the surface in Dex-absent osteogenic differentiation medium |
| NFM_5%Dex | Non-functionalized PCL NFMs loaded with 5% of Dex in Dex-absent osteogenic differentiation medium |
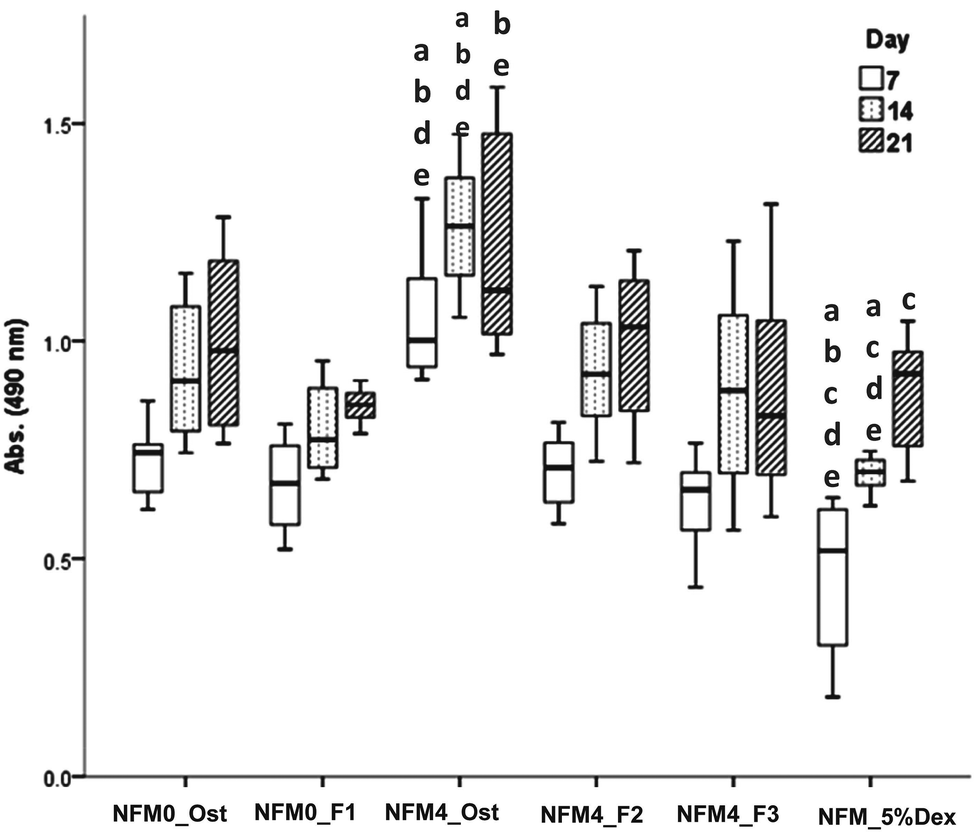 | ||
| Fig. 6 Box plot of hBMSCs’ viability (i.e. Abs. (490 nm)) when cultured on NFM0_Ost, NFM0_F1, NFM4_Ost, NFM4_F2, NFM4_F3 and NFM_5%Dex during 7, 14 and 21 days. a denotes significant differences compared to NFM0_Ost, b denotes significant differences compared to NFM0_F1, c denotes significant differences compared to NFM4_Ost, d denotes significant differences compared to NFM4_F2 and e denotes significant differences compared to NFM4_F3. | ||
In terms of cell proliferation (Fig. 7), on the 7th day under the hBMSC culture conditions Dex-loaded liposomes without cholesterol immobilized on the surface of UV–ozone treated NFMs (NFM4_F3) and the NFM_5%Dex displayed significantly lower DNA concentration values than all other culture conditions (p < 0.001). The same culture conditions (i.e. the NFM4_F3 and the NFM_5%Dex) also displayed significantly lower DNA concentration values on the 14th and 21st day of culture than the NFM4_Ost and the NFM4_F2 (p < 0.001). Inversely, hBMSCs cultured on the NFM4_Ost displayed significantly higher proliferation than on NFM0_Ost, NFM4_F3 and NFM_5%Dex (p < 0.001) on the 14th and 21st day. This result suggests that an effective release of Dex on the surface of the NFMs may interfere with the metabolic activity of the cells under those conditions. The supplementation of the medium or the release of Dex from liposomes in suspension seems not to have a similar effect over the proliferation of cells.
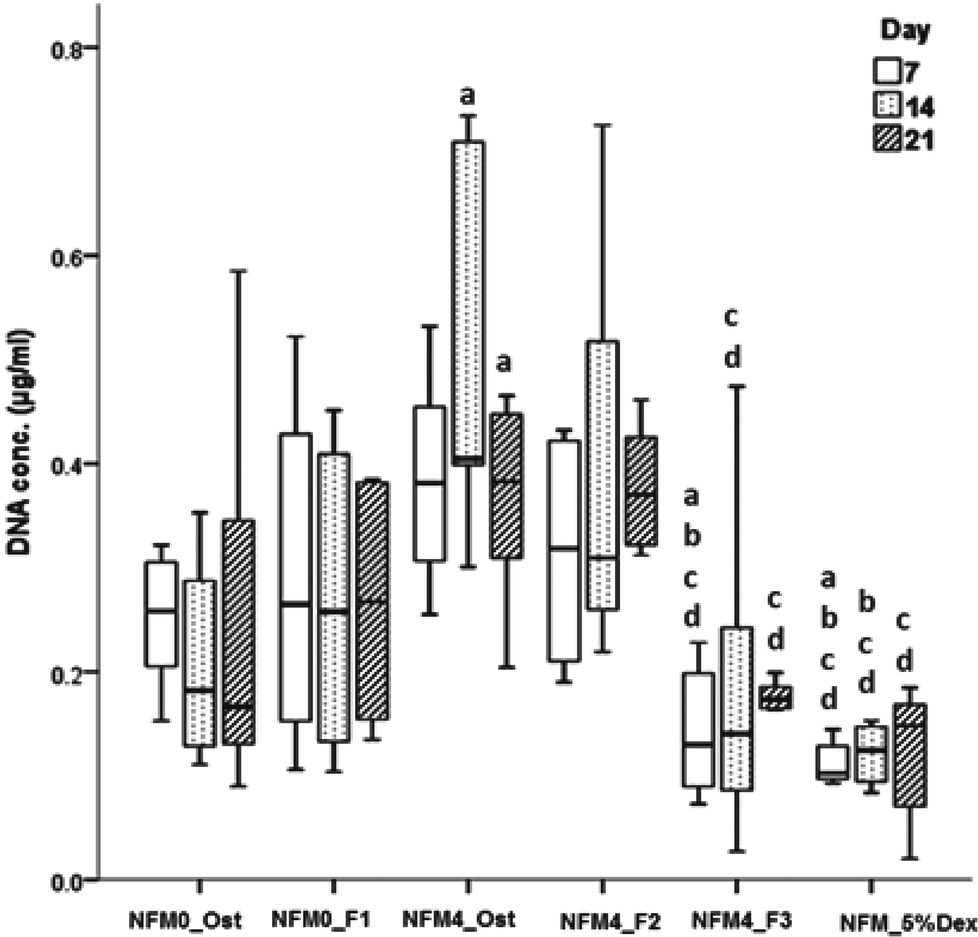 | ||
| Fig. 7 Box plot of the hBMSCs’ proliferation (i.e. DNA conc. (μg ml−1)) when cultured on NFM0_Ost, NFM0_F1, NFM4_Ost, NFM4_F2, NFM4_F3 and NFM_5%Dex during 7, 14 and 21 days. a denotes significant differences compared to NFM0_Ost, b denotes significant differences compared to NFM0_F1, c denotes significant differences compared to NFM4_Ost, d denotes significant differences compared to NFM4_F2. | ||
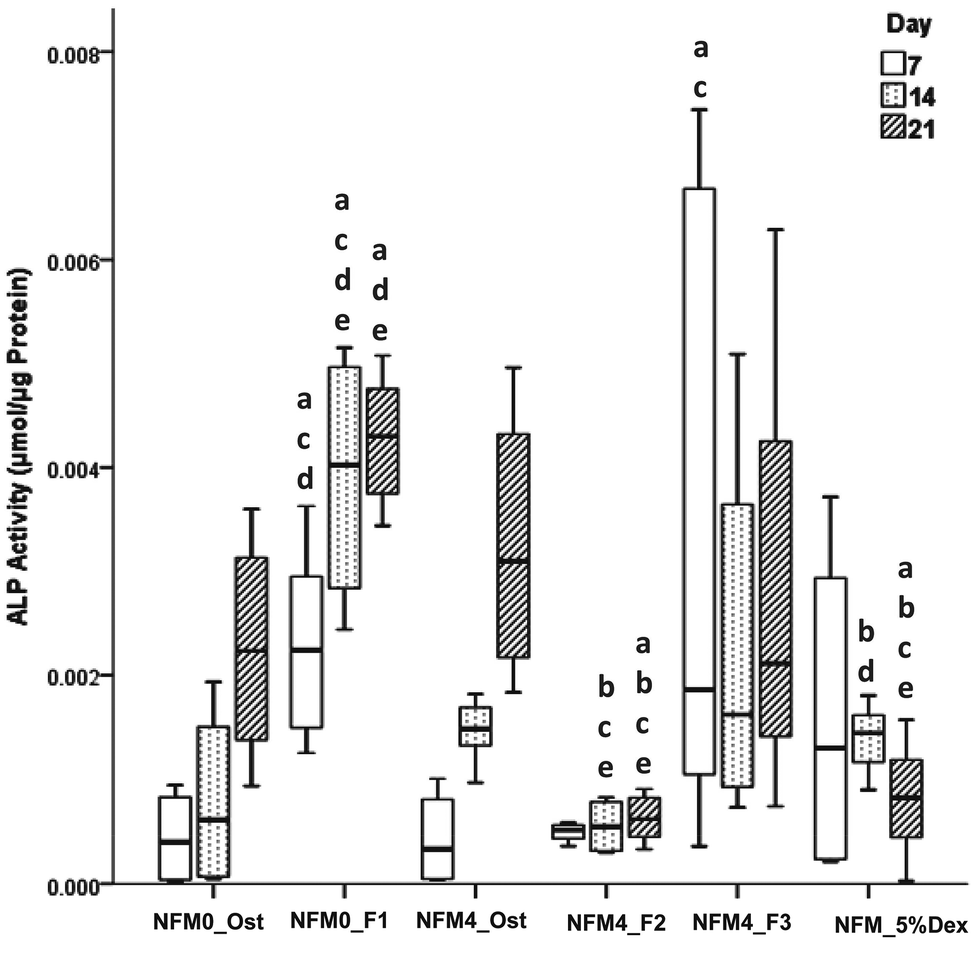 | ||
| Fig. 8 Box plot of the ALP activity (μmol h−1 μg−1 protein) from the hBMSCs cultured on NFM0_Ost, NFM0_F1, NFM4_Ost, NFM4_F2, NFM4_F3 and NFM_5%Dex during 7, 14 and 21 days. a denotes significant differences compared to NFM0_Ost, b denotes significant differences compared to NFM0_F1, c denotes significant differences compared to NFM4_Ost, d denotes significant differences compared to NFM4_F2 and e denotes significant differences compared to NFM4_F3. | ||
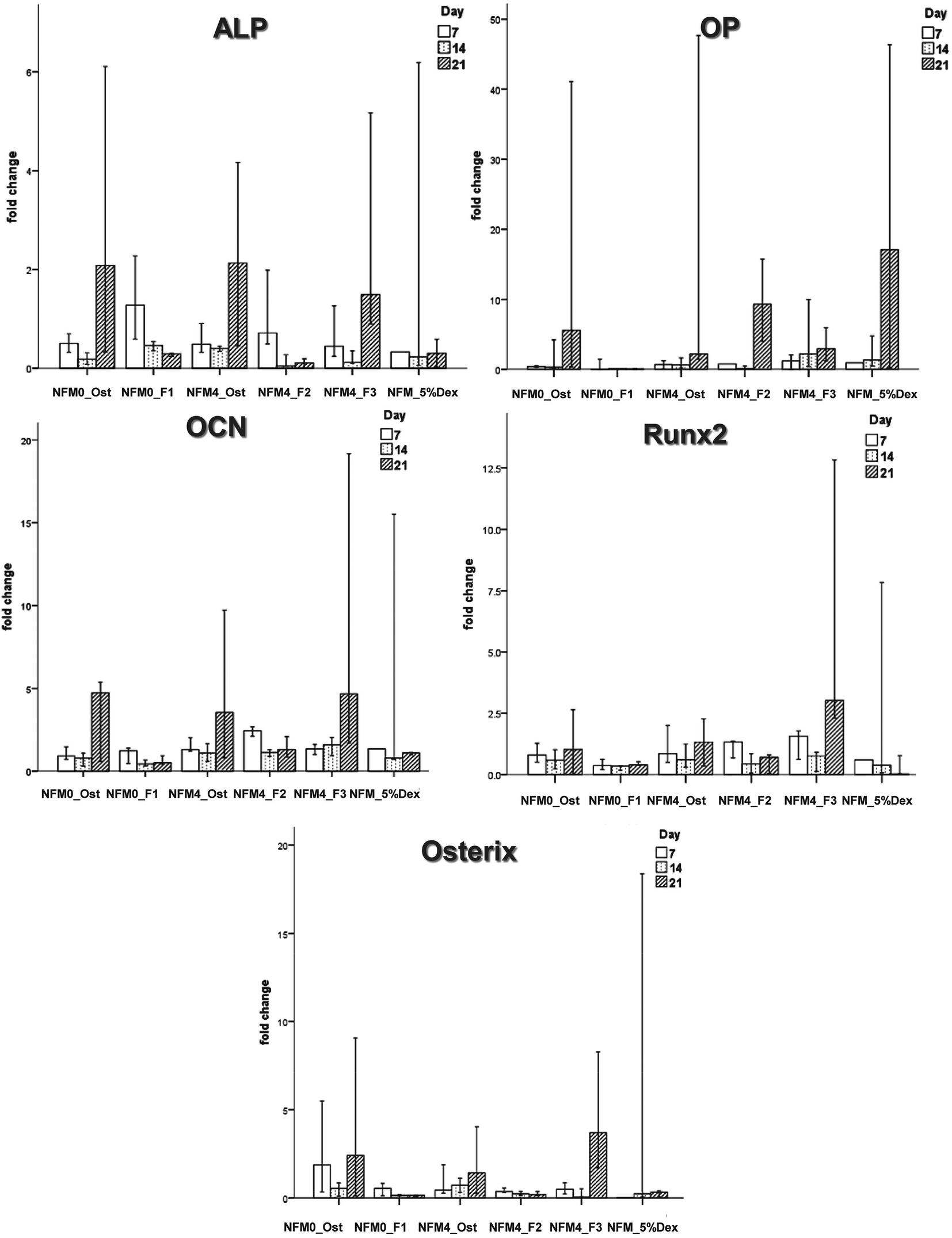 | ||
| Fig. 9 Bar plot of the ALP, OP, OCN, Runx2, and Osterix in NFM0_Ost, NFM0_F1, NFM4_Ost, NFM4_F2, NFM4_F3 and NFM_5%Dex. | ||
| Conditions | ALP | OP | OCN | Runx2 | Osterix |
|---|---|---|---|---|---|
| NFM0_Ost | 0.023 | 0.283 | 0.190 | 0.584 | 0.296 |
| NFM0_F1 | 0.018 | 0.888 | 0.126 | 0.117 | 0.231 |
| NFM4_Ost | 0.105 | 0.034 | 0.500 | 0.551 | 0.905 |
| NFM4_F2 | 0.049 | 0.036 | 0.041 | 0.176 | 0.184 |
| NFM4_F3 | 0.013 | 0.397 | 0.044 | 0.034 | 0.020 |
| NFM_5%Dex | 0.852 | 0.757 | 0.400 | 0.543 | 0.311 |
3. Discussion
Indeed, we4 and others have already reported the loading of dexamethasone (as a model drug) in electrospun meshes, showing its efficacy in differentiating stem cells. The present manuscript describes a very different concept, in which we immobilize on the surface of the PCL electrospun substrate liposomes, which release dexamethasone on the surface locally. This innovative method is needed since it enables functionalization of the surface of polymeric substrates (any polymeric substrate where we can find or insert amine groups) enabling the release of a bioactive agent on the surface where the cells are attached. As such, this work shows much enhanced versatility in the range of substrate materials where it can be applied and the liposomes were already shown to be able to carry many different types of drugs (including hydrophilic and hydrophobic), supporting the claim that this strategy may be applied for many different strategies. The advantages of our system, besides its versatility, include a substantial reduction of the total amount of drug used, maximizing its efficacy. The drug is not affected by the electrospinning process, not requiring exposure of the drugs to the solvents used for processing.3.1. Development and characterization
Herein, we report the chemical modification of electrospun PCL NFMs enabling the immobilization of Dex-loaded liposomes onto their surfaces. To achieve this purpose, initial UV–ozone irradiation was used to generate reactive free radicals that were immediately subjected to aminolysis. These modified surfaces were reacted with 2IT to generate sulfide (SH) pendant groups. Dex-loaded liposomes were covalently bonded to the SH groups present at the surface of electrospun NFMs. The availability of the drug-release vehicle at the surface of the NFMs (where initial cellular contact occurs) enables a sustained release of the loaded drug in the vicinity of the cells in culture, consequently increasing its efficacy and bioavailability. The activation and functionalization study demonstrated that the concentration of free SH groups at the surface of NFMs increases with the UV–ozone irradiation time (Table 2). The optimum SH concentration is achieved at 4 min of UV–ozone irradiation. Moreover, the effect of 2IT and DTNB reaction times on the grafting of SH groups was determined. Analyzing the effect of both factors on the quantification of SH groups (Table 2), the impact of the variation in t of DTNB is more relevant than t of 2IT. Therefore, in accordance with the data presented in the SEM micrographs (Fig. 2), the UV–ozone irradiation and aminolysis have a significant effect on the integrity of the electrospun PCL NFM causing shortening of the fibers. The damage to the nanofibers is mainly due to the aminolysis reaction. In fact, this effect has also been detected and previously reported in the literature44 in amorphous biodegradable aliphatic polyester nanofibers. However, the morphology of the nanofibers present in the bulk of the mesh underneath the surface is significantly affected neither by the irradiation nor by the chemical treatment. A BODIPY fluorescent dye was covalently bound to the SH groups present at the surface of electrospun NFMs, and their spatial distribution was characterized by fluorescence microscopy as uniform throughout the NFM surfaces. Fig. 3C shows the homogeneous distribution of SH groups at the surface of the irradiated and chemically functionalized electrospun NFMs.The ζ-potential analysis revealed that the liposomes are negatively charged due to the presence of the terminal carboxylic groups in the lipid DSPE-PEG (Table 4). Indeed, the negative ζ-potential of PEGylated liposomes is positive since it avoids agglomeration and stabilizes the liposome suspension.45 The size of the Dex-loaded liposomes showed a monodisperse distribution, with diameters ranging between 103.70 (with PE-Rho) and 119.10 nm (without PE-Rho). The DSPE-PEG-Mal lipids comprising the outer layer of the liposomes facilitates the binding to the SH groups grafted onto the surface of electrospun NFMs. Specifically, the maleimide group (Mal) reacts with SH groups at pH ranging from 6.5 to 7.5, forming a non-reversible and stable thioether link. The particular efficiency of surface-bonded PEG chains is explained by the steric repulsive barrier around the liposomes provided by the covalently bonded PEG.46 Some methods were previously proposed to determine the incorporated amounts of PEG-derivatized lipids in liposomes for the physicochemical characterization of PEG-coated liposomes.47 PEGylated liposome formulations were developed for cancer treatment, showing to be efficient in stabilizing drug-loaded liposomes, allowing to bind to proteins and to increase their stability in the blood circulation.48 The EDS analysis (Fig. 4E) confirms the presence of the element phosphorus that is derived from the characteristic phosphate group of the phospholipids used to produce the liposomes. The spatial distribution of the liposomes on the surface of electrospun NFMs was evaluated from fluorescence emitted by the rhodamine dye (Fig. 4F). This analysis showed that the immobilization on the surface of Dex-loaded liposomes was successful and evenly distributed throughout the surface of the NFM.
The effect of the presence of cholesterol (Chol) in the liposome formulation, washing time, liposome size and volume of solution to obtain the maximum capacity of Dex-loaded liposomes immobilized on the surface of electrospun PCL NFMs was studied (Table 5). Our data demonstrated that the presence of Chol has a major negative effect on the encapsulation efficiency of Dex. Specifically, the concentration of Dex increased when Chol was removed from the liposome formulation, which is in accordance with the results from previous reports in the literature (e.g.ref. 49). Moreover, one washing step was sufficient to remove the liposomes not covalently linked to the reactive SH groups. Liposomes produced with 400 nm size were shown to enable an increase of the loaded Dex. Previous reported data show that the extrusion process of the liposomes reduces the efficiency of drug encapsulation.50
The release behavior of Dex from the liposomes immobilized on the surface of NFMs showed an initial burst release, which ended after 12 h (Fig. 5). Afterwards, a slower but steady Dex release was observed during the remaining period of study. Jaiswal et al.51 suggested that the effective concentration of Dex for the osteogenic differentiation of MSCs should be kept within the range of 100–1000 nM, showing toxic effects above 1000 nM. Our results are consistent with the availability of Dex at the surface of electrospun NFMs (where the initial cell attachment occurs) at a concentration relevant for inducing the osteogenic differentiation of MSCs. To confirm this hypothesis, further biological assays with human bone marrow-derived mesenchymal stem cells (hBMSCs) were conducted.
3.2. Biological activity
The effect of Dex-loaded liposomes immobilized on the surface of PCL NFMs on the viability, proliferation, protein synthesis and osteogenic differentiation of hBMSCs was assessed by the use of standard protocols. Biological data show that immobilized Dex-loaded liposomes (NFM4_F2 and NFM4_F3) are not cytotoxic to hBMSCs (Fig. 6). Thus, the performance of the PCL NFMs when the liposomes are immobilized on the surface does not affect the performance of the meshes. Moreover, they are able to induce an earlier osteogenic differentiation of hBMSCs since the 7th day of culture as depicted by the results of ALP activity (Fig. 8) and by gene expression patterns (Fig. 9) for NFM4_F3. This significant result is in accordance with our previous report where Dex-loaded liposomes were used in solution to induce the osteogenic differentiation of hBMSCs on tissue culture plates.52 This observation is of statistically significant relevance when compared to the control culture condition NFM_Ost or to the incorporation of Dex into electrospun NFM (NFM_5%Dex), for the in vitro induction of hBMSC osteogenic differentiation. Those previously explored strategies need 14–21 days to get the hBMSCs fully differentiated into the osteogenic lineage. In a previous study, Martins et al.4 tested the bioactivity of the released Dex by cultivating hBMSCs on 15% Dex-loaded PCL NFMs, in dexamethasone-absent osteogenic differentiation medium formulation. The early osteogenic differentiation of the hBMSCs on the 7th day of culture was not observed, as well as under the control condition NFM_5%Dex. Somehow this relevant effect may be related to the release of Dex by the liposomes in the vicinity of the cells in culture, contributing to the direct interaction between the liposomes and the cells, and even to the possible uptake of the liposomes by the cells into the intracellular space. Dex-loaded liposomes in suspension, i.e. NFM0_F1, showed a better biological performance than the control condition NFM0_Ost regarding the ALP activity. However, the early osteogenic differentiation of the hBMSCs on the 7th day of culture was also not observed under that condition.The biological results show a positive physicochemical effect of the activation by irradiation and further chemical functionalization of electrospun NFMs under osteogenic differentiation conditions (NFM4_Ost) when compared with untreated NFMs. The surface activation and functionalization improved the biological performance of PCL NFMs in the osteogenic differentiation of hBMSCs when compared to the control supplemented osteogenic medium culture conditions (NFM0_Ost). It is well described in the literature that some functional groups, such as amines and thiols, influence positively the adhesion, proliferation and differentiation of stem cells.53,54 Our data further confirm that hypothesis.
The concentration of Dex in the culture medium and the mode of release are important factors in the successful differentiation of hBMSCs.9,51 By using the liposomes immobilized on the surface of electrospun PCL NFMs, we hypothesized a benefit by releasing the loaded bioactive agent in the vicinity of the cells, increasing its bioavailability without the need for further supplementation of the culture medium. Above, we described that the concentration of Dex decreases when cholesterol is used in the liposome formulation. This explains the low ALP activity observed when hBMSCs were cultured on NFM4_F2. Also, this result can explain the low ALP activity observed when hBMSCs were cultured on NFM_5%Dex, which is at the lower level of the Dex concentration required to differentiate hBMSCs.51 By giving repeatedly Dex-loaded liposomes in suspension, NFM0_F1, the ALP activity increased as described above. However, for implantation in vivo into a bone defect as a release system, it would not be practical to use this modality. By using NFM4_F3, we could implant our functionalized NFM into a bone defect as an autonomous device, and the released Dex would be sufficient to promote the in situ osteogenic differentiation of attracted MSCs minimizing the risk of undesirable negative side effects caused by systemic administration.
Liposomes have been used with some success in tissue engineering, such as in an animal model for cartilage repair, avoiding the limited efficacy of the direct injection of growth factors into the synovial cavity.55 Moreover, the conjugation of liposomes with a scaffold could improve the bioactive agent release efficiency.55 Also, the nanofibers with embedded growth factor-loaded liposomes showed an enhancement of MCS proliferation.38 Therefore, this multi-functionalized and versatile carrier of drugs or different growth/differentiation factors may also be used to transfect cells in vivo.
4. Conclusions
The present work proposes the use of surface immobilized Dex-loaded liposomes in a strategy to provide electrospun PCL NFMs with the autonomous capability to support the osteogenic differentiation of hBMSCs. The PCL NFM surface was activated and functionalized by UV–ozone irradiation and aminolysis, respectively. Optimal functionalization was obtained with 4 min UV–ozone irradiation time and 1 h of subsequent aminolysis. Dex-loaded liposomes were covalently immobilized on the surface of electrospun NFMs, and the release kinetics showed a steady release of Dex (until 21 days) after an initial burst period of 12 h. The developed system is intended to act as a drug carrier that can be designed to operate as a synthetic support for MSC growth and, simultaneously, to allow a direct supply of the differentiation factor (e.g. Dex) to the adherent cells. To validate this hypothesis, an osteogenic differentiation study was conducted using human bone marrow-derived mesenchymal stem cells (hBMSC). Biological data showed that the Dex-loaded liposomes immobilized on the surface of electrospun PCL NFMs exhibited no cytotoxicity and promoted the effective osteogenic differentiation of hBMSCs when compared to the osteogenic supplementation of the culture medium. In conclusion, we herein validate the concept of using liposomes immobilized on the surface of a nanostructured system to be used as an autonomous medical device for the local and sustained release of growth/differentiation factors relevant for bone regenerative strategies.5. Experimental section
5.1. Production and characterization
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 volume ratio) was electrospun at 8–10 kV, using a needle-to-ground collector distance of 20 cm, and a flow rate of 1.0 mL h−1. The NFMs were collected in a flat vegetal foil, and the solvents were evaporated under vacuum. Dex-loaded PCL NFMs were produced by adding 5 wt% Dex/PCL to the polymeric solution and electrospun, as described elsewhere.4 Those Dex-releasing nanofiber meshes were used as controls in the biological assays.
151 M−1 cm−1 was used. Optimization of the thiol insertion was performed. The UV–ozone irradiation was maintained constant at 4 min, while the 2IT and DTNB reaction time was varied. We evaluated the impact of these two parameters on the SH-functionalization reaction and on the determination of the SH concentration at the NFM surface. The morphologies of the activated and chemically functionalized NFMs were characterized by scanning electronic microscopy (SEM; NanoSEM, Nova 200). Prior to this analysis, the samples were coated with gold/palladium using a Cressington Sputter Coater 208 HR device. To assess the distribution of SH groups at the surface of electrospun NFMs, three samples of SH-functionalized PCL NFMs were immersed in a pre-prepared fluorescent BODIPY (493/503 FL N-(2-aminoethyl)maleimide) solution (0.4 mM BODIPY in PBS at pH 7). The SH-functionalized PCL NFMs were incubated for 4 hours at room temperature. As negative control, untreated samples were immersed in an equivalent solution. Afterwards, both types of NFMs were washed three times for 5 minutes with Tween 20 washing solution (0.5% (v/v) Tween 20 and phosphate buffer at pH 7) by gentle shaking. As the BODIPY is sensitive to light, all the steps were carried out under light protection. Binding of the fluorescent dye to the NFMs was further analyzed by fluorescence microscopy (AXIOIMAGE RZ1M, ZEISS, Germany).
:3 volume ratio) was electrospun at 8–10 kV, using a needle-to-ground collector distance of 20 cm, and a flow rate of 1.0 mL h−1. The NFMs were collected in a flat vegetal foil, and the solvents were evaporated under vacuum. Dex-loaded PCL NFMs were produced by adding 5 wt% Dex/PCL to the polymeric solution and electrospun, as described elsewhere.4 Those Dex-releasing nanofiber meshes were used as controls in the biological assays.
151 M−1 cm−1 was used. Optimization of the thiol insertion was performed. The UV–ozone irradiation was maintained constant at 4 min, while the 2IT and DTNB reaction time was varied. We evaluated the impact of these two parameters on the SH-functionalization reaction and on the determination of the SH concentration at the NFM surface. The morphologies of the activated and chemically functionalized NFMs were characterized by scanning electronic microscopy (SEM; NanoSEM, Nova 200). Prior to this analysis, the samples were coated with gold/palladium using a Cressington Sputter Coater 208 HR device. To assess the distribution of SH groups at the surface of electrospun NFMs, three samples of SH-functionalized PCL NFMs were immersed in a pre-prepared fluorescent BODIPY (493/503 FL N-(2-aminoethyl)maleimide) solution (0.4 mM BODIPY in PBS at pH 7). The SH-functionalized PCL NFMs were incubated for 4 hours at room temperature. As negative control, untreated samples were immersed in an equivalent solution. Afterwards, both types of NFMs were washed three times for 5 minutes with Tween 20 washing solution (0.5% (v/v) Tween 20 and phosphate buffer at pH 7) by gentle shaking. As the BODIPY is sensitive to light, all the steps were carried out under light protection. Binding of the fluorescent dye to the NFMs was further analyzed by fluorescence microscopy (AXIOIMAGE RZ1M, ZEISS, Germany).
The amount of Dex and lipids present in the NFMs was quantified to evaluate their affinity to the NFM surfaces. After liposome immobilization, 12 PCL NFMs were immersed in a solution of ethanol for 24 h to dissolve the lipids. The solutions were analyzed by high performance liquid chromatography (HPLC, KNAUER, Germany) using an Atlantis T3 5 μm C-18 column; a flow rate of 1 mL min−1; 0.2% phosphoric acid–acetonitrile (55:45) as the mobile phase, and UV detection at 247 nm. Dex was quantified using a calibration curve obtained with standard solutions, ranging from 4 × 10−4 to 0.1 mM. The total lipid concentration was assessed by the Bartlett colorimetric assay, as described elsewhere.56 The principle of the Bartlett assay is based on the colorimetric determination of inorganic phosphate. The phospholipid content of liposomes can be determined after the destruction of the phospholipid with perchloric acid to obtain the inorganic phosphate. The inorganic phosphate is converted to phospho-molybdic acid by the addition of ammonium molybdate, which is reduced to a blue colored complex by 4-amino-2-naphthyl-4-sulfonic acid during heating. This compound can be determined colorimetrically at 830 nm. For the maximum capacity of Dex-loaded liposomes immobilized on the surface of the NFMs, the effect of the presence of cholesterol, the washing time, the liposome size and the volume of liposomes in the solution was all studied. The experiments were performed in triplicate, and each specimen was also analyzed by HPLC (KNAUER, Germany).
5.2. Biological activity
Cell proliferation was quantified by the total amount of double-stranded DNA, along the culturing time. Quantification was performed using the Quant-iT™ Pico- Green dsDNA Assay Kit (Invitrogen™, Molecular Probes™, Oregon, USA), according to the instructions of the manufacturer. Briefly, hBMSCs within the construct were lysed by osmotic and thermal shock, and the supernatant was used for the DNA quantification assay. The fluorescence of the dye was measured at an excitation wavelength of 485/20 nm and at an emission wavelength of 528/20 nm, in a microplate reader (Synergie HT, Bio-Tek, USA). Quadruplicates were made for each sample per culturing time. The DNA concentration for each sample was calculated using a standard curve (DNA concentration ranging from 0.0 to 1.5 μg mL−1) relating the amount of DNA to the fluorescence intensity.
All data concerning cell viability, proliferation and ALP activity were independently measured and normalized against the cell number for each sample. For that, a standard calibration curve was obtained using known hBMSC cell numbers at passage 4, ranging from 0 to 5 × 105 cells, with n = 12. The dsDNA concentration of these samples was determined according to the Quant-iT™ PicoGreen® dsDNA Reagent, previously described. The following equation was obtained: y = 0.0054x + 86.68 with R2 = 0.998, where y is the measured fluorescence value and x is the cell number, and used to estimate the cell numbers for each measurement.
For the quantification of total proteins synthesized by the hBMSCs in culture, the Micro BCA™ Protein Assay kit (Thermo Scientific, Pierce, Rockford, USA) was used according to the manufacturer's instructions. This is a colorimetric detection and quantification method which utilizes bicinchoninic acid (BCA) as the detection reagent for Cu1+ formed when Cu2+ is reduced by proteins in an alkaline environment. Quadruplicates of lysed cells at each culturing time were incubated at 37 °C for 2 h. A purple-colored reaction product was measured at 562 nm in a microplate reader (Synergie HT, Bio-Tek, USA) and calculated based on an albumin standard curve, ranging from 0 to 40 μg mL−1.
Reverse transcriptase (RT)-PCR was performed according to the protocol from the iScript™ cDNA synthesis kit (Quanta BioSciences™, Gaithersburg , MD). Briefly, a reaction mixture consisting of 1X iScript Reaction Mix, 1 μL iScript Reverse Transcriptase, 100 ng RNA template and nuclease-free water was prepared of 20 μL total volume. The single-strand cDNA synthesis occurred by incubating the complete reaction mixture for 5 min at 22 °C, followed by 30 min at 42 °C and terminated by incubation at 85 °C for 5 min.
Amplification of the target cDNA for real-time PCR quantification was performed according to manufacturer's instructions, using 2 μL RT cDNA products, 250 nM each primer (bone-specific primer sets listed4) 1X PerfeCta® SYBR® Green FasterMix® (Quanta BioSciences™, Gaithersburg, MD) and nuclease-free water, in a final volume of 25 μL. Forty-four cycles of denaturation (95 °C, 10 s), annealing (temperature dependent on the gene, 30 s) and extension (72 °C, 30 s) were carried out in the Mastercycler® epgradient S realplex thermocycler (Eppendorf, Hamburg, Germany) for all genes. The transcript expression data were normalized to the housekeeping gene Glyceraldehyde-3-phosphate-dehydrogenase (GAPDH) and the quantification was performed according to the Livak method (2−ΔΔCt method), considering the standard osteogenic differentiation medium (TCPS_osteo) as a calibrator.
5.3. Statistical analysis
Data were statistically analyzed using the IBM SPSS software (version 20, SPSS Inc.). We first applied the Shapiro–Wilk test to test the assumption of normality and the results showed that the data did not follow a normal distribution. Consequently, the non-parametric Mann–Whitney U and Kruskal–Wallis tests were applied. P values lower than 0.01 were considered statistically significant in the analysis of the results.Acknowledgements
The authors thank the Portuguese Foundation for Science and Technology for the Ph.D. grant of N. S. Monteiro (SFRH/BD/62465/2009), the Post-Doc grants of A. Martins (SFRH/BPD/70669/2010) and R. A. Pires (SFRH/BPD/39333/2007), and the OsteoGraphy project (PTDC/EME-MFE/2008). This work was partly supported by the FIND & BIND (NMP4-SL-2009-229292) and MaxBone (PTDC/SAU-ENB/115179/2009/FCOMP-01-0124-FEDER-015729) projects.References
- S. F. Zhang and H. Uludag, Pharm. Res., 2009, 26, 1561–1580 CrossRef CAS PubMed.
- M. P. Lutolf, P. M. Gilbert and H. M. Blau, Nature, 2009, 462, 433–441 CrossRef CAS PubMed.
- M. Kulkarni, U. Greiser, T. O'Brien and A. Pandit, Trends Biotechnol., 2010, 28, 28–36 CrossRef CAS PubMed.
- A. Martins, A. R. C. Duarte, S. Faria, A. P. Marques, R. L. Reis and N. M. Neves, Biomaterials, 2010, 31, 5875–5885 CrossRef CAS PubMed.
- A. R. C. Duarte, J. F. Mano and R. L. Reis, Acta Biomater., 2009, 5, 2054–2062 CrossRef CAS PubMed.
- V. E. Santo, M. E. Gomes, J. F. Mano and R. L. Reis, Nanomed., 2012, 7, 1045–1066 CrossRef CAS PubMed.
- J. S. Park, H. N. Yang, D. G. Woo, H. M. Chung and K. H. Park, Tissue Eng., Part A, 2009, 15, 2163–2175 CrossRef CAS PubMed.
- L. Longobardi, L. O'Rear, S. Aakula, B. Johnstone, K. Shimer, A. Chytil, W. A. Horton, H. L. Moses and A. Spagnoli, J. Bone Miner. Res., 2006, 21, 626–636 CrossRef CAS PubMed.
- E. Anitua, M. Sanchez, G. Orive and I. Andia, Trends Pharmacol. Sci., 2008, 29, 37–41 CrossRef CAS PubMed.
- V. E. Santo, K. Sato, J. Ratanavaraporn, M. E. Gomes, J. F. Mano, R. L. Reis and Y. Tabata, J. Tissue Eng. Regen. Med., 2012, 6, 330–330 Search PubMed.
- A. Martins, R. L. Reis and N. M. Neves, Int. Mater. Rev., 2008, 53, 257–274 CrossRef CAS PubMed.
- A. K. Ghag, J. E. Gough and S. Downes, Biomater. Sci., 2014, 2, 233–241 RSC.
- L. A. Smith, X. Liu and P. X. Ma, Soft Matter, 2008, 4, 2144–2149 RSC.
- C. P. Barnes, S. A. Sell, E. D. Boland, D. G. Simpson and G. L. Bowlin, Adv. Drug Delivery Rev., 2007, 59, 1413–1433 CrossRef CAS PubMed.
- W. Mattanavee, O. Suwantong, S. Puthong, T. Bunaprasert, V. P. Hoven and P. Supaphol, ACS Appl. Mater. Interfaces, 2009, 1, 1076–1085 CAS.
- A. Martins, E. D. Pinho, S. Faria, I. Pashkuleva, A. P. Marques, R. L. Reis and N. M. Neves, Small, 2009, 5, 1195–1206 CAS.
- F. Darain, W. Y. Chan and K. S. Chian, Soft Mater., 2011, 9, 64–78 CrossRef CAS.
- M. Mori, Y. Uyama and Y. Ikada, J. Polym. Sci., Polym. Chem., 1994, 32, 1683–1690 CrossRef CAS.
- W. He, T. Yong, W. E. Teo, Z. W. Ma and S. Ramakrishna, Tissue Eng., 2005, 11, 1574–1588 CrossRef CAS PubMed.
- N. Bölgen, İ. Vargel, P. Korkusuz, Y. Z. Menceloğlu and E. Pişkin, J. Biomed. Mater. Res., Part B, 2007, 81B, 530–543 CrossRef PubMed.
- H. Q. Cao, X. Jiang, C. Chai and S. Y. Chew, J. Controlled Release, 2010, 144, 203–212 CrossRef CAS PubMed.
- M. D. Schofer, P. P. Roessler, J. Schaefer, C. Theisen, S. Schlimme, J. T. Heverhagen, M. Voelker, R. Dersch, S. Agarwal, S. Fuchs-Winkelmann and J. R. J. Paletta, PLoS One, 2011, 6, e25462 CAS.
- J. Suk Choi and H. Sang Yoo, J. Bioact. Compat. Polym., 2007, 22, 508–524 CrossRef PubMed.
- W.-E. Teo and S. Ramakrishna, Compos. Sci. Technol., 2009, 69, 1804–1817 CrossRef CAS PubMed.
- Y.-Y. Wang, L.-X. Lü, Z.-Q. Feng, Z.-D. Xiao and N.-P. Huang, Biomed. Mater., 2010, 5, 054112 CrossRef PubMed.
- J. Shi, A. R. Votruba, O. C. Farokhzad and R. Langer, Nano Lett., 2010, 10, 3223–3230 CrossRef CAS PubMed.
- F. Ruggeri, A. Akesson, P.-Y. Chapuis, C. A. S. Nielsen, M. P. Monopoli, K. A. Dawson, T. G. Pomorski and M. Cardenas, Soft Matter, 2013, 9, 8862–8870 RSC.
- M. E. Lynge, B. M. Teo, M. B. Laursen, Y. Zhang and B. Stadler, Biomater Sci, 2013, 1, 1181–1192 RSC.
- M. Goldberg, R. Langer and X. Jia, J. Biomater. Sci., Polym Ed., 2007, 18, 241–268 CrossRef CAS PubMed.
- Y. Liu, Z. Li and D. Liang, Soft Matter, 2012, 8, 4517–4523 RSC.
- S. Pulavendran and G. Thiyagarajan, Biotechnol. Bioprocess Eng., 2011, 16, 393–399 CrossRef CAS PubMed.
- Q. Tan, H. Tang, J. G. Hu, Y. R. Hu, X. M. Zhou, Y. M. Tao and Z. S. Wu, Int. J. Nanomedicine, 2011, 6, 929–942 CrossRef CAS PubMed.
- S. Mourtas, M. Kastellorizios, P. Klepetsanis, E. Farsari, E. Amanatides, D. Mataras, B. R. Pistillo, P. Favia, E. Sardella, R. d'Agostino and S. G. Antimisiaris, Colloids Surf., B: Biointerfaces, 2011, 84, 214–220 CrossRef CAS PubMed.
- S. S. Wang, M. C. Yang and T. W. Chung, Drug Delivery, 2008, 15, 149–157 CrossRef CAS PubMed.
- S. Meyenburg, H. Lilie, S. Panzner and R. Rudolph, J. Controlled Release, 2000, 69, 159–168 CrossRef CAS.
- T. W. Chung, M. C. Yang and W. J. Tsai, Int. J. Pharm., 2006, 311, 122–129 CrossRef CAS PubMed.
- G. Wang, M. E. Babadagli and H. Uludag, Mol. Pharm., 2011, 8, 1025–1034 CrossRef CAS PubMed.
- A. Mickova, M. Buzgo, O. Benada, M. Rampichova, Z. Fisar, E. Filova, M. Tesarova, D. Lukas and E. Amler, Biomacromolecules, 2012, 13, 952–962 CrossRef CAS PubMed.
- D. M. Huang, Y. Hung, B. S. Ko, S. C. Hsu, W. H. Chen, C. L. Chien, C. P. Tsai, C. T. Kuo, J. C. Kang, C. S. Yang, C. Y. Mou and Y. C. Chen, FASEB J., 2005, 19, 2014–2020 CrossRef CAS PubMed.
- D. Ami, T. Neri, A. Natalello, P. Mereghetti, S. M. Doglia, M. Zanoni, M. Zuccotti, S. Garagna and C. A. Redi, Biochim. Biophys. Acta, 2008, 1783, 98–106 CrossRef CAS PubMed.
- X. Yu, L. Wang, Z. Xia, L. Chen, X. Jiang, D. Rowe and M. Wei, Biomater. Sci., 2014, 2, 242–251 RSC.
- A. H. C. Poulsson, S. A. Mitchell, M. R. Davidson, A. J. Johnstone, N. Emmison and R. H. Bradley, Langmuir, 2009, 25, 3718–3727 CrossRef CAS PubMed.
- P. Tyllianakis, S. Kakabakos, G. Evangelatos and D. Ithakissios, Appl. Biochem. Biotechnol., 1993, 38, 15–25 CrossRef CAS.
- T. G. Kim and T. G. Park, Macromol. Rapid Commun., 2008, 29, 1231–1236 CrossRef CAS.
- A. Yousefi, F. Esmaeili, S. Rahimian, F. Atyabi and R. Dinarvand, Sci. Pharm., 2008, 77, 453–464 CrossRef PubMed.
- K. Hristova, A. Kenworthy and T. J. McIntosh, Macromolecules, 1995, 28, 7693–7699 CrossRef CAS.
- K. Shimada, S. Matsuo, Y. Sadzuka, A. Miyagishima, Y. Nozawa, S. Hirota and T. Sonobe, Int. J. Pharm., 2000, 203, 255–263 CrossRef CAS.
- J. N. Moreira, T. Ishida, R. Gaspar and T. M. Allen, Pharm. Res., 2002, 19, 265–269 CrossRef CAS.
- N. Monteiro, A. Martins, D. Ribeiro, S. Faria, N. A. Fonseca, J. N. Moreira, R. L. Reis and N. M. Neves, J. Tissue Eng. Regen. Med., 2013 DOI:10.1002/term.1817.
- U. Bhardwaj and D. J. Burgess, Int. J. Pharm., 2010, 388, 181–189 CrossRef CAS PubMed.
- N. Jaiswal, S. E. Haynesworth, A. I. Caplan and S. P. Bruder, J. Cell. Biochem., 1997, 64, 295–312 CrossRef CAS.
- N. Monteiro, A. Martins, D. Ribeiro, S. Faria, N. A. Fonseca, J. N. Moreira, R. L. Reis and N. M. Neves, J. Tissue Eng. Regen. Med., 2012, 6, 33–33 Search PubMed.
- R. Thire, T. O. Meiga, S. Dick and L. R. Andrade, Macromol. Symp., 2007, 258, 38–44 CrossRef CAS.
- J. M. Curran, R. Chen and J. A. Hunt, Biomaterials, 2006, 27, 4783–4793 CrossRef CAS PubMed.
- P. Giannoni and E. B. Hunziker, Biotechnol. Bioeng., 2003, 83, 121–123 CrossRef CAS PubMed.
- V. P. Torchilin and V. Weissig, Liposomes: A Practical Approach, Oxford University Press, 2nd edn, 2003 Search PubMed.
- B. Delorme and P. Charbord, Methods Mol. Med., 2007, 140, 67–81 CAS.
| This journal is © The Royal Society of Chemistry 2014 |