Utilization of star-shaped polymer architecture in the creation of high-density polymer brush coatings for the prevention of platelet and bacteria adhesion†
Masayasu
Totani
a,
Tsuyoshi
Ando
*a,
Kayo
Terada
a,
Takaya
Terashima
b,
Ill Yong
Kim
c,
Chikara
Ohtsuki
c,
Chuanwu
Xi
d,
Kenichi
Kuroda
*e and
Masao
Tanihara
a
aGraduate School of Materials Science, Nara Institute of Science and Technology, 8916-5 Takayama, Ikoma 630-0192, Japan. E-mail: tando@ms.naist.jp
bDepartment of Polymer Chemistry, Graduate School of Engineering, Kyoto University, Katsura, Nishikyo-ku, Kyoto 615-8510, Japan
cGraduate School of Engineering, Nagoya University, Furo-cho, Chikusa-ku, Nagoya 464-8603, Japan
dDepartment of Environmental Health Sciences, School of Public Health, University of Michigan, Ann Arbor, Michigan 48109, USA
eDepartment of Biologic and Materials Sciences, School of Dentistry, University of Michigan, Ann Arbor, Michigan 48109, USA. E-mail: kkuroda@umich.edu
First published on 22nd May 2014
Abstract
We demonstrate utilization of star-shaped polymers as high-density polymer brush coatings and their effectiveness to inhibit the adhesion of platelets and bacteria. Star polymers consisting of poly(2-hydroxyethyl methacrylate) (PHEMA) and/or poly(methyl methacrylate) (PMMA) were synthesized using living radical polymerization with a ruthenium catalyst. The polymer coatings were prepared by simple drop casting of the polymer solution onto poly(ethylene terephthalate) (PET) surfaces and then dried. Among the star polymers prepared in this study, the PHEMA star polymer (star-PHEMA) and the PHEMA/PMMA (mol. ratio of 71/29) heteroarm star polymer (star-H71M29) coatings showed the highest percentage of inhibition against platelet adhesion (78–88% relative to the non-coated PET surface) and Escherichia coli (94–97%). These coatings also showed anti-adhesion activity against platelets after incubation in Dulbecco's phosphate buffered saline or surfactant solution for 7 days. In addition, the PMMA component of the star polymers increased the scratch resistance of the coating. These results indicate that the star-polymer architecture provides high polymer chain density on PET surfaces to prevent adhesion of platelets and bacteria, as well as coating stability and physical durability to prevent exposure of bare PET surfaces. The star polymers provide a simple and effective approach to preparing anti-adhesion polymer coatings on biomedical materials against the adhesion of platelets and bacteria.
Introduction
Biomedical synthetic materials, such as poly(ethylene terephthalate) (PET) and silicone, are prone to adhesion of proteins, cells, and bacteria, causing functional failures in implants, artificial organs, catheters, and diagnostic devices, and increasing the risk of secondary infections.1–3 A common strategy to prevent protein and microbial adhesion is to modify the surfaces of these materials using hydrophilic polymers, including non-ionic poly(ethylene glycol),4–6 poly(2-hydroxyethyl methacrylate) (PHEMA), triblock copolymers consisting of PHEMA and hydrophobic polystyrene (PSt) (PHEMA-b-PSt-b-PHEMA),7 poly(2-methoxyethyl acrylate),8 and polyethylene oxide (PEO)-poly(propylene oxide) block copolymers.9,10 Recently, zwitterionic polymers, including poly(2-methacryloyloxyethyl phosphorylcholine),11,12 poly(sulfobetaine methacrylate),13 and poly(carboxybetaine methacrylate),14 have been utilized as new materials. When used on surfaces, these polymers prevent the non-specific hydrophobic binding of proteins, cells, and bacteria, which is the primary driving force in the initial stage of the adhesion mechanism.Polymer chains anchored onto surfaces also provide physical barriers against protein adhesion because of the exclusion volume around polymer chains.15,16 These polymer effects of hydrophilic layers and exclusion volumes are enhanced further when polymer chains are packed densely and form brush structures on the surfaces (Fig. 1).17,18 A common method for preparing polymer brushes is the polymerization of monomers from initiators that are covalently fixed onto surfaces (graft polymerization).17 Another method is to attach pre-existing polymer chains covalently onto the plastic surfaces.10 Although polymer brushes can be prepared by these methods, achieving a high density of polymer brushes on plastics is challenging because chemically inert plastic surfaces are difficult to modify covalently with a high density of initiators or to attach with pre-existing polymers. In addition, the modification of plastic surfaces needs substrate-specific chemical treatment, and some organic solvents/agents for surface chemistries and polymerization may not be compatible with existing biomedical plastics. Therefore, a simple and versatile method for preparing high-density polymer brush coatings compatible with biomedical synthetic materials would be beneficial for biomedical applications.
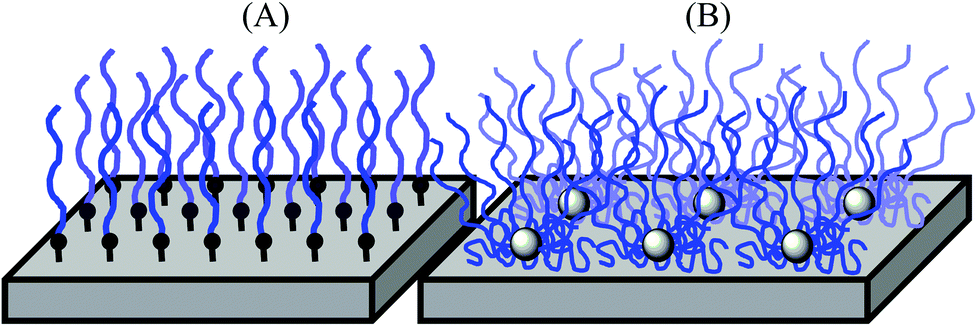 | ||
| Fig. 1 Polymer brush structures on surfaces by (A) graft polymers vs. (B) star polymers. | ||
In this report, we demonstrate a new design strategy for preparing stable high-density coatings of hydrophilic polymer chains on plastic surfaces. We utilize star-shaped polymers preassembled with a number of hydrophilic PHEMA polymer chains. This star-polymer architecture intrinsically provides high polymer chain density when coated on surfaces. Star polymers with PEG chains have been previously used for anti-fouling coatings.19,20 These applications demonstrate the effective anti-fouling effects against proteins and bacteria. However, these water-soluble PEG star polymers require covalent attachment to surfaces for stable coatings. To that end, the hydrophilic, but water-insoluble, PHEMA star polymers studied in this report will be packed tightly with the highly entangled polymer chains, providing physical cross-linking of star polymers and increasing the coating stability (Fig. 1). This allows for a simple coating method of solvent casting or dip coating on pre-existing plastic materials. This method also minimizes the use of organic solvents and chemical treatment, facilitating coating preparation. In addition, we further extended the polymer design to include heteroarm star polymers having both hydrophilic PHEMA and hydrophobic poly(methyl methacrylate) (PMMA) polymer chains. In general, PMMA has higher hydrophobicity, hardness, and adhesiveness to plastic surfaces compared with PHEMA.21 Therefore, we expect that the star-polymer PMMA arms will anchor the hydrophilic PHEMA chains onto plastic surfaces, increasing the stability of the polymer coatings in water and the physical durability of the coatings. It has been previously reported that hydrophobic modification of star-polymers consisting of poly(ethylene glycol) methacrylate provided stable polymer coating on membranes due to the decreased water-solubility and association of hydrophobic polymers, providing high density of polymer chains, and exhibit potent anti-fouling activity.22–24 In contrast to these previous reports, our strategy is to use a mixture of hydrophilic PHEMA and hydrophobic PMMA arms, which allows controlling the hydrophilic/hydrophobic balance of star polymers without changing the anti-fouling properties of each PHEMA polymer chain.
In this study, we report polymer synthesis using a living radical polymerization method to prepare monodispersed star-polymer architectures.25 These surface structures and morphologies of the polymer coatings were examined by scanning electron microscopy (SEM) and atomic force microscopy (AFM). The mechanical stability of coatings was also examined by quantifying the resistance of the coatings against physical scratching. The anti-adhesion activity of the star-polymer coatings was determined using platelets and the model bacterium Escherichia coli (E. coli). The anti-adhesion properties of the coatings were also examined for coating stability after soaking the coatings in buffer or surfactant solution for 7 days.
Experimental section
Materials
Methyl methacrylate (MMA, Tokyo Chemical Industry Co., Ltd, Tokyo (TCI), Tokyo, Japan, purity >99%), tributylamine (n-Bu3N; TCI, Tokyo, Japan, purity >98%), toluene (Aldrich, St. Louis, MO, purity >99%), and ethylene glycol dimethacrylate (EGDMA, Aldrich, St. Louis, MO, purity >98%) were purified by distillation over calcium hydride before use. Chloro(indenyl)bis(triphenylphosphine)ruthenium (Ru(Ind)Cl(PPh3)2 (Ru), STREM, purity >98%) and triethylamine (TCI, Tokyo, Japan, purity >98%) were used without purification. The water was deionized water from a Milli-Q (18 MΩ cm) system. Ethyl α-chloro-α-phenylacetate (ECPA), methyl α-chloro-α-phenylacetate (MCPA),26 and 2-(trimethylsilyloxy)ethyl methacrylate (TMSOEMA)27 were prepared according to the literature. A PET film (FS2000, Futamura Kagaku K.K., Osaka, Japan) was cleaned by sonication in 0.2 μm-filtered ethanol for 30 min and then dried overnight under vacuum.Polymer characterization
The molecular weights Mn, Mw, and the molecular weight distribution (Mw/Mn) of the polymers were measured by size exclusion chromatography (SEC) in N,N-dimethylformamide (DMF) containing 10 mM LiBr at 40 °C (flow rate: 1 mL min−1) on three linear-type poly(2-hydroxyethyl methacrylate) gel columns (Shodex® OHpak SB-806M × 3; exclusion limit = 2 × 107; 0.8 cm i.d. × 30 cm) connected to a Jasco PU-2080 precision pump, a Jasco RI-2031 refractive index detector, and a Jasco UV-2075 UV/vis detector set at 270 nm. Mn and Mw were determined from a calibration curve prepared by 10 standard PMMA samples. The 1H nuclear magnetic resonance (1H NMR) spectra of each sample were measured using a JNM-ECP 500 spectrometer (JEOL Ltd, Tokyo, Japan). The absolute Mw and Mw/Mn of the star polymers were determined by multiangle laser light scattering (MALLS) in DMF containing 10 mM LiBr at 40 °C on a Dawn E instrument (Wyatt Technology Corp., Ga–As laser, λ = 690 nm). The concentration of residual ruthenium in the star polymers was measured from microwave-induced plasma mass spectra (MIP–MS) (P-6000, HITACHI, Tokyo, Japan). The hydrodynamic diameter of the star polymers was measured using a dynamic light scattering (DLS) spectrometer equipped with a He–Ne laser at 633 nm (Zetasizer Nano-ZS, Malvern, UK).Synthesis of living PMMA (lin-PMMA 10k)
Polymerization of MMA was carried out under argon (Ar) in a 1000 mL round-bottomed flask equipped with a three-way stopcock. ECPA (4.46 mL, 26.0 mmol), MMA (278 mL, 2600 mmol), n-Bu3N solution (30.6 mL, 26.0 mmol, 850 mM in toluene), and Ru(Ind)Cl(PPh3)2 (2.24 g, 2.60 mmol) were added to toluene (335 mL). Immediately after mixing, the polymer solution was separated into nine aliquots in a 100 mL flask at 25 °C under Ar. The polymer solution was then degassed by bubbling with Ar for 10 min. The mixtures were placed in an oil bath with the temperature controlled at 80 °C. The polymerization was terminated by cooling the mixtures in an ice bath after 19 h. The monomer conversion was determined by 1H NMR analysis, the solvent was removed under reduced pressure, and the crude polymer was precipitated in hexane to remove any unreacted monomers. The Ru-complex was removed by silica gel and alumina column chromatography eluted with toluene. After removing the solvent, the resultant PMMA was dissolved in 1,4-dioxane and lyophilized to give a white powder. Mn = 8300, Mw = 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 400, Mw/Mn = 1.25 (SEC). 1H NMR (500.16 MHz, CDCl3, Si(CH3)4 = 0 ppm): δ (ppm) = 4.21–3.94 (–O–CH2–CH3), 3.80–3.42 (–OCH3), 2.22–1.34 (–CH2–), 1.32–0.64 (–CH3).
400, Mw/Mn = 1.25 (SEC). 1H NMR (500.16 MHz, CDCl3, Si(CH3)4 = 0 ppm): δ (ppm) = 4.21–3.94 (–O–CH2–CH3), 3.80–3.42 (–OCH3), 2.22–1.34 (–CH2–), 1.32–0.64 (–CH3).
Synthesis of living poly(2-(trimethylsilyloxy)ethyl methacrylate)
The precursor poly(2-(trimethylsilyloxy)ethyl methacrylate) (PTMSOEMA) was prepared using TMSOEMA and the procedure described for PMMA except for the removal of unreacted TMSOEMA, where the unreacted TMSOEMA was removed by precipitation of crude PTMSOEMA in a MeOH/H2O (80/20 v/v) mixed solvent. Mn = 16300, Mw = 21400, Mw/Mn = 1.31. 1H NMR (500.16 MHz, CDCl3, Si(CH3)4 = 0 ppm): δ (ppm) = 4.14–3.84 (–CH2–CH2–OSi(CH3)3), 3.84–3.63 (–CH2–CH2–OSi(CH3)3), 3.60–3.50 (–CH), 2.60–1.43 (–CH2–), 2.16–0.56 (–CH3), 0.12–0.08 (–Si(CH3)3).
Synthesis of heteroarm star polymers: star-H71M29
The heteroarm star polymers are denoted as star-HXMY, where X and Y indicate the mole percentage of the PHEMA and PMMA arms, respectively. They were determined by 1H NMR analysis of purified heteroarm star polymers (see the ESI†).In a 100 mL round-bottomed flask, PTMSOEMA (8.13 g, 0.502 mmol, Mn = 16200, Mw/Mn = 1.30), PMMA (1.39 g, 0.167 mmol, Mn = 8300, Mw/Mn = 1.25), Ru(Ind)Cl(PPh3)2 (0.156 g, 0.134 mmol), toluene (68.8 mL), n-Bu3N (0.32 mL, 1.34 mmol), and EGDMA (1.26 mL, 6.68 mmol) were added sequentially in this order at 25 °C under Ar. Immediately after degassing by three freeze–pump–thaw cycles, the mixtures were placed in an oil bath at 80 °C. After 52 h, the reaction was terminated by cooling the mixtures in an ice bath. The obtained star polymer was dissolved in toluene (20 wt%) and precipitated by the addition of quintuple volume of hexane to remove the unreacted PTMSOEMA. Then the polymer was dried under reduced pressure. The polymer was dissolved in acetone (20 wt%), and 0.2 times the volume with water was poured into this polymer solution to remove the unreacted PMMA. The Ru-complex was removed by silica gel and alumina column chromatography eluted with toluene. The TMS protecting group was removed by addition of a small volume of 1.5 N HCl aq. in ethanol/acetone (1/1 v/v). The resulting solution was poured into hexane to precipitate a star polymer and was separated by suction filtration and dried under vacuum overnight at room temperature. Mw = 227000, Mw/Mn = 1.17 (SEC–MALLS), HEMA/MMA = 70/30 (mol%). 1H NMR (500.16 MHz, CDCl3/CD3OD = 1/1, Si(CH3)4 = 0 ppm): δ (ppm) = 4.27–3.90 (–CH2–CH2–OH), 3.88–3.72 (–CH2–CH2–OH), 3.72–3.53 (–OCH3), 2.31–1.40 (–CH2–), 1.40–0.80 (–CH3).
Synthesis of other star polymers
Synthesis of other star polymers was carried out using the same procedure described for the star-H71M29 polymer using various ratios of the precursors, PTMSOEMA and PMMA, and the precursor PTMSOEMA by itself (see the ESI† for details).Star-PHEMA: Mw = 286000, Mw/Mn = 1.25 (SEC–MALLS). 1H NMR (500.16 MHz, CD3OD, Si(CH3)4 = 0 ppm): δ (ppm) = 4.16–3.85 (–CH2–CH2–OH), 3.85–3.59 (–CH2–CH2–OH), 2.19–1.42 (–CH2–), 1.42–0.67 (–CH3).
Star-PMMA: Mw = 209000, Mw/Mn = 1.15 (SEC–MALLS). 1H NMR (500.16 MHz, CDCl3, Si(CH3)4 = 0 ppm): δ (ppm) = 3.70–3.40 (–OCH3), 1.88–1.30 (–CH2–), 1.30–0.38 (–CH3).
Star-H47M53: Mw = 291000, Mw/Mn = 1.17 (SEC–MALLS), HEMA/MMA = 46/54 (mol%). 1H NMR (500.16 MHz, CDCl3/CD3OD = 1/1, Si(CH3)4 = 0 ppm): δ (ppm) = 4.33–3.91 (–CH2–CH2–OH), 3.88–3.72 (–CH2–CH2–OH), 3.72–3.55 (–O–CH3), 2.31–1.40 (–CH2–), 1.39–0.48 (–CH3).
Star-H22M78: Mw = 250000, Mw/Mn = 1.23 (SEC–MALLS). HEMA/MMA = 22/78 (mol%), 1H NMR (500.16 MHz, CDCl3/CD3OD = 1/1, Si(CH3)4 = 0 ppm): δ (ppm) = 4.33–3.95 (–CH2–CH2–OH), 3.91–3.74 (–CH2–CH2–OH), 3.74–3.54 (–O–CH3), 2.24–1.38 (–CH2–), 1.38–0.48 (–CH3).
Synthesis of linear polymers
Linear polymers were synthesized according to previously reported methods28 (see the ESI† for details).Lin-PHEMA 27k: Mn = 19800, Mw = 26700, Mw/Mn = 1.34 (SEC). 1H NMR (500.16 MHz, CD3OD, Si(CH3)4 = 0 ppm): δ (ppm) = 4.18–3.91 (–CH2–CH2–OH), 3.91–3.67 (–CH2–CH2–OH), 2.20–1.44 (–CH2–), 1.44–0.71 (–CH3).
Lin-PHEMA 290k: Mn = 161000, Mw = 286000, Mw/Mn = 1.77 (SEC). 1H NMR (500.16 MHz, CD3OD, Si(CH3)4 = 0 ppm): δ (ppm) = 4.18–3.90 (–CH2–CH2–OH), 3.91–3.67 (–CH2–CH2–OH), 2.20–1.44 (–CH2–), 1.44–0.71 (–CH3).
PHEMA/PMMA diblock copolymer (lin-Block): Mn = 26300, Mw = 32500, Mw/Mn = 1.23 (SEC). HEMA/MMA = 52/48 (mol%). 1H NMR (500.16 MHz, CD3OD/CDCl3 = 1/1, Si(CH3)4 = 0 ppm): δ (ppm) = 4.35–3.90 (–CH2–CH2–OH), 3.90–3.72 (–CH2–CH2–OH), 3.72–3.44 (–OCH3), 2.37–1.40 (–CH2–), 1.40–0.55 (–CH3).
HEMA/MMA random copolymer (lin-Random): Mn = 24500, Mw = 29200, Mw/Mn = 1.19 (SEC), HEMA/MMA = 51/49 (mol%). 1H NMR (500.16 MHz, CD3OD/CDCl3 = 1/1, Si(CH3)4 = 0 ppm): δ (ppm) = 4.20–3.97 (–CH2–CH2–OH), 3.90–3.72 (–CH2–CH2–OH), 3.72–3.51 (–OCH3), 2.19–1.45 (–CH2–), 1.35–0.70 (–CH3).
Polymer coatings
Lin-PHEMA 27k, lin-PHEMA 290k, and star-PHEMA were dissolved in methanol; a methanol/acetone mixture (1/1 v/v) was used to dissolve all PHEMA/PMMA heteroarm star polymers (star-H71M29, star-H47M53, star-H22M78), lin-Block, and lin-Random; acetone was used to dissolve lin-PMMA 10k and star-PMMA. All polymer solution samples were filtered using a PTFE filter (pore size = 0.45 μm) before casting. The polymer solutions (25 μL, 0.10 mg mL−1) were dropped on PET films (1.0 cm × 1.0 cm), evaporated at room temperature, and then dried under reduced pressure overnight. These samples were used for the microscopic surface characterization (AFM and SEM), contact angle measurement, and bacterial adhesion assay. For the platelet adhesion assay, the coated films were cut into four pieces (0.5 cm × 0.5 cm).Surface characterization
AFM images were recorded using an SPI-400 system (Seiko Instruments Inc., Chiba, Japan) in a tapping mode using an RTESP7 tip (Veeco Inst.). The static contact angles of the polymer surfaces were measured at room temperature with a contact angle goniometer DM-501 (Kyowa Interface Science Co., Ltd, Saitama, Japan) by dropping Milli-Q water (2.0 μL) on the polymer-coated surface using a microsyringe, and the angle was monitored with a microscope after 30 s. The contact angle of the air bubbles (7.5–8.0 μL) on the polymer-coated surfaces in water was determined at room temperature. The polymer-coated surfaces were incubated in water at 37 °C for 12 h prior to the measurement. The presented data are the average values of three samples. Errors were determined through evaluation of the standard deviation of the measurements.Platelet adhesion
Blood was drawn from a healthy volunteer. The fresh blood containing 0.1% sodium citrate as an anti-coagulant was centrifuged at 800 rpm (152g) for 5 min, and the obtained supernatant, platelet-rich plasma (PRP), was diluted three times with Dulbecco's phosphate buffered saline (PBS). The platelet concentration (5 × 105 cells μL−1) in diluted PRP was determined using a counting chamber. The 12 polymer-coated substrates on PET (0.5 cm × 0.5 cm) were fixed on the bottom of a glass Petri dish (diameter 3.3 cm) using a small amount of silicon adhesive compound (bath-bond Q, Konishi co., Ltd, Osaka, Japan). The polymer-coated surface was washed with Milli Q water three times and finally immersed in PBS at 37 °C for 12 h for hydration. The diluted PRP (2.0 mL) was added to the Petri dish, and the solution was incubated at 37 °C for 30 min under humid conditions. The PRP solution was removed, and the substrates were washed three times with PBS. The adhered platelets were fixed in 2% glutaraldehyde in PBS solution at 4 °C for 2 h. The samples were washed three times with PBS and once with water, and then dried under vacuum overnight. All samples were sputter-coated with gold using a VPS-020 Quick Coater (ULVAC KIKO, Ltd, Miyazaiki, Japan) prior to SEM (S-4800, Hitachi, Tokyo, Japan) observation. SEM images were obtained at an accelerating voltage of 15 kV, and the magnifications were 4 × 102 and 1.5 × 103. The number of adhered platelets on the polymer-coated surfaces was determined from the SEM images. At least three readings on three different parts of a sample were measured. The data and errors presented are the average values and standard deviation of the three samples.Bacterial adhesion
A polymer-coated film was fixed to the bottom of wells in a 24-well culture plate using a small amount of silicon adhesive compound and dried under reduced pressure overnight. The polymer-coated surfaces were washed three times with Milli-Q water and finally immersed in PBS at 37 °C for 12 h for hydration. E. coli (ATCC® 25922™) was grown in Muller–Hinton II (MH) broth (5 mL, pH = 7.4) at 37 °C overnight. The cell culture was diluted with MH broth to give an OD600 of 0.1 and was incubated at 37 °C and 180 rpm for 90 min. The bacterial culture in the mid-logarithmic phase (OD600 = 0.5–0.6) was washed three times in MH broth by centrifuging 5 mL of the culture at 3700 rpm for 5 min, and resuspended in 10% MH broth in distilled water adjusted to an OD600 of 0.003. The bacterial suspension (2.0 mL) was added to each well and incubated at 37 °C for 20 h. After incubation, the OD590 of the supernatants was measured using a microplate reader as a measure of bacterial growth. The supernatant was removed from the well, and the polymer-coated substrates were rinsed three times with PBS buffer solution to remove non-adherent planktonic bacteria. Substrates with adhered bacteria were transferred to a new 24-well plate to quantify only the bacteria adherent to the substrate, because bacteria might adhere non-specifically to a well wall of an assay plate incubated with bacteria. After removing the PBS, 10% Bac Titer-Glo™ in PBS (500 μL) was added to the bacteria adhered to the coatings and incubated for 5 min at room temperature. The incubated Bac Titer-Glo™ solution was transferred to a 96-well white microplate, and the luminescence from the solutions was measured to determine the viability of the adherent bacteria.SEM images of the adherent bacteria
Adherent bacteria on the polymer-coated surfaces were prepared using the same method as the bacterial adhesion assay. The polymer coatings were incubated with bacteria at 37 °C for 20 h, and the adhered bacteria were fixed with 2% glutaraldehyde in PBS solution at 4 °C for 2 h. The samples were washed three times with PBS and water, and were dried under vacuum overnight. All samples were observed in the same procedure as the platelet adhesion.Scratch test
The scratch resistance of the star-PHEMA and star-H71M29 coatings was evaluated using a continuous-loading-type scratch intensity tester (HEIDON Tribogear Type 18, Shinto Scientific Co., Ltd, Tokyo, Japan). All samples were scratched using a sapphire scratcher (60 μm tip diameter) with a constant load of 4.9 mN (0.5 g), 19.6 mN (2.0 g), and 49.0 mN (5.0 g) at a testing speed of 600 mm min−1. After testing, the scratch width was measured by SEM. An average of five spots measuring a scratch width was reported, and a comparative analysis was done using a Student's t test.Stability test
Each polymer-coated PET substrate was fixed on the bottom of a 50 mL vial. PBS (20 mL) or 0.5 wt% Triton X-100 in PBS (20 mL) was added into the vials. The vials were closed with caps and incubated at 37 °C while shaking at 180 rpm for 7 days. The substrates were washed five times with Milli-Q (20 mL). The sample's stability was evaluated by the platelet adhesion assay described above.Results and discussion
Polymer design and syntheses
A series of star polymers with different ratios of PHEMA and PMMA was prepared using the arm-mixing method.28,29 We first prepared linear PTMSOEMA (Mw = 21400, Mw/Mn = 1.31, DPn = 79) and PMMA (Mw = 10400, Mw/Mn = 1.25, DPn = 81) as precursor polymers with almost the same star-polymer arm lengths by living radical polymerization using a Ru catalyst (Scheme 1). TMS groups protected the hydroxyl group of HEMA prior to the polymerization, which facilitates the polymer preparation in non-polar organic solvents and avoids undesired interactions with the Ru catalyst during polymerization. The growing end groups of these precursor polymers were cross-linked with EGDMA using a Ru catalyst, giving a core–shell, star-shaped structure. An SEC curve showed the formation of star polymers, as a new peak appeared in the higher molecular weight region (MW ∼ 105), and only a trace amount of precursor polymers (MW ∼ 104) was observed (Fig. 2A). The molecular weight distributions of the resulting star polymers were relatively narrow (Mw/Mn = 1.15–1.39). These results suggest that the star polymers were prepared in a controlled and quantitative manner. The crude star polymers contained approximately 22 μmol g−1-polymer of residual Ru, as determined by MIP–MS. The purification of polymers by column chromatography using both silica gel and alumina columns reduced the amount of Ru significantly to approximately 0.53 μmol g−1 polymer.
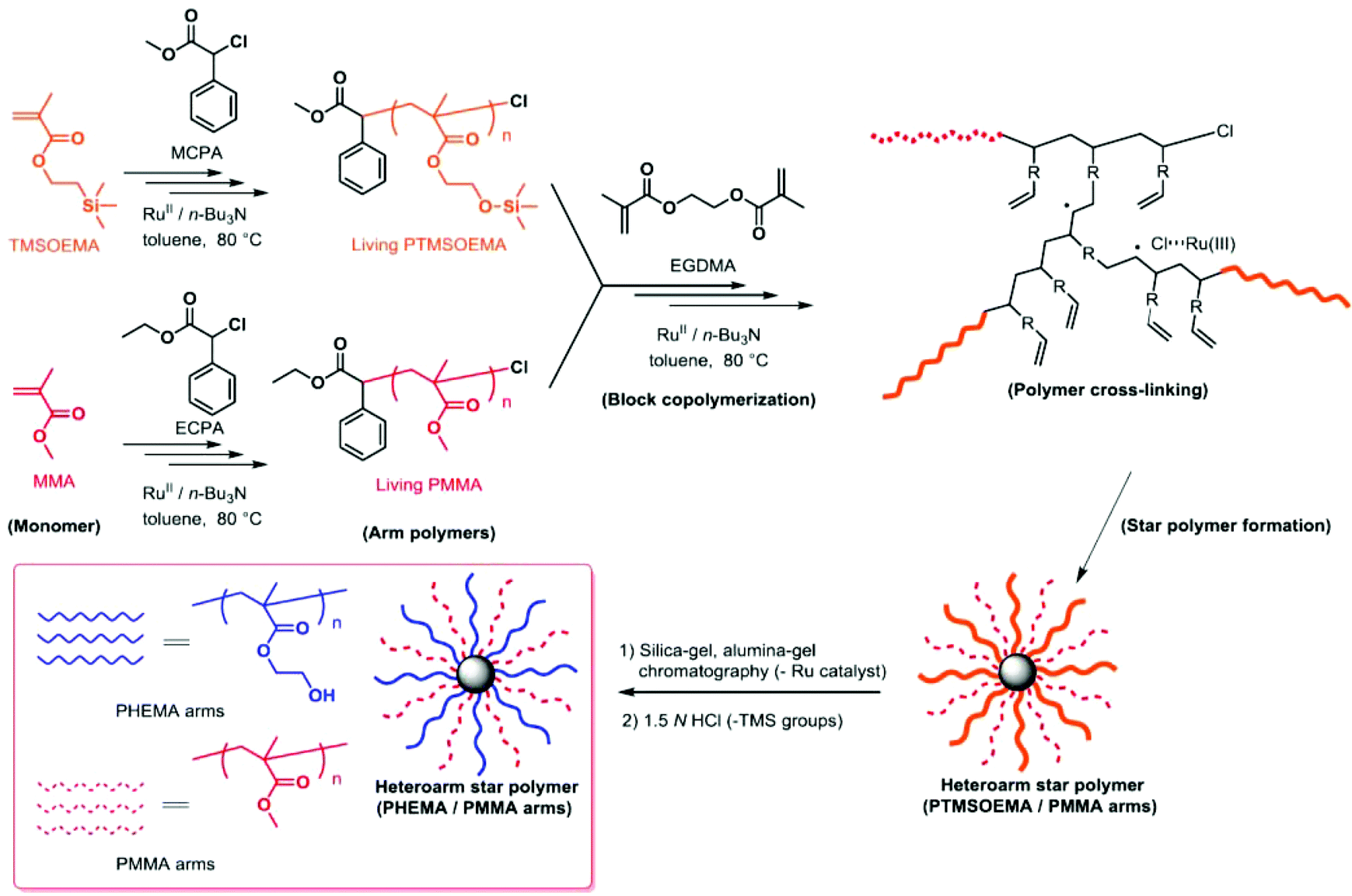 | ||
| Scheme 1 Synthesis of PHEMA/PMMA heteroarm star polymers by Ru(II)-catalyzed living radical polymerization. | ||
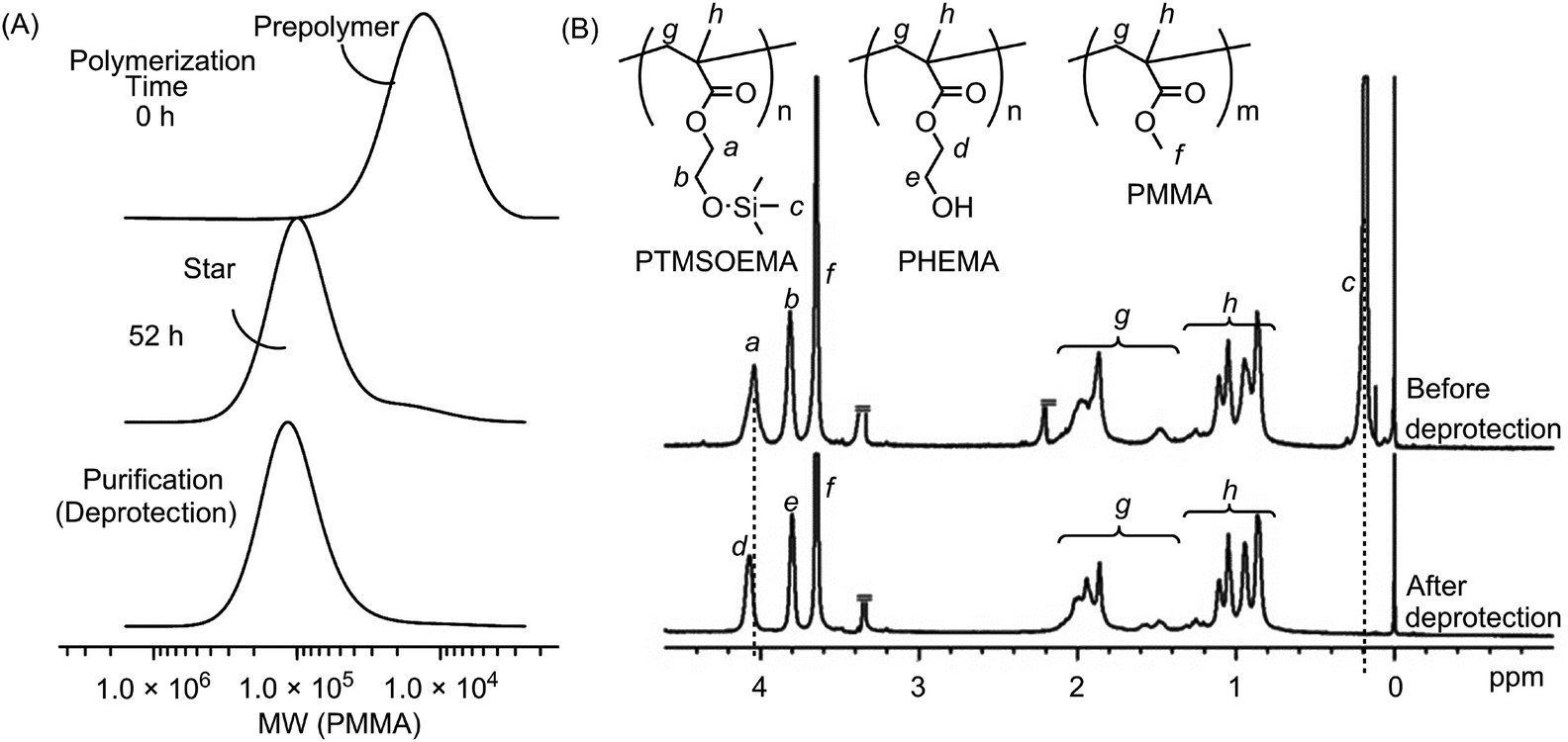 | ||
| Fig. 2 (A) SEC curves of the star-H47M53 polymer at 0 and 52 h after the cross-linking reaction. As the star polymers formed, a new peak appeared in the higher molecular weight (MW) region (MW ∼ 105), and only a trace amount of precursor polymers (MW ∼ 104) was observed. After purification by precipitation and TMS-deprotection, the unreacted precursor polymers were removed. (B) 1H NMR spectra of the heteroarm star PTMSOEMA–PMMA (before deprotection) and star-H47M53 polymers (after deprotection). The peak of the TMS of PTMSOEMA at 0.2 ppm disappeared after HCl treatment, indicating the complete removal of TMS groups to give hydroxyl groups. | ||
The TMS protecting groups of PTMSOEMA were removed quantitatively by HCl treatment to give deprotected star polymers with PHEMA, as the peak at 0.2 ppm (TMS group) disappeared completely in the 1H NMR spectrum (Fig. 2B). The number of arms of the star polymers was 18–20 for all star polymers, except for star-H71M29 that had 14 arms (Table 1) (see the ESI† for calculation). The PHEMA and PMMA star polymers are referred to as star-PHEMA and star-PMMA, respectively. We also prepared linear homopolymers lin-PHEMA 27k (Mw = 26700) and 290k (Mw = 286000), as well as an amphiphilic diblock copolymer lin-Block (51/49 mol%, Mw = 32500) and a random copolymer lin-Random (51/49 mol%, Mw = 29200) for comparison.
| Samples | Polymer structure | M w (g mol−1) | M w/Mn | PHEMA/PMMAe (mol. ratio) | No. of armsg (PHEMA/PMMA) | Contact angles,h (°) | |
|---|---|---|---|---|---|---|---|
| Sessile drop | Captive bubble | ||||||
| a The weight-average molecular weight (Mw) was determined by SEC. b The weight-average molecular weight (Mw) was determined by SEC–MALLS. c M w/Mn was determined by SEC. d M w/Mn was determined by SEC–MALLS. e The molar ratios of PHEMA to PMMA units of star polymers. See ESI for calculation. f The HEMA and MMA monomer units of lin-Block and lin-Random were 0.52/0.48 and 0.51/0.49, respectively. g The total number of arms in a star polymer. The numbers of PHEMA and PMMA arms are given in parentheses. See ESI for calculation. h Contact angles of the water droplets (sessile drop) and air-in-water (captive bubble drop). i Not determined because of non-adhesion of air bubbles. | |||||||
| Lin-PHEMA 27k | Linear | 26700a |
1.34c | 1.00/0.00 | 1 (1:0) |
21 ± 4 | 153 ± 10 |
| Lin-PHEMA 290k | Linear | 286000a |
1.77c | 1.00/0.00 | 1 (1:0) |
43 ± 2 | 159 ± 5 |
| Star-PHEMA | Star | 286000b |
1.24d | 1.00/0.00 | 20 (20:0) |
39 ± 1 | —i |
| Star-H71M29 | Star | 227000b |
1.17d | 0.71/0.29 | 14 (10:4) |
45 ± 3 | —i |
| Star-H47M53 | Star | 291000b |
1.39d | 0.47/0.53 | 19 (9:10) |
47 ± 2 | —i |
| Star-H22M48 | Star | 250000b |
1.23d | 0.22/0.78 | 18 (4:14) |
53 ± 4 | —i |
| Star-PMMA | Star | 209000b |
1.15d | 0.00/1.00 | 16 (0:16) |
73 ± 2 | 137 ± 10 |
| Lin-PMMA 10k | Linear | 10400a |
1.25c | 0.00/1.00 | 1 (0:1) |
71 ± 2 | 125 ± 2 |
| Lin-Block | Linear | 32500a |
1.23c | —f | 2 (1:1) |
43 ± 5 | 155 ± 11 |
| Lin-Random | Linear | 29200a |
1.19c | —f | 1 | 45 ± 3 | 126 ± 5 |
Coating substrates and preparation of polymer-coated surfaces
To test our strategy for preparing anti-fouling polymer coatings, we used poly(ethylene terephthalate) (PET) as an initial model substrate for the biomedical polymeric materials in this study. We chose PET because it has been widely used as a biomedical material for implants and artificial organs, including artificial blood vessels30 and heart valves,31 which have been also prone to adhesion of proteins, cells and bacteria, causing functional failures.1,2 PET films were coated by drop casting a polymer solution in organic solvents onto the PET film surface (Fig. 3A). We used methanol for the star-PHEMA polymer or a mixture of methanol/acetone (1/1 v/v) for the PHEMA–PMMA heteroarm star polymers because of the low solubility of PMMA arms with methanol. The coating solvent was first evaporated at room temperature, and then the coatings were dried under reduced pressure overnight.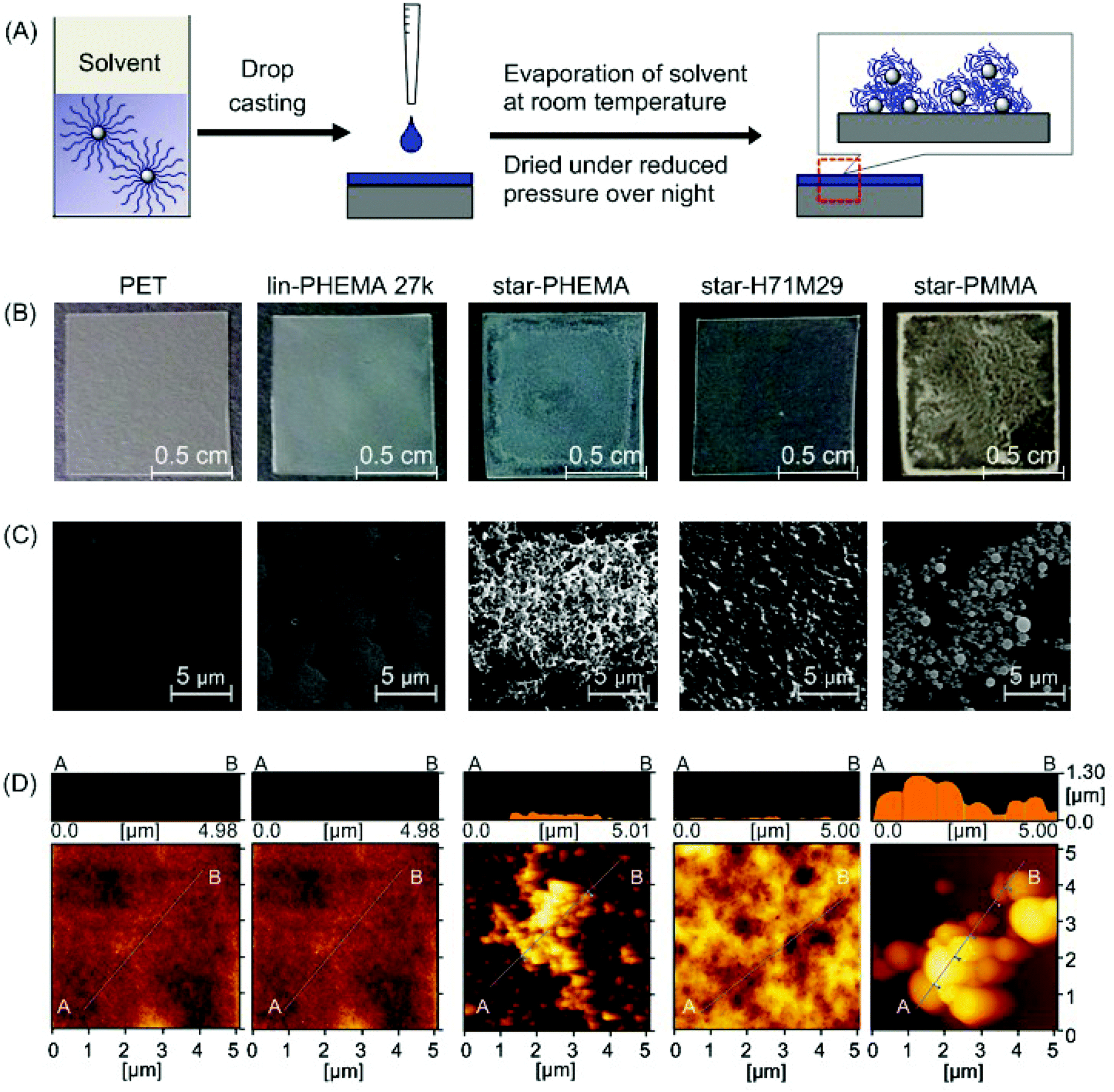 | ||
| Fig. 3 Polymer-coated surfaces. (A) Preparation of polymer coatings by drop casting. The polymer solution in a methanol or methanol/acetone mixture was dropped onto a PET surface and dried under reduced pressure overnight. (B) Pictures of polymer coatings. An unmodified PET film is transparent; the star-PHEMA and star-PMMA coatings appear to be heterogeneous. (C) SEM images of polymer-coated surfaces, and (D) AFM topographic images. See ESI† for surface images of other polymers. | ||
Surface characterization of coated surfaces
The surface morphology and topographical structures are key determinants for the anti-adhesion properties of polymer coatings against proteins, cells, and bacteria.32–34 We first examined the polymer-coated surfaces by SEM (Fig. 3C). The PET surface coated with lin-PHEMA 27k was relatively smooth. Interestingly, star-PHEMA formed fibrous aggregates, resulting in a network structure covering the entire substrate surface, which is likely reflected by the translucency of coated films (Fig. 3B). The difference in the surface structures between the linear and star polymers indicates that the star-polymer architecture of star-PHEMA is responsible for the formation of aggregates, possibly because of the high density of polymer chains, enhancing the polymer packing and entanglement. It has been reported previously that star-shaped poly(L-lactic acid) polymers self-assemble to nanofibrous structures forming hollow microspheres.35In contrast, the coating of star-H71M29 showed relatively rough surfaces, but no distinctive topographical structure was observed, giving transparent films. The star-PMMA showed clustering of small aggregates with a relatively uniform size, forming an island–sea structure on the surface (Fig. 3C). On the other hand, a number of small aggregates were scattered on the coating of lin-PMMA 10k (Fig. S2 in the ESI†). These results indicate that the surface structure of coatings depends on the polymer structures (star vs. linear), as well as on the properties (PHEMA vs. PMMA). The homo-star polymers (PHEMA and PMMA star polymers) tended to form aggregate structures on surfaces, likely because of the high density of polymer chains. However, the heteroarm star polymers containing both PMMA and PHEMA polymer chains rendered the coatings more homogeneous, indicating that the polymer aggregation and surface morphology can be controlled by the polymer-arm composition of the star polymers.
The microscopic structures of the polymer coatings were examined by AFM (Fig. 3D). The PET surface and polymer coatings displayed some roughness, giving a root-mean-square roughness of 4.69 nm (PET), 0.428 nm (lin-PHEMA 27k), 60.3 nm (star-PHEMA), 36.5 nm (star-H71M29), and 328 nm (star-PMMA). Interestingly, all surfaces coated with the star polymers had spherical structures 36–78 nm in diameter and approximately 17 nm in height, while the linear polymer PHEMA 27k did not show any specific surface structures (see Fig. S1† for AFM images of star-H47M53 and star-H22M78). We wonder whether the spherical structures consisted of individual star polymers or possibly aggregates of multiple star polymers. To that end, the size of the star polymers in the casting solvents was determined by DLS. The values of the hydrodynamic radius of the star polymers in the coating solvents were 19.0 nm (star-PHEMA), 17.3 nm (star-H71M29), 39.3 nm (star-H47M53), 17.2 nm (star-H22M78), and 14.8 nm (star-PMMA), with a relatively narrow distribution. This suggests that the multiple star polymers form spherical aggregates on the coatings during the casting and drying processes.
Wettability of polymer coatings
To determine the wettability of the coated surfaces, we measured the static contact angles of water droplets and air bubbles at the polymer coating. The wettability of surfaces plays an important role in the adhesion mechanisms of proteins and cells. In general, hydrophilic surfaces reduce the non-specific hydrophobic adhesion of proteins and cells, which is the initial key step in the biofouling mechanism. However, the wettability of surfaces does not relate directly to their ability to inhibit adhesion of biomolecules and cells, and it is known that the anti-adhesion activity of surfaces is also affected by polymer architectures,5 freezing bound water on surfaces, and the functionality of surface groups.36,37 Using the sessile drop method, the contact angle of non-coated PET was 65°. The contact angles of 39° for star-PHEMA and 43° for lin-PHEMA 290k are similar, although their polymer structures (star vs. linear) and surface morphologies (network vs. smooth surface) are significantly different. The contact angle increased as the PMMA ratio of star polymers increased from 52% to 71% (Table 1). This indicates that the hydrophobic PMMA increased the hydrophobicity of coating surfaces.The static water contact angles that were determined reflect the wettability of dry surfaces.38 However, adhesion of proteins and bacteria occurs generally in aqueous environments, and the surface properties in water are likely more related to the anti-adhesion activity of coatings. To that end, we determined the contact angle of air bubbles adherent on the coatings incubated in water at 37 °C for 12 h. The contact angles of the lin-PHEMA 27k and 290k coatings were 153° and 159°, respectively. On the other hand, the contact angles of the star-PHEMA and heteroarm star polymers could not be measured because air bubbles were not adsorbed on the coating surfaces, indicating that these coatings are highly hydrophilic. This suggests that the hydrophilic PHEMA star-polymer arms are hydrated and expand into water during incubation, increasing the hydrophilicity.39 Since the polymer chains are preassembled into the star polymer architectures, the polymer coatings are likely to develop polymer brush-like structures as the polymers are highly hydrated. The anti-adhesion activity of coatings against platelets and bacteria appears to reflect the hydrophilicity of the coatings and the polymer chain density in aqueous media, which is discussed below.
Scratch test
Durability of the coatings is imperative because the shear force is a common cause for the failed medical device surface coatings. To examine the physical durability of coatings, we evaluated the scratch resistance in the polymer coatings by measuring the scratch width caused by different loadings (Fig. 4A). The scratch width of the star-PHEMA coating was 40 μm at 4.9 mN loading and increased to 50 μm as the loading strength was increased to 19.6 mN and 49 mN (Fig. 4B). Conversely, the star-H71M29 displayed no scratches at 4.9 mN, and the scratch width was 22 μm at 19.6 mN, which is significantly smaller than that of the star-PHEMA coating. These results indicate that the star-H71M29 coating is not scratched readily compared with the star-PHEMA coating. In general, the scratch resistance reflects the mechanical strength and adhesiveness of coatings to substrates. This result indicates that the star-H71M29 coating provides a relatively homogeneous coating structure with higher mechanical strength and adhesiveness to a PET surface than the heterogeneous coating of the PHEMA star polymer. This may be because of the properties of PMMA, which displays good adhesiveness to plastic materials and hardness in general. The physical durability of the star-H71M29 coating will facilitate the handling of coated materials and will be useful for applications such as coating catheters and devices where physical strength in a coating is desirable.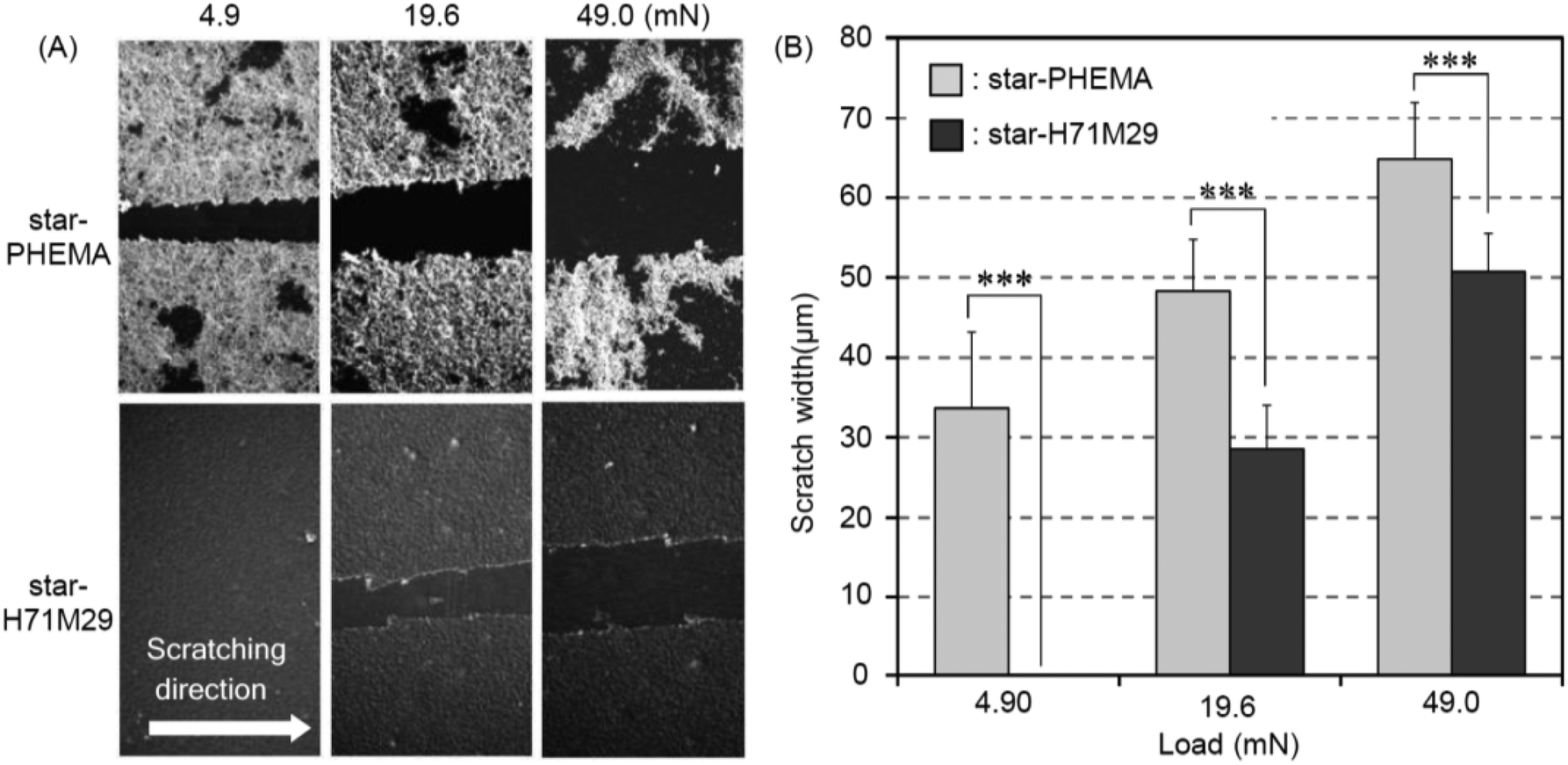 | ||
| Fig. 4 Scratch resistance test. (A) SEM of star-PHEMA and star-H71M29 coatings at the different scratch loads. (B) Scratch widths on star-PHEMA and star-H71M29 coating surfaces after loading (mean ± standard deviation, n = 3). ***p < 0.001. | ||
To examine the microstructures of coating layers in more detail, the marginal portion of the scratch was examined further by AFM (Fig. 5). The star-PHEMA coating has a homogeneous coating layer with a thickness of approximately 11 nm and aggregate structures with a height of approximately 250 nm, which is likely to be a part of the macroscopic fibrous network structure of coating observed in the SEM image (Fig. 2). This indicates that the star-PHEMA coating consists of a three-dimensional network structure on the underlying homogeneous coating layer. It should be noted that the non-coated PET surface did not show any scratches at 49 mN loading. Therefore, the bottom layer of the star-PHEMA coating is not an artifact due to the scratched PET. On the other hand, the star-H71M29 coating had one layer with a thickness of approximately 140 nm, indicating that the heteroarm star polymers provide homogeneous coating surfaces.
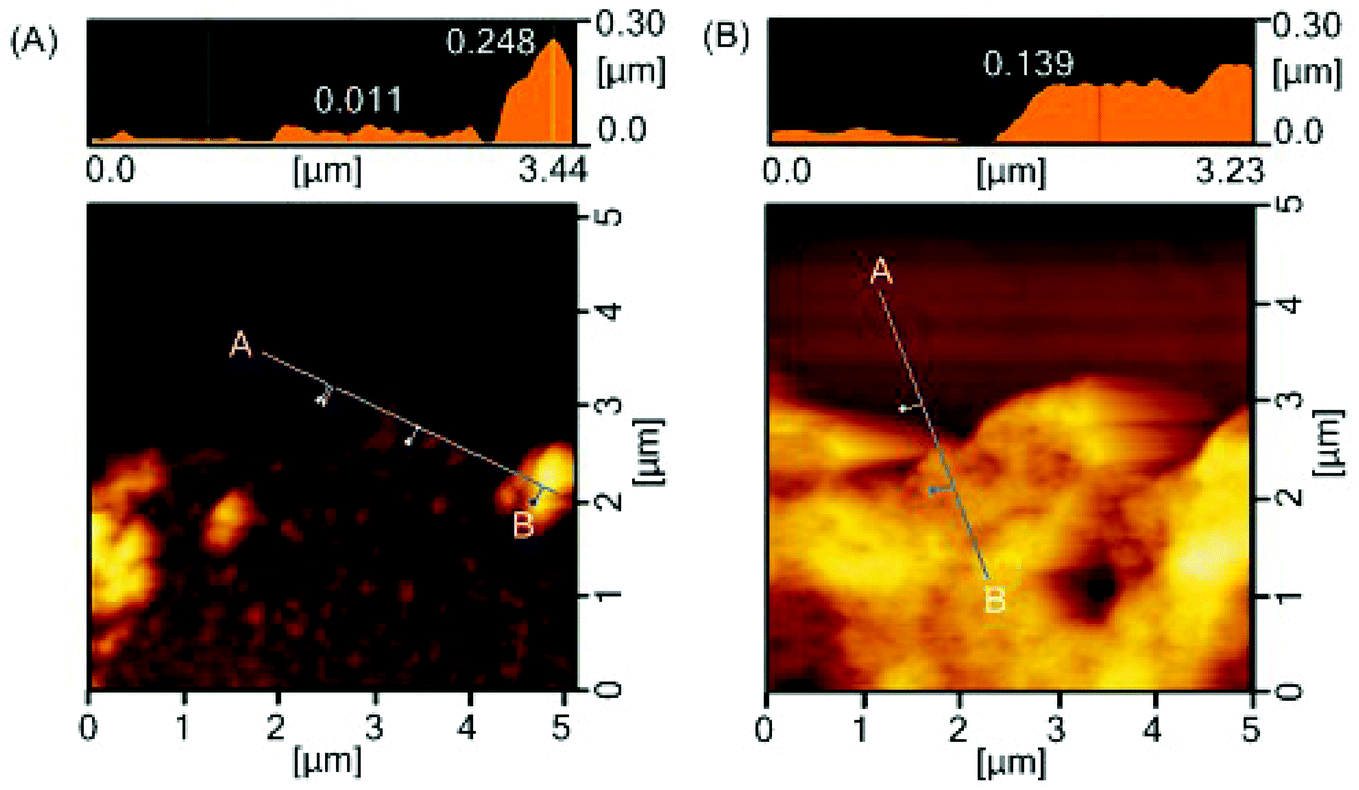 | ||
| Fig. 5 Surface characterization after scratch testing. AFM images of edge of scratch on (A) star-PHEMA- and (B) star-H71M29-coated surfaces. | ||
Platelet adhesion
To assess the anti-adhesion effectiveness of the star polymers on PET surfaces, we examined in vitro platelet adhesion to the coated surfaces. PET is a conventional biomedical synthetic material for artificial blood vessels and heart valves. However, the PET surface is prone to platelet adhesion, which triggers fibrin production, resulting in blood clot formation. Accordingly, we used PET as a model substrate to test the effectiveness of star polymer coatings to prevent platelet adhesion, and the results were compared with non-coated PET as a positive control. The coated PET substrates were soaked in water for 12 h prior to the platelet adhesion assay to hydrate the polymer coatings and to increase the surface hydrophilicity, as indicated by the air-bubble contact angle measurement discussed above. The polymer-coated PET substrates were incubated with PRP at 37 °C for 30 min, based on the standard protocol in the literature.40 The platelet adhesion on the coated surfaces was characterized by SEM. The number of platelets that adhered to star-PHEMA and star-H71M29 coatings was significantly smaller than that for the non-coated PET and other star and linear polymers (Fig. 6A). The magnified SEM images indicated that the morphology of platelets adherent on surfaces depends on the coatings. Some platelets formed lamellipodia on the non-coated PET surface, which induced aggregation of platelets to adhere firmly onto the surface. On the other hand, each platelet seems to be isolated rather than aggregated for most of the polymer coatings.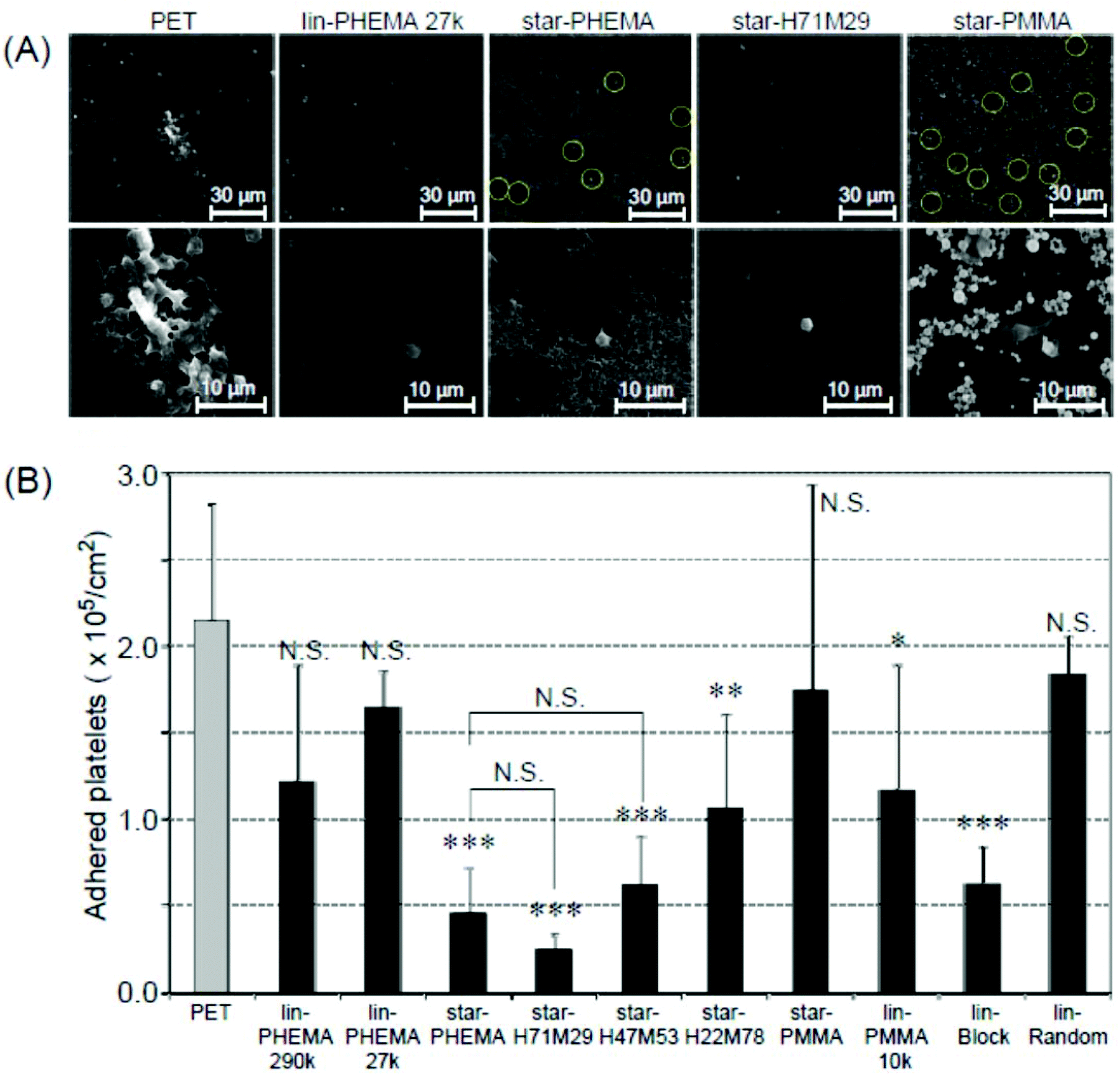 | ||
| Fig. 6 Platelet adhesion to polymer-coated surfaces. (A) SEM images of adherent platelets on the polymer-coated surfaces. The platelets on the star-PHEMA and star-PMMA polymers are highlighted by circles for clarification. Image magnification: ×400 (top) and ×1500 (bottom). Images of samples are shown in Fig. S3 and S4 in the ESI.† (B) The number of adherent platelets on polymer coatings. The number of platelets was determined from the SEM images (mean ± standard deviation, n = 3). ***p < 0.0001, **p < 0.001, *p < 0.005 vs. PET. | ||
To quantify the platelet adhesion, the number of adherent platelets on the coatings was determined from the SEM images (Fig. 6A). It is evident that the number of platelets (Fig. 6B) on the coating of star polymers, except for the star-PMMA coating, was significantly smaller than those for the non-coated PET. The percentages of inhibition were 78% for the star-PHEMA, 23% for the lin-PHEMA 27k, and 43% for the lin-PHEMA 290k coatings (Table 2). These results indicate that star-PHEMA prevented platelet adhesion more effectively than the linear PHEMA polymers. This seems to reflect the higher hydrophilicity of star-PHEMA, determined from the air-bubble contact angle in water, compared with lin-PHEMA. These results also suggest that the star-shaped polymer architecture plays an important role in resistance to platelet adhesion. Since the polymer chains (∼20 arms) are assembled into one core to give star-shaped polymers, the polymer coatings provide polymer brush-like structures on the coating surface, which are likely to expand in water, increasing the hydrophilicity of the coatings and the exclusion volume of polymer brushes, thus expelling platelets more effectively than linear polymers. However, rough surfaces generally favour platelet adhesion because of increased areas available for adhesion, as well as geometrical niches for adhesion mechanisms.33,41 In addition, because the coatings consist of multiple layers, platelets may also adhere to not only the surface, but also the coating layers or structures. Therefore, the fibrous network structure of the star-PHEMA coating could rather enhance the platelet adhesion. We speculate that the fibrous network is hydrated and swollen with water, which increases the coverage of coatings and reduces the surface roughness and contributes to high hydrophilicity and thus high anti-adhesion activity.
| Polymer coatings | Inhibition percentagea (%) | |||
|---|---|---|---|---|
| Platelet | E. coli | |||
| Standard testb | Stability test PBSc (7 days) | Stability test Triton X solnd (7 days) | Standard test | |
| a The percentage of inhibition was calculated by the following equation: % of adherent platelet inhibition = (adherent platelet on PET − adherent platelet on polymer coatings) × 100/adherent platelet on PET. % of adherent bacteria inhibition = (luminescence of adherent bacteria on PET–luminescence of adherent bacteria) × 100/luminescence of adherent bacteria on PET. b Coatings incubated in PBS for 12 hours prior to the assay, calculated based on the data from Fig. 6. c Coatings incubated in PBS for 7 days prior to the assay. d Coatings incubated in 0.5 wt% Triton X-100 in PBS for 7 days prior to the assay. | ||||
| Lin-PHEMA 290k | 43 ± 27 | 71 ± 16 | 71 ± 3 | 53 ± 2 |
| Lin-PHEMA 27k | 23 ± 8 | 35 ± 48 | 26 ± 23 | 87 ± 8 |
| Star-PHEMA | 78 ± 11 | 97 ± 3 | 92 ± 5 | 92 ± 3 |
| Star-H71M29 | 88 ± 1 | 96 ± 1 | 88 ± 3 | 94 ± 1 |
| Lin-Block | 71 ± 7 | 56 ± 27 | 63 ± 21 | 93 ± 5 |
The coatings of the heteroarm star polymers also showed an inhibitory effect against platelet adhesion. Star-H71M29 showed an anti-adhesion effect similar to star-PHEMA despite containing PMMA although the hydrophobic properties of coating surfaces generally enhance adhesion of proteins and cells.42 This may be due to the effect of aggregation of hydrophobic PMMA, which might increase the density of star polymers, and thus increase the density of hydrophilic polymer chains.22 We also speculate that the PMMA polymer chains may adhere to the PET surface and sequester from the surfaces, and the coating surface could be mostly covered by the PHEMA chains. The scratch tests and surface images also indicate that the star-H71M29 coating is stable and has a smooth surface (Fig. 4 and 3D), which decreases roughness and defect formation, reducing niches for platelet adhesion. These effects could contribute to the anti-adhesion properties of star-H71M29 against platelets. In addition, the number of adhered platelets increased as the percentage of PMMA arms in the star polymers increased. Since the average number of polymer arms is similar for all star polymers, this result indicates that the polymers containing more hydrophobic PMMA or less hydrophilic PHEMA are less resistant to platelet adhesion, which is consistent with the previous reports that the hydrophobicity of coatings increases platelet adhesion.43
For comparison, the block copolymer lin-Block showed an anti-adhesion effect similar to that of the star-polymer coatings, although the random copolymer lin-Random did not show any significant anti-adhesion properties. The PMMA block segment of the block copolymer is likely to increase the adhesion of polymer chains to the PET surface, anchoring the hydrophilic PHEMA segments, which provide polymer brush structures and prevent platelet adhesion.11 It has also been previously reported that polymer coatings on a glass surface with amphiphilic copolymers, including PHEMA-b-PSt-b-PHEMA, effectively prevent adhesion of platelets and filopodium.7 Block copolymers with fluoroalkyl components also showed anti-fouling and fouling-release activities against proteins, bacteria, and marine organisms.44–46 These studies suggested that formation of phase-separated domains by the hydrophilic and hydrophobic (fluorinated) polymers are responsible for the anti-adhesion effect because these domains may disrupt settlement of protein and microbial adhesion.47,48 It is not clear in the AFM images whether the block copolymer and heteroarm star polymers studied in this report form segregated domains in their coatings. However, microstructures or domains of star polymers might also contribute to resistance to platelet adhesion, although a more detailed study is necessary.
Bacterial adhesion
Biomedical synthetic materials suffer from bacterial adhesion and subsequent biofilm formation, causing adverse infections and complications. To evaluate the resistance of polymer coatings to bacterial adhesion, we used E. coli as an initial model bacterium. E. coli is one of the pathogens causing adverse device- and implant-associated infections.49 In general, the results are similar to those of platelet adhesion. E. coli formed dense bacterial clusters on the unmodified PET surface (Fig. 7A and S5†). These bacterial clusters are considered to be adhered strongly on the surfaces because the non-adherent and lightly adherent bacteria on the surface were washed away with PBS 3 times after the incubation, and only strongly adhered bacteria can remain on the surface. The star-PHEMA and star-H71M29 coatings showed few bacteria. The E. coli appears to adhere on the top of the network structure of the star-PHEMA coating rather than being trapped or in varied positions in the network. This may support the notion that the polymer network is swollen with water and covers the coating surface, preventing bacterial adhesion to the empty spaces in the network. The adherent bacteria were quantified by the luminescence assay (Fig. 7B). It should be noted that there is no significant difference in the OD of the bacterial assay solutions incubated with the coatings, indicating that these polymer coatings did not inhibit bacterial growth in the solution. This suggests that inhibiting bacterial growth or killing bacteria is not the primary mechanism of these coatings’ resistance to bacterial adhesion. Star-PHEMA and star-H71M29, and lin-PHEMA 27k and lin-Block showed a similar level of bacterial adhesion inhibition, although the lin-PHEMA 290k polymer did not prevent platelet adhesion (Fig. 6B). These results indicate that the high-density polymer brushes on the coating surface effectively prevent E. coli adhesion. Similar to the platelet adhesion, the bacterial adhesion increased as the percentage of PMMA in the star polymers was increased, indicating that more bacteria adhere to the coating with higher hydrophobicity. The electronically neutral and hydrophilic polymer blush exhibits the effective inhibition of the bacterial adhesion.50 PHEMA chains of the star polymer coated on the surface are also electronically neutral and hydrophilic, and they thus showed the effective resistance to E. coli adhesion. In addition, the PHEMA chain does not have a D-mannose-like structure, which is the ligand of the adhesion in the fimbriae, and it prevents the adhesion mediated by the fimbriae.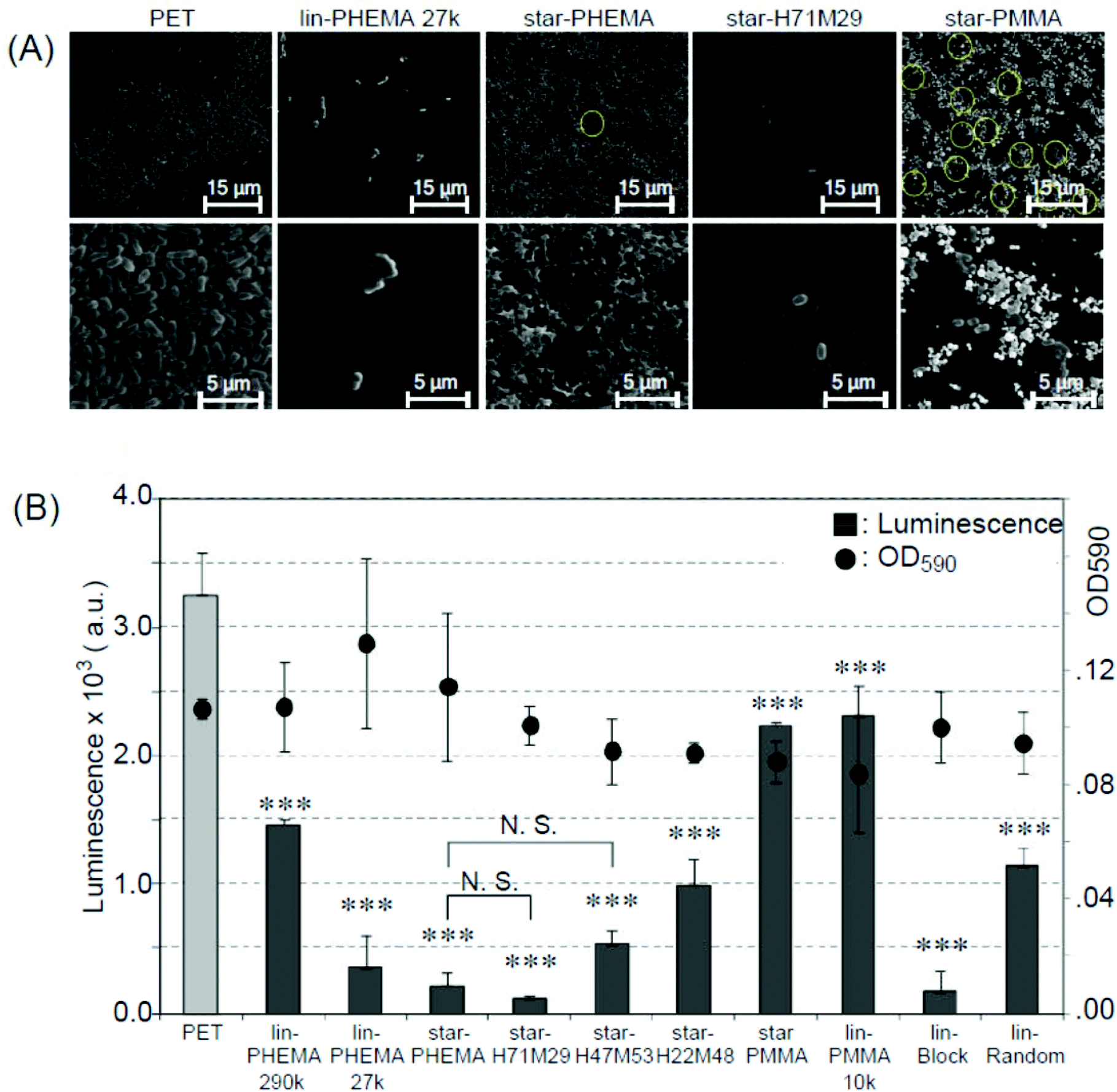 | ||
| Fig. 7 Bacterial adhesion to polymer-coated surfaces. (A) SEM images of adherent E. coli (ATCC 25922) on polymer-coated surfaces. Image magnification ×400 (top) and ×2500 (bottom). Images of samples are shown in Fig. S5 and S6 in the ESI.† (B) Bacterial adhesion and growth in solution (mean ± standard deviation, n = 3). Adhesion of E. coli on to polymer-coated surface was quantified using the luminescence assay. The growth of E. coli was determined by OD590 (•) after 20 h, 37 °C incubation. ***p < 0.0001 vs. PET. | ||
Stability of coatings in an aqueous environment
Because the polymers studied are not attached covalently onto the PET surfaces, the polymers could be released into water or the coatings delaminated, exposing the bare PET surface after a prolonged period. This would compromise the anti-adhesion effect of coatings against platelets. To that end, the coating stability was tested by incubating the coatings in PBS or surfactant (Triton X-100) solution for 7 days at 37 °C with gentle shaking at 180 rpm prior to the platelet adhesion assay. The star-PHEMA and star-H71M29 coatings retained a good anti-adhesion effect against platelets even after the surfactant challenge (Fig. 8). Lin-PHEMA 290k and 27k and lin-Block showed the same level of percentages of inhibition as the same samples tested previously, presented in Fig. 8. The percentage of inhibition by the star polymer coatings (96–97%) was slightly higher than those of the same samples without incubation (78–88%) (Table 2). The slight enhancement of the anti-adhesion properties may be related to the hydration of the coated polymer chains in water during the incubation for 7 days, increasing the hydrophilicity of coatings. In this experiment, the polymer-coated surfaces achieved the durable coatings. This is an important factor for medical application as anti-thrombogenic and anti-microbial coating materials. Although the polymer coatings retained the anti-fouling effects, it is not clear at this point that the polymer coatings are physically intact or underwent any changes in surface structures after incubation in water or with the surfactant. Although the detailed investigation on the coating stability is beyond the scope of this report, quantitative analysis of the coating stability such as thickness changes and defect formation and potential leachables in aqueous media and physiological fluids would be necessary for the use of the polymers as anti-fouling coatings on biomedical materials.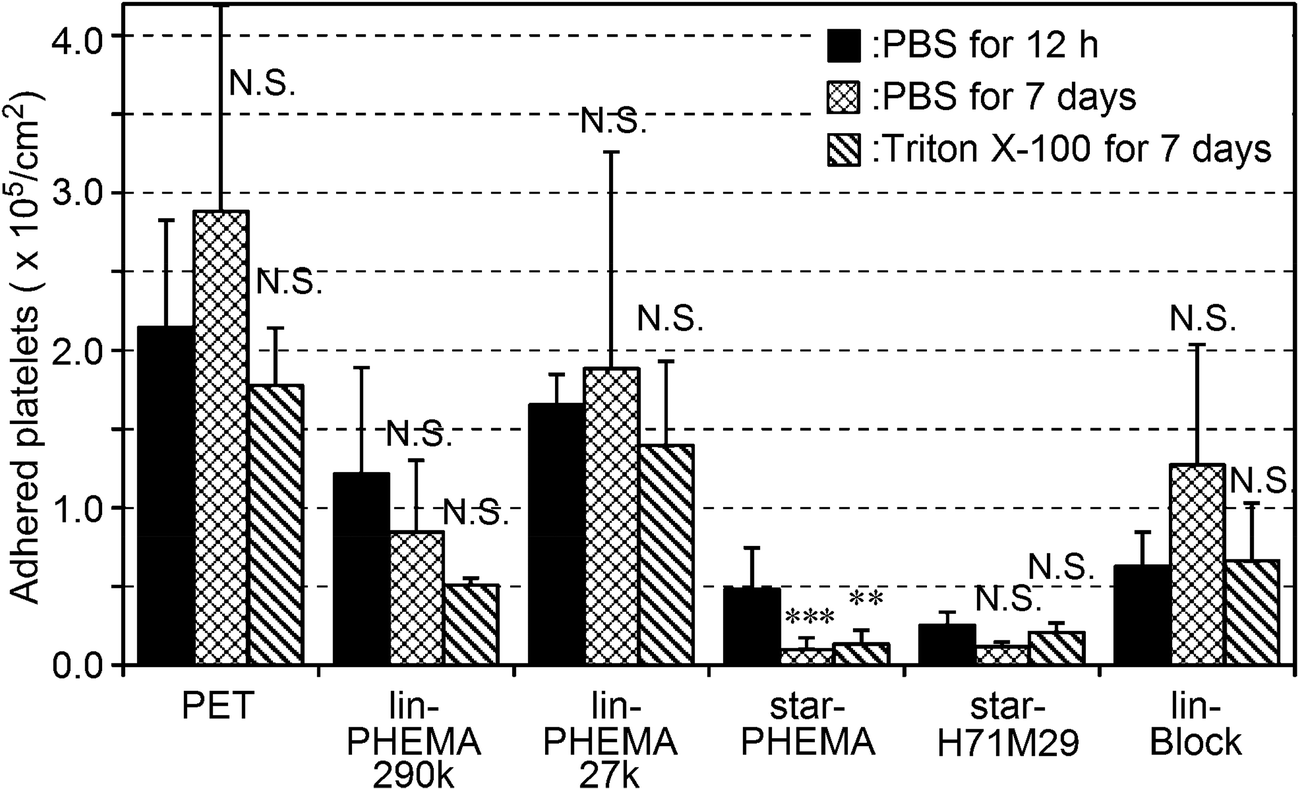 | ||
| Fig. 8 Stability test of polymer-coated surface by platelet adhesion to the coatings after incubation in PBS for 12 h, and PBS or 0.5 wt% Triton X-100 solution with gentle shaking for 7 days. ***p < 0.0001, **p < 0.001, *p < 0.005 vs. PET. | ||
Conclusion
In summary, we synthesized star-shaped polymers with hydrophilic PHEMA and hydrophobic PMMA polymer arms using Ru-catalyzed living radical polymerization. PET films were coated by drop casting the star-polymer solution and drying. SEM and AFM analyses indicated that star-PHEMA aggregated on PET films to form a fibrous network but that star-H71M29 provided relatively smoother surfaces compared with star-PHEMA and star-PMMA. Among the star polymers, star-PHEMA and star-H71M29 inhibited adhesion of platelets and E. coli by 78–88% relative to the non-coated PET surface. These coatings retained the anti-adhesion properties after incubation in PBS or surfactant solution for a week, suggesting that the star-polymer architecture provided high polymer chain density on the surfaces to prevent adhesion of platelets and bacteria, as well as coating stability to prevent exposure of the bare PET surfaces. In addition, the PMMA component of the star polymers increased the scratch resistance, providing physical durability for potential applications of coatings on medical implants and devices.The results indicate that star polymers can provide an effective approach to the preparation of highly dense polymer brushes on PET surfaces for anti-adhesion coatings against platelets and E. coli. Although the presented study showed the promising results, detailed studies on their molecular mechanism and anti-fouling effectiveness against a panel of healthcare-related bacterial pathogens would be necessary to determine their usefulness as anti-fouling coatings. The polymer preparation and coating methods are simple and cost-effective. PET was used as a model substrate in this report, but this star-polymer coating could be used for a wide range of biomedical synthetic materials. To that end, our on-going work includes testing the star-polymer coatings on a variety of abiotic surfaces such as metals and plastics to determine the versatility of this approach. It has been reported previously that dopamine derivatives have been utilized to modify inert plastic and metal surfaces with chemically labile groups for polymer modifications, providing versatile methods for surface modification and anti-fouling coatings.51 Similarly, we envision that the star-polymer coatings may provide an effective strategy using polymer brush coatings for many different types of surfaces.
Acknowledgements
We thank NIH for financial support (1R21DE020908-1 to KK and CX) and NSF for the NSF CAREER Award (DMR-0845592 to KK). We would also like to thank Dr Xiuwan Lan and Dr Jianfeng Wu at the University of Michigan for helpful discussion. We also thank Prof. Mitsuo Sawamoto at Kyoto University for the use of the SEC–MALLS, and Dr Kazuma Yasuhara at the Nara Institute of Science and Technology for the use of DLS instrument.References
- P. Didisheim, ASAIO J., 1994, 40, 230–237 CrossRef CAS PubMed
.
- R. O. Darouiche, N. Engl. J. Med., 2004, 350, 422–429 CrossRef PubMed
- D. Mack, A. P. Davies, L. G. Harris, H. Rohde, M. A. Horstkotte and J. K.-M. Knobloch, Anal. Bioanal. Chem., 2007, 387, 399–408 CrossRef CAS PubMed
- Z. Zhang, J. Wang, Q. Tu, N. Nie, J. Sha, W. Liu, R. Liu, Y. Zhang and J. Wang, Colloids Surf., B, 2011, 88, 85–92 CrossRef CAS PubMed
- S. Nagaoka and A. Nakao, Biomaterials, 1990, 11, 119–121 CrossRef CAS
- I. Banerjee, R. C. Pangule and R. S. Kane, Adv. Mater., 2011, 23, 690–718 CrossRef CAS PubMed
- M. Shimada, M. Unoki, N. Inaba, H. Tahara, I. Shinohara, T. Okano, Y. Sakurai and K. Kataoka, Eur. Polym. J., 1983, 19, 929–933 CrossRef CAS
- M. Tanaka, T. Motomura, M. Kawada, T. Anzai, Y. Kasori, T. Shiroya, K. Shimura, M. Onishi and A. Mochizuki, Biomaterials, 2000, 21, 1471–1481 CrossRef CAS
- J. H. Lee, Y. M. Ju and D. M. Kim, Biomaterials, 2000, 21, 683–691 CrossRef CAS
- M. R. Nejadnik, H. C. van der Mei, W. Norde and H. J. Busscher, Biomaterials, 2008, 29, 4117–4121 CrossRef CAS PubMed
- Y. Iwasaki and K. Ishihara, Anal. Bioanal. Chem., 2005, 381, 534–546 CrossRef CAS PubMed
- J. H. Seo, S. Kakinoki, Y. Inoue, K. Nam, T. Yamaoka, K. Ishihara, A. Kishida and N. Yui, Biomaterials, 2013, 34, 3206–3214 CrossRef CAS PubMed
- G. Cheng, Z. Zhang, S. Chen, J. D. Bryers and S. Jiang, Biomaterials, 2007, 28, 4192–4199 CrossRef CAS PubMed
- G. Cheng, G. Li, H. Xue, S. Chen, J. D. Bryers and S. Jiang, Biomaterials, 2009, 30, 5234–5240 CrossRef CAS PubMed
- N. Ayres, Polym. Chem., 2010, 1, 769–777 RSC
- S. Chen, L. Li, C. Zhao and J. Zheng, Polymer, 2010, 51, 5283–5293 CrossRef CAS PubMed
- C. Yoshikawa, A. Goto, Y. Tsujii, T. Fukuda, T. Kimura, K. Yamamoto and A. Kishida, Macromolecules, 2006, 39, 2284–2290 CrossRef CAS
- G. R. Llanos and M. V. Sefton, J. Biomater. Sci., Polym. Ed., 1993, 4, 381–400 CrossRef CAS PubMed
- S. J. Sofia, V. Premnath and E. W. Merrill, Macromolecules, 1998, 31, 5059–5070 CrossRef CAS PubMed
- K. Bruellhof, J. Fiedler, M. Möller, J. Groll and R. E. Brenner, Int. J. Artif. Organs, 2010, 33, 646–653 Search PubMed
- S. Bauer, P. Schmuki, K. von der Mark and J. Park, Prog. Mater. Sci., 2013, 58, 261–326 CrossRef CAS PubMed
- D.-G. Kim, H. Kang, S. Han and J.-C. Lee, J. Mater. Chem., 2012, 22, 8654–8661 RSC
- D.-G. Kim, H. Kang, Y.-S. Choi, S. Han and J.-C. Lee, Polym. Chem., 2013, 4, 5065–5073 RSC
- D.-G. Kim, H. Kang, S. Han, H. J. Kim and J.-C. Lee, RSC Adv., 2013, 3, 18071–18081 RSC
- M. Ouchi, T. Terashima and M. Sawamoto, Chem. Rev., 2009, 109, 4936–5050 CrossRef PubMed
- K. Y. Baek, M. Kamigaito and M. Sawamoto, J. Polym. Sci., Part A: Polym. Chem., 2002, 40, 1937–1944 CrossRef CAS
- A. Hirao, H. Kato, K. Yamaguchi and S. Nakahama, Macromolecules, 1986, 19, 1294–1299 CrossRef CAS
- M. Kamigaito, T. Ando and M. Sawamoto, Chem. Rec., 2004, 4, 159–175 CrossRef CAS PubMed
- H. Gao and K. Matyjaszewski, Macromolecules, 2008, 41, 4250–4257 CrossRef CAS
- S. Ramakrishna, J. Mayer, E. Wintermantel and K. W. Leong, Compos. Sci. Technol., 2001, 61, 1189–1224 CrossRef CAS
- S. Tokunaga and R. Tominaga, J. Artif. Organs, 2010, 13, 77–87 CrossRef PubMed
- L. Mei, H. J. Busscher, H. C. van der Mei and Y. Ren, Dent. Mater., 2011, 27, 770–778 CrossRef CAS PubMed
- L. B. Koh, I. Rodriguez and S. S. Venkatraman, Biomaterials, 2010, 31, 1533–1545 CrossRef CAS PubMed
- M. L. B. Palacio, S. R. Schricker and B. Bhushan, J. R. Soc. Interface, 2011, 8, 630–640 CrossRef CAS PubMed
- X. Liu, X. Jin and P. X. Ma, Nat. Mater., 2011, 10, 398–406 CrossRef CAS PubMed
- M. Tanaka and A. Mochizuki, J. Biomed. Mater. Res., 2004, 68, 684–695 CrossRef PubMed
- E. Ostuni, R. G. Chapman, R. E. Holmlin, S. Takayama and G. M. Whitesides, Langmuir, 2001, 17, 5605–5620 CrossRef CAS
- T. Tsukagoshi, Y. Kondo and N. Yoshino, Colloids Surf., B, 2007, 54, 101–107 CrossRef CAS PubMed
- H. Kitano, M. Imai, T. Mori, M. Gemmei-Ide, Y. Yokoyama and K. Ishihara, Langmuir, 2003, 19, 10260–10266 CrossRef CAS
- G. C. Xu, Y. Hibino, Y. Suzuki, M. Tanihara, Y. Imanishi and K. Awazu, J. Biomater. Sci., Polym. Ed., 2001, 12, 503–514 CrossRef CAS PubMed
- M. Hulander, A. Lundgren, L. Faxälv, T. L. Lindahl, A. Palmquist, M. Berglin and H. Elwing, Colloids Surf., B, 2013, 110, 261–269 CrossRef CAS PubMed
- D. Rana and T. Matsuura, Chem. Rev., 2010, 110, 2448–2471 CrossRef CAS PubMed
- M.-X. Hua, Q. Yanga and Z.-K. Xua, J. Membr. Sci., 2006, 285, 196–205 CrossRef PubMed
- T. Merian and J. M. Goddard, J. Agric. Food Chem., 2012, 60, 2943–2957 CrossRef CAS PubMed
- J. A. Callow and M. E. Callow, Nat. Commun., 2011, 2, 1–10 Search PubMed
- S. Krishnan, C. J. Weinman and C. K. Ober, J. Mater. Chem., 2008, 18, 3405–3413 RSC
- C. S. Gudipati, J. A. Finlay, J. A. Callow, M. E. Callow and K. L. Wooley, Langmuir, 2005, 21, 3044–3053 CrossRef CAS PubMed
- E. Martinelli, S. Agostini, G. Galli, E. Chiellini, A. Glisenti, M. E. Pettitt, M. E. Callow, J. A. Callow, K. Graf and F. W. Bartels, Langmuir, 2008, 24, 13138–13147 CrossRef CAS PubMed
- M. Melzer and C. Welch, Lancet Infect. Dis., 2012, 12, 103–104 CrossRef
- E. E. MacKintosh, J. D. Patel, R. E. Marchant and J. M. Anderson, J. Biomed. Mater. Res., Part A, 2006, 78, 836–842 CrossRef PubMed
- H. Lee, S. M. Dellatore, W. M. Miller and P. B. Messersmith, Science, 2007, 318, 426–430 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available: Syntheses of star polymers. Calculation of the number of arms. SEM and AFM images of coatings. SEM images of platelet adhesion. SEM images of bacterial adhesion. See DOI: 10.1039/c4bm00034j |
| This journal is © The Royal Society of Chemistry 2014 |