Peptoids for biomaterials science
King Hang Aaron
Lau
Department of Pure and Applied Chemistry, University of Strathclyde, Thomas Graham Building, 295 Cathedral Street, Glasgow, G1 1XL, UK. E-mail: aaron.lau@strath.ac.uk; Tel: +44 (0)141 548 2162
First published on 16th January 2014
Abstract
Poly(N-substituted glycine) “peptoids” have conventionally been exploited for drug discovery and therapeutics due to their structural similarity to peptides, protease resistance, and relative ease of synthesis. This mini-review highlights recent reports of peptoid self-assembled nanostructures and macromolecular interfaces relevant to biomaterials science. The results illustrate how the versatility of peptoid design and synthesis could be exploited to generate multifunctional, modular and precisely tunable biointerfaces and biomaterials.
 King Hang Aaron Lau | Dr K. H. Aaron Lau is a lecturer in the Department of Pure and Applied Chemistry at the University of Strathclyde. He received his Sc.B. and Sc.M. from Brown University and completed his Ph.D. at the Max Planck Institute for Polymer Research. He was previously a US National Institutes of Health National Research Service Award postdoctoral fellow with Prof. Phillip B. Messersmith at Northwestern University. Aaron's core interests are in bioinspired materials and biointerfaces, and he currently directs projects in peptoid biomaterials and bioinspired nanopore applications. |
1. Introduction
Materials that can be conveniently tuned to present the biochemical, morphological and mechanical features of native biological systems are of special interest to biomaterials science.1–3 The incorporation of targeting elements minimizes the potential of negative systemic effects, an important translational consideration.1 These ideas have inspired significant research in peptidic biomaterials3,4 as well as the chemical functionalization of both naturally-derived and synthetic polymers.2,5 Poly(N-substituted glycine) “peptoids” (Fig. 1A), a class of highly customizable peptidomimetic macromolecules, could also potentially enable significant advances in biomaterials science.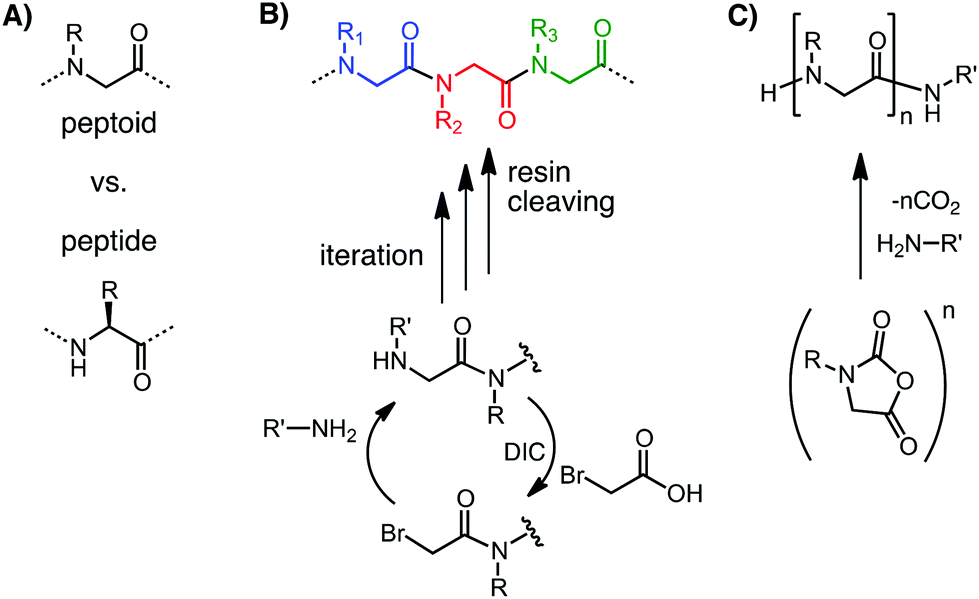 | ||
| Fig. 1 Peptoid chemical structure and synthesis. (A) A shift in the sidechain connection from the α-carbon to the amide nitrogen, accompanied by loss of main chain chirality and amide hydrogens, differentiates peptoids from peptides (L-amino acid residue shown). (B) Submonomer solid phase synthesis: residue coupling is split into elongating the chain via acylation with a haloacetic acid and sidechain introduction via displacement with a primary amine submonomer. DIC: N,N′-diisopropylcarbodiimide. (C) NCA peptoid polymerization. | ||
Peptoids† were developed in the late 1980s for combinatorial drug discovery as synthetically convenient, modular, peptidomimetic molecules.6,7 They are structural isomers of peptides with sidechains connected to the amide nitrogens instead of the α-carbons (Fig. 1A). This structural change confers resistance to protease degradation.7–9 The sidechain shift results in tertiary amide linkages, leading to a backbone that is achiral and that has no H-bond donors. These properties confer substantial conformational flexibility in the main chain.10–14 Chirality and control over secondary and higher order structure formation can be reintroduced by sidechain chemistry and sequence designs.10,12,13,15
Synthetic convenience and modularity in the generation of sequences with monomer-level programmability over sidechain chemistry and chain length derive largely from the introduction of the submonomer solid phase synthesis by Zuckermann et al. (Fig. 1B).7,16 No backbone protection is required and diverse primary amines, many of which are commercially available, can be incorporated as sidechains in an iterative protocol. Over 250 different residues, including analogs or close mimics of all the canonical amino acids, have been demonstrated.17,18 Peptoids of up to 100 residues in length have been demonstrated by coupling together two submonomer-synthesized 50-mers.19 Alternatively peptoids can be synthesized with high degrees of polymerization >100,20 but with limitations in sequence control, by the living polymerization of N-carboxy anhydride (NCA) monomers (Fig. 1C).21–23 The polymer of sarcosine, which is the analog of alanine and the simplest peptoid, was actually known from early investigations into living polymerization.21 The list of peptoid monomers available to NCA polymerization has been significantly expanded in recent years.21,22
The biorecognition abilities of peptoids have been demonstrated through two decades of peptoid therapeutics research, protein-binding sequence discovery, and secondary structure design, which have been recently reviewed elsewhere.10,15,24–27 As originally envisioned,7 many bioactive sequences have been discovered by screening peptoid libraries.8,13 These include antimicrobial peptoids—a significant area of peptoid research discussed in recent reviews.10,25,27 Recent studies have also demonstrated peptoid sequences potentially useful in diagnosing or treating amyloid diseases.28
This mini-review highlights the recent developments in peptoids research which extend into the field of biomaterials science. A number of recent reviews have focused on how the synthetic accessibility and sequence programmability in chemistry and chain length of peptoids have generated broad potential in macromolecular and nanostructure engineering.12,13,21,22 This mini-review first discusses the biomaterial applications of these self-assembled structures. Attention is then turned to peptoid macromolecular interfaces and platforms that could offer unique possibilities. The aim is to illustrate how the versatility in peptoid design and synthesis can lead to macromolecular architectures with high potential in biomaterials science.
2. Self-assembled systems
2.1. Molecular (hydro)gelators
A pair of studies by Wu et al.29 and Mangunuru et al.30 demonstrate that low molecular weight peptoid sequences could be utilized as gelators. Wu et al. picked a sequence of four peptoid analogs of phenylalanine (Phe) as the gelation motif (Fig. 2; peptoid phenylalanine, i.e. N-benzylglycine, is shortened to NPhe below). The tetrapeptoid (NPhe)4 was linked via a glycine residue to tripeptides with demonstrated bioactivity (RGD, YSV and VPP), as well as a triglycine GGG control. Gels of all four sequences could be formed in phosphate buffered saline at a concentration of 10 mg mL−1 at pH 7.4. However the behavior of standalone (NPhe)4 and (NPhe)4G were not reported. The hydrogels were composed of either nanosheets or nanofibrils (Fig. 2A–C), and the highest shear storage modulus was observed for (NPhe)4GGGG (750 Pa over 0.1–100 rad s−1). The viabilities of several cell lines in solutions of the peptoids were not seriously affected (tested up to 200 μM). Fibroblasts also cultured well on the (NPhe)4GRGD and, especially, on the (NPhe)4GYSV gels. Finally, it was demonstrated that the (NPhe)4 sequence, but not (D-Phe)4 or (L-Phe)4, was effective in protecting the pendant RGD sequences from degradation by proteinase K. | ||
| Fig. 2 Peptoid hydrogels. TEM images of hydrogels composed of (A) (NPhe)4GGGG, (B) (NPhe)4GRGD, (C) (NPhe)4GYSV, and (D) (NPhe)4GVPP. The first two hydrogels were composed of sheet-like microstructures while the latter two were formed from filamentous structures. Adapted from ref. 29 with permission from The Royal Society of Chemistry. | ||
In a different approach, Mangunuru et al. scanned for gelators among a small library synthesized by Ugi one-pot four-component reactions.30 Single peptoid residues functionalized at both the C and N “termini” were generated. Many of the sidechain functional groups investigated have previously been demonstrated using the submonomer solid phase protocol.17 Although Mangunuru et al. reported only gels formed in water mixed with ≥33% ethanol or dimethyl sulfoxide (DMSO), this work highlighted the chemical tunability possible with peptoids and the chemical groups that could promote peptoid gelation.
The best gelators reported by Mangunuru et al. exhibited substituted aryl, cyclohexyl or protected glucosamine groups, while combinations with unsubstituted benzyl groups did not gel well. This is in apparent contrast to the gelation of the (NPhe)4 hybrids reported by Wu et al. However, the multiple unsubstituted benzyls in the (NPhe)4 motif could have induced a larger hydrophobic interaction between the peptoids. Juxtaposition of (NPhe)4 with the hydrophilic peptide sequences could also have induced amphiphile self-assembly. In fact, the combination of hydrophobic and amphiphile interactions feature prominently in the self-assembly of many peptoid nanostructures, as described below.
2.2. Nano and micro-structures
Nanosheets which are two molecular layers thick and water soluble have recently been applied as an antibody-mimetic platform by Zuckermann et al. (Fig. 3).31 The researchers had earlier found that nanosheets can be assembled at the air–water interface from sequences that laterally align to form two opposing layers (Fig. 3B).32,33 Assembly required the electrostatic interaction between oppositely charged peptoid residues, and the alternation of hydrophilic and hydrophobic residues such that a hydrophobic core can be formed between the opposing layers (Fig. 3A–C). Overall sequence lengths of 12 or more residues were required to build up a sufficiently attractive interaction. Zuckermann et al. then inserted oligopeptide segments with known recognition properties between peptoid sequences possessing the periodic self-assembly motif (Fig. 3D). These sequences also assembled into nanosheets, with the oligopeptides “squeezed out” as dangling loops. The nanosheets thus acted as a structural scaffold for the presentation of biorecognition loops and mimicked the conceptual structure of antibodies (Fig. 3E).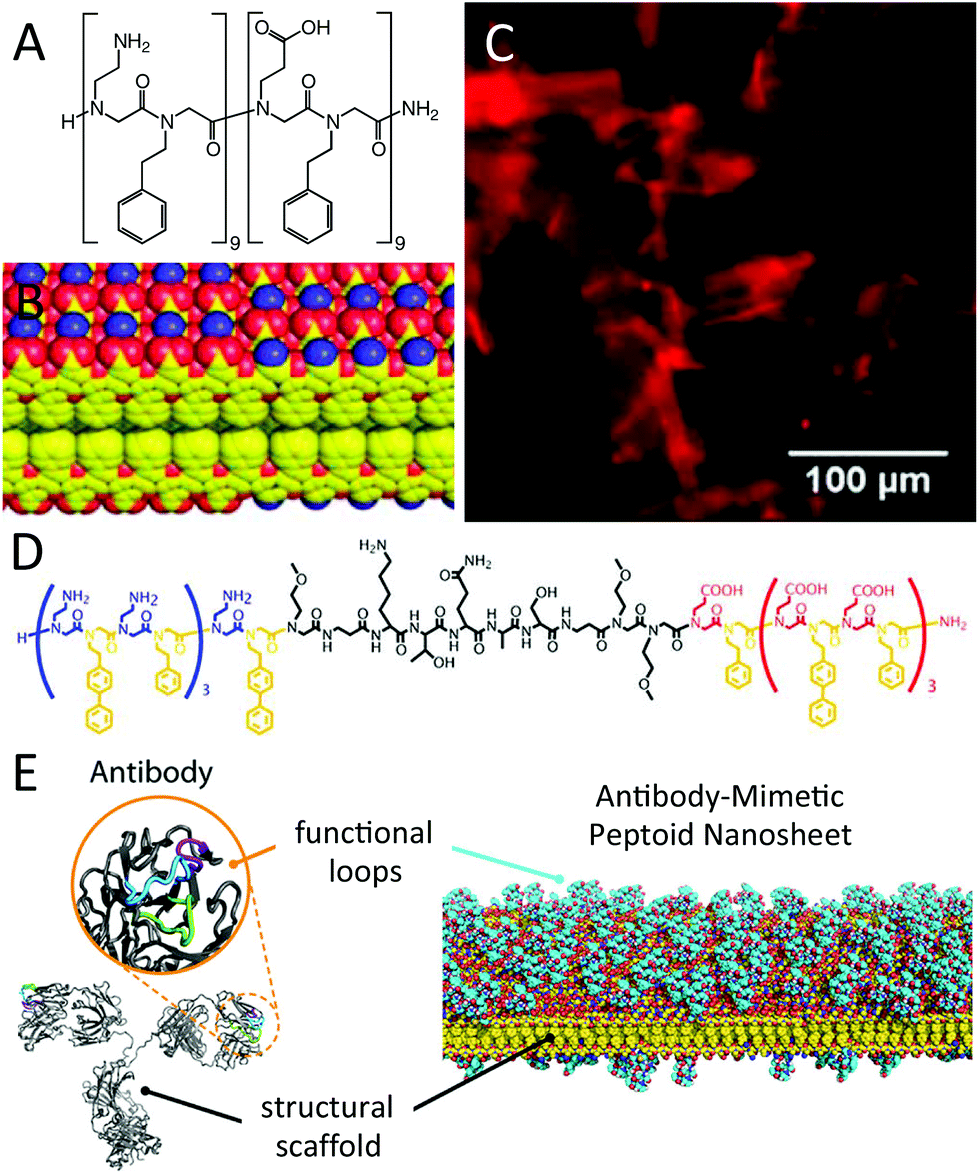 | ||
| Fig. 3 Peptoid nanosheets. (A) Chemical structure of a model sequence that can self-assemble into nanosheets, and (B) partial view of the nanosheet molecular model showing the sequestration of hydrophobic phenyl-terminated sidechains and the outward presentation of the amine- and carboxylic acid-terminated sidechains (red: oxygen; blue: nitrogen; yellow: carbon). (C) Free-floating sheets imaged by fluorescence optical microscopy, stained by Nile Red that had sequestered into the hydrophobic interior of the bilayers. (D) Chemical structure of a sequence that formed antibody-mimetic nanosheets, as illustrated in (E). Reprinted with permission from ref. 31 (Copyright 2013 American Chemical Society) and from ref. 33 (Copyright 2011 Wiley Periodicals). | ||
Molecular recognition was demonstrated by three examples: (i) the serine of a consensus peptide of casein kinase II presented on the nanosheets could be phosphorylated by the enzyme; (ii) proteases were able to digest the presented peptides without disrupting the nanosheets; and (iii) gold nanocrystals could be grown from E. coli gold-binding peptides presented on the nanosheets. The peptoid nanosheets do not aggregate in buffer, possibly due to the zwitterionic nature of the nanosheet surface (Fig. 3B). There is no critical peptoid concentration once the nanosheets have been assembled but the sheets can be designed to re-dissolve at a certain pH by tuning the sequence arrangement.33 Being 2D objects, the nanosheets possess a high surface area to mass ratio, and could provide a high degree of cargo loading using either the hydrophobic core or a functionalized surface. Together with the commercial availability of the peptoid submonomer precursors used in the self-assembly motif and the modularity of the sequence design, the nanosheets hold significant promise for a range of delivery and sensing applications.
Nano and micro-fibril self-assembly have been demonstrated for peptoids synthesized by both the submonomer solid phase and NCA polymerization routes. The Zhang group investigated34 NCA diblock copolypeptoids composed of blocks with methylene (i.e. sarcosine—hydrophilic21,35) and hydrophobic decyl sidechains. These block copolymers were found to self-assemble into spherical micelles that, over the course of several days, transitioned into cylindrical micelles with core diameters of 12 nm.34 Zuckermann et al. found solid phase synthesized amphiphilic diblock sequences that could self-assemble, first into nanosheets that then rolled (also over the course of several days) into microfibrillar helices.36 The ability to dial in specific block lengths and arrangements with the submonomer protocol was used to show that self-assembly required both hydrophobic and ionic interactions of amphiphilic sequences.
Nanoparticles have been associated with the earliest peptoid research. Sequences with repeating triplet cationic–hydrophobic–hydrophobic motifs were found to form nanoparticulate DNA–peptoid complexes that were efficient gene transfection agents.37 Similar sequences, as well as polyamphoteric ones, have recently been shown to form microspheres.38 In a different line of research, hydrophilic polysarcosine synthesized by NCA polymerization have been exploited for controlling the water solubility of amphiphilic peptoid–peptide/polymer block copolymers that self-assemble into (nano)particles for controlled release and imaging applications.39
3. Macromolecular interfaces and platforms
3.1. Antifouling peptoids
Peptoid chains grafted on solid surfaces as polymer brushes have been demonstrated to confer excellent resistance against protein adsorption and cell attachment.35,40–43 Several peptoid sidechains and chain lengths have been investigated. Intriguingly, polymer brushes composed of sarcosine, the peptoid with the simplest sidechain (a single methylene: Fig. 4), exhibited antifouling performance that is as excellent as peptoids with methoxyethyl sidechains that resemble the repeating unit of PEG.35,40,42 In both cases, protein adsorption could essentially be prevented at chain lengths of just 20 repeating units, and long term fibroblast attachment could also be prevented (Fig. 4).35,40 The resistance against bacteria attachment ranged from 75% to 99% (1 d), depending on the combination of the sidechain and the bacteria strain being tested.35,41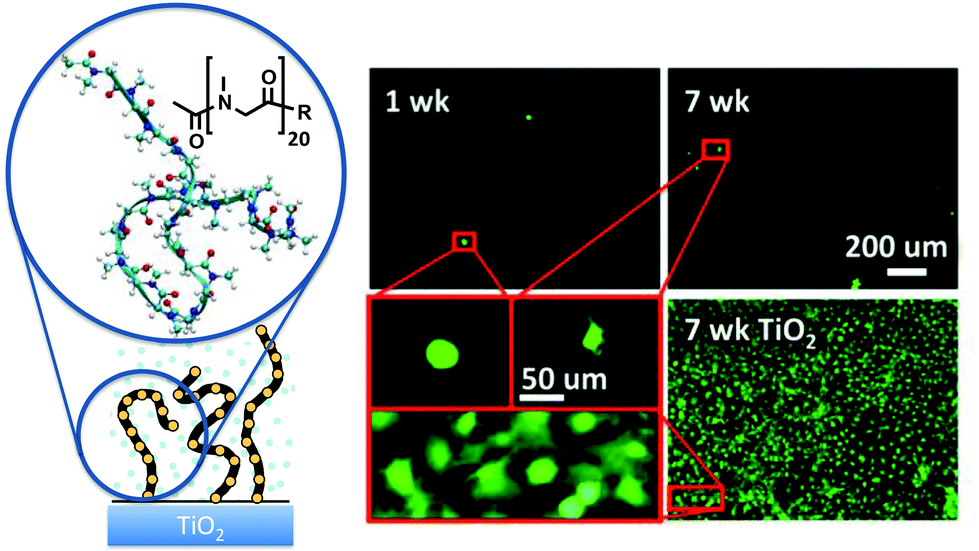 | ||
| Fig. 4 Antifouling polysarcosine surface grafted peptoid brush. The left shows the schematic, chemical structure and molecular dynamics snapshot of the polysarcosine 20-mer used in antifouling experiments. The fluorescence micrographs on the right show that the number of 3T3 mouse fibroblasts attached on the polysarcosine-coated surface was very low through 7 weeks, while a confluent layer was achieved on the uncoated TiO2 control surface during the same period (only 7 weeks data shown). Fresh fibroblasts were reseeded on the samples twice a week. Although some cells were observed on the polysarcosine surface, their morphologies were rounded, exhibiting few filopodia, and were poorly attached. Reprinted with permission from ref. 35. Copyright 2012 American Chemical Society. | ||
According to molecular theory, the chain length and grafting density of a well-solubilized, flexible polymer brush dominate its antifouling performance.42,44 As such the short persistence length and inherent flexibility of the peptoid backbone,11 conferred by its achirality and the isomerization between trans and cis conformations,14 render hydrophilic peptoids an attractive antifouling platform. Consistent with this observation, excellent antifouling performance was also shown for β-peptoids with methyl and ethyl sidechains.45 Last but not least, the flexibility in peptoid synthesis enables convenient functionalization of antifouling peptoid biointerfaces, as demonstrated by peptoid brushes decorated with saccharides46 and antimicrobial peptides.41
3.2. Clickable multivalent scaffolds
Using the submonomer protocol, Kirshenbaum et al. demonstrated that azide–alkyne cycloaddition on alkyne- or azide-terminated sidechains can be used to attach, in a sequence-specific manner, multiple and multifarious functional groups that are otherwise difficult to incorporate.47 These included estradiol, a hormone, and the electrochemically active ethynylferrocene. Polysaccharide mimics that can efficiently bind Concanavalin A have been demonstrated by clicking on mannose sidechains to a peptoid scaffold.48 The easy and precise chain length control by submonomer peptoid synthesis was exploited to determine that efficient binding to Concanavalin A required peptoid–mannose sequences of five or more residues. Similarly, control in the attachment positions of aminoglycoside sidechains clicked onto a peptoid backbone, separated by “spacer” residues with propyl sidechains, was used to tailor the specificity in the aminoglycoside targeting of RNAs that cause myotonic muscular dystrophy.49 Furthermore polyglycerated and polyglycosylated peptoids have been demonstrated by thiol–ene click addition to a NCA polymerized peptoid with allyl sidechains.50 Peptoid macrocycles with alkyne-terminated sidechains have also been synthesized by both the submonomer protocol51 and NCA polymerization.52 Depending on the sequence, the macrocycles could crystallize and adopt two interconvertible conformations and reversibly sequester water.513.3. Peptoid libraries for materials discovery
Peptoid combinatorial libraries have been fruitfully used for therapeutics and protein-binding sequence discovery (see Introduction). Recent reports highlight the utility of the approach for materials applications. The concept has been applied to the discovery of small molecule gelators from a small library (see section 2.1). Kodadek et al. have developed53 a cell-based assay for screening vascular endothelial growth factor receptor 2 (VEGFR2)-binding peptoids that could potentially be adapted for biomaterials discovery. Cells expressing VEGFR2 were fluorescently labelled and exposed to PEG-co-PS solid phase synthesis resin beads (140–170 μm in diameter), each displaying one of over 250![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 possible peptoid 9-mers generated by split–pool combinatorial synthesis from 8 different peptoid residues in 8 reaction vessels. Beads on which VEGFR2-expressing cells attached were selected for automated Edman sequencing of the “hit” peptoids for further affinity refinement. In effect, each bead acted as a test well in a multi-well plate cell attachment assay for parallel evaluation of cell behavior.
000 possible peptoid 9-mers generated by split–pool combinatorial synthesis from 8 different peptoid residues in 8 reaction vessels. Beads on which VEGFR2-expressing cells attached were selected for automated Edman sequencing of the “hit” peptoids for further affinity refinement. In effect, each bead acted as a test well in a multi-well plate cell attachment assay for parallel evaluation of cell behavior.
In the area of (bio)mineralization, library investigation of amphiphilic peptoids with specific sequences of hydrophobic and acidic sidechains discovered sequences that could accelerate calcite crystal growth by 23-fold at only 50 nM peptoid concentration (compared with enhancement factors of 1.64 or less by acidic peptides of similar molecular weights).54 In a different study, trimers of a hydroxylated peptoid residue (analog of serine) were discovered to exhibit dual-action antifreeze effects—enhanced ice growth inhibition and melting temperature reduction.55 Both studies found that the sequence of peptoid additives could be used to control the crystal morphology, and that specific sequences likely altered the interactions of water molecules with the crystallizing surfaces. These reports also illustrate that libraries of sequence-specific peptoids enable the mechanistic investigation of how variations in chemical structure alter performance.
4. Summary and outlook
The studies reviewed indicate the wide range of potential biomaterial and biointerfacial applications enabled by peptoids. Peptoid hydrogelators that are compatible with cell culture have been found. The modular design of self-assembled peptoid nanosheets with bioactive sequences has been demonstrated, opening up potential applications in drug delivery and biosensing. Peptoid nano and micro-fibrils can also be self-assembled, and peptoid nanoparticles have been explored for gene transfection and controlled release applications. Biointerfaces in the guise of antifouling peptoid polymer brushes that can be further functionalized with saccharides and antimicrobial peptides have been demonstrated. Clickable multivalent peptoid scaffolds lower the synthetic barrier to the introduction of complex sidechains and polysaccharide mimics. In addition, peptoid libraries hold significant promise for biomaterials discovery.The knowledge gained from two decades of peptoid peptidomimetics research10,13,24–26,56 could potentially be quickly translated for biomaterials applications. Design rules for dialing in secondary structure and peptoid folding by sidechain design and sequence control have emerged.10,13,15 A catalogue of bioactive peptoids has been included in a recent review.17 Peptoids have also been shown to exhibit enhanced cell penetration compared to the corresponding peptide sequences,57 but they can also be tuned, as in the case of some antimicrobial peptoids, to target bacterial membranes.58 Although the increased conformational flexibility of the peptoid backbone is expected to decrease binding affinity24 (e.g. integrin receptor binding by direct peptoid analogs of RGD has either not been reported59 or was not elicited60), certain peptoid sequences lacking secondary structure appear to possess activities superior to those of their structured counterparts.10,58,61 The resistance of peptoids against protease degradation8,9,18,31,41 is also potentially beneficial for protecting a pharmacological cargo,37 enhancing stability and bioavailability,6,7,24 and long-term biomedical applications, such as antifouling coatings.40
A continuing catalyst for peptoids research is the relative synthetic ease with which the sidechain chemistry and sequence can be precisely tuned to achieve the desired property. Stimuli-responsiveness encoded by the sequence design would undoubtedly be advantageous, and pH,33 temperature62 and solvent condition19 effects have been reported. Peptoids could be used to mediate nanoparticle assembly63 and to enhance the stability of nanoparticles in difficult ionic conditions.64 Biodegradability could potentially be reintroduced by inserting suitable peptide sequences. An interesting challenge remains in the routine synthesis of long, sequence-specific peptoids (e.g. >100 residues, as in many protein amino acid sequences). Ultimately, a bright future awaits the application of peptoids in biomaterials science as researchers further exploit the knowledge gained through peptoid therapeutics research and the highly accessible chemical versatility, self-assembly capabilities, and other intrinsic properties of peptoids.
Acknowledgements
KHAL acknowledges support from the University of Strathclyde Strategic Academic Investment Scheme (SAIS).Notes and references
- E. T. Pashuck and M. M. Stevens, Sci. Transl. Med., 2012, 4, 160sr4 CrossRef PubMed.
- R. A. Petros and J. M. DeSimone, Nat. Rev. Drug Discov., 2010, 9, 615 CrossRef CAS PubMed; O. Z. Fisher, A. Khademhosseini, R. Langer and N. A. Peppas, Acc. Chem. Res., 2010, 43, 419 CrossRef PubMed.
- D. Sengupta and S. C. Heilshorn, Tissue Eng., Part B, 2010, 16, 285 CrossRef CAS PubMed; J. P. Jung, J. Z. Gasiorowski and J. H. Collier, Biopolymers, 2010, 94, 49 CrossRef PubMed.
- S. L. Kuan, Y. Z. Wu and T. Weil, Macromol. Rapid Commun., 2013, 34, 380 CrossRef CAS PubMed; R. L. DiMarco and S. C. Heilshorn, Adv. Mater., 2012, 24, 3923 CrossRef PubMed; M. Zelzer and R. V. Ulijn, Chem. Soc. Rev., 2010, 39, 3351 RSC.
- M. Malmsten, Curr. Opin. Colloid Interface Sci., 2013, 18, 468 CrossRef CAS PubMed; O. F. Khan and M. V. Sefton, Trends Biotechnol., 2011, 29, 379 CrossRef PubMed.
- R. J. Simon, R. S. Kania, R. N. Zuckermann, V. D. Huebner, D. A. Jewell, S. Banville, S. Ng, L. Wang, S. Rosenberg, C. K. Marlowe, D. C. Spellmeyer, R. Y. Tan, A. D. Frankel, D. V. Santi, F. E. Cohen and P. A. Bartlett, Proc. Natl. Acad. Sci. U. S. A., 1992, 89, 9367 CrossRef CAS.
- R. N. Zuckermann, Biopolymers, 2011, 96, 545 CrossRef CAS PubMed.
- S. M. Miller, R. J. Simon, S. Ng, R. N. Zuckermann, J. M. Kerr and W. H. Moos, Bioorg. Med. Chem. Lett., 1994, 4, 2657 CrossRef CAS.
- S. M. Miller, R. J. Simon, S. Ng, R. N. Zuckermann, J. M. Kerr and W. H. Moos, Drug Dev. Res., 1995, 35, 20 CrossRef CAS.
- S. A. Fowler and H. E. Blackwell, Org. Biomol. Chem., 2009, 7, 1508 CAS.
- A. M. Rosales, H. K. Murnen, S. R. Kline, R. N. Zuckermann and R. A. Segalman, Soft Matter, 2012, 8, 3673 RSC.
- A. M. Rosales, R. A. Segalman and R. N. Zuckermann, Soft Matter, 2013, 9, 8400 RSC.
- J. Sun and R. N. Zuckermann, ACS Nano, 2013, 7, 4715 CrossRef CAS PubMed.
- K. Kirshenbaum, A. E. Barron, R. A. Goldsmith, P. Armand, E. K. Bradley, K. T. V. Truong, K. A. Dill, F. E. Cohen and R. N. Zuckermann, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 4303 CrossRef CAS; Q. Sui, D. Borchardt and D. L. Rabenstein, J. Am. Chem. Soc., 2007, 129, 12042 CrossRef PubMed.
- B. Yoo and K. Kirshenbaum, Curr. Opin. Chem. Biol., 2008, 12, 714 CrossRef CAS PubMed; A. M. Czyzewski and A. E. Barron, AIChE J., 2008, 54, 2 CrossRef.
- R. N. Zuckermann, J. M. Kerr, S. B. H. Kent and W. H. Moos, J. Am. Chem. Soc., 1992, 114, 10646 CrossRef CAS.
- A. S. Culf and R. J. Ouellette, Molecules, 2010, 15, 5282 CrossRef CAS PubMed.
- J. A. W. Kruijtzer, L. J. F. Hofmeyer, W. Heerma, C. Versluis and R. M. J. Liskamp, Chem.–Eur. J., 1998, 4, 1570 CrossRef CAS.
- H. K. Murnen, A. R. Khokhlov, P. G. Khalatur, R. A. Segalman and R. N. Zuckermann, Macromolecules, 2012, 45, 5229 CrossRef CAS.
- M. Schneider, C. Fetsch, I. Amin, R. Jordan and R. Luxenhofer, Langmuir, 2013, 29, 6983 CrossRef CAS PubMed; N. Gangloff, C. Fetsch and R. Luxenhofer, Macromol. Rapid Commun., 2013, 34, 997 CrossRef PubMed.
- R. Luxenhofer, C. Fetsch and A. Grossmann, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 2731 CrossRef CAS.
- D. H. Zhang, S. H. Lahasky, L. Guo, C. U. Lee and M. Lavan, Macromolecules, 2012, 45, 5833 CrossRef CAS.
- H. R. Kricheldorf, Angew. Chem., Int. Ed., 2006, 45, 5752 CrossRef CAS PubMed.
- R. M. J. Liskamp, D. T. S. Rijkers, J. A. W. Kruijtzer and J. Kemmink, ChemBioChem, 2011, 12, 1626 CrossRef CAS PubMed.
- M. T. Dohm, R. Kapoor and A. E. Barron, Curr. Pharm. Des., 2011, 17, 2732 CrossRef CAS.
- T. Kodadek, Curr. Opin. Chem. Biol., 2010, 14, 713 CrossRef CAS PubMed.
- S. Lohan and G. S. Bisht, Mini-Rev. Med. Chem., 2013, 13, 1073 CrossRef CAS.
- Y. Luo, S. Vali, S. Y. Sun, X. S. Chen, X. Liang, T. Drozhzhina, E. Popugaeva and I. Bezprozvanny, ACS Chem. Neurosci., 2013, 4, 952 CrossRef CAS PubMed; A. Y. Yam, X. Wang, C. M. Gao, M. D. Connolly, R. N. Zuckermann, T. Bleu, J. Hall, J. P. Fedynyshyn, S. Allauzen, D. Peretz and C. M. Salisbury, Biochemistry, 2011, 50, 4322 CrossRef PubMed; M. M. Reddy, R. Wilson, J. Wilson, S. Connell, A. Gocke, L. Hynan, D. German and T. Kodadek, Cell, 2011, 144, 132 CrossRef PubMed; C. M. Gao, A. Y. Yam, X. Wang, E. Magdangal, C. Salisbury, D. Peretz, R. N. Zuckermann, M. D. Connolly, O. Hansson, L. Minthon, H. Zetterberg, K. Blennow, J. P. Fedynyshyn and S. Allauzen, PLoS One, 2010, 5, e15725 Search PubMed; R. C. Elgersma, G. E. Mulder, J. A. W. Kruijtzer, G. Posthuma, D. T. S. Rijkers and R. M. J. Liskamp, Bioorg. Med. Chem. Lett., 2007, 17, 1837 CrossRef PubMed.
- Z. D. Wu, M. Tan, X. M. Chen, Z. M. Yang and L. Wang, Nanoscale, 2012, 4, 3644 RSC.
- H. P. R. Mangunuru, H. Yang and G. J. Wang, Chem. Commun., 2013, 49, 4489 RSC.
- G. K. Olivier, A. Cho, B. Sanii, M. D. Connolly, H. Tran and R. N. Zuckermann, ACS Nano, 2013, 7, 9276 CrossRef CAS PubMed.
- K. T. Nam, S. A. Shelby, P. H. Choi, A. B. Marciel, R. Chen, L. Tan, T. K. Chu, R. A. Mesch, B. C. Lee, M. D. Connolly, C. Kisielowski and R. N. Zuckermann, Nat. Mater., 2010, 9, 454 CrossRef CAS PubMed; B. Sanii, R. Kudirka, A. Cho, N. Venkateswaran, G. K. Olivier, A. M. Olson, H. Tran, R. M. Harada, L. Tan and R. N. Zuckermann, J. Am. Chem. Soc., 2011, 133, 20808 CrossRef PubMed.
- R. Kudirka, H. Tran, B. Sanii, K. T. Nam, P. H. Choi, N. Venkateswaran, R. Chen, S. Whitelam and R. N. Zuckermann, Biopolymers, 2011, 96, 586 CrossRef CAS.
- C. U. Lee, T. P. Smart, L. Guo, T. H. Epps and D. H. Zhang, Macromolecules, 2011, 44, 9574 CrossRef CAS PubMed.
- K. H. A. Lau, C. Ren, T. S. Sileika, S. H. Park, I. Szleifer and P. B. Messersmith, Langmuir, 2012, 28, 16099 CrossRef CAS PubMed.
- H. K. Murnen, A. M. Rosales, J. N. Jaworski, R. A. Segalman and R. N. Zuckermann, J. Am. Chem. Soc., 2010, 132, 16112 CrossRef CAS PubMed.
- J. E. Murphy, T. Uno, J. D. Hamer, F. E. Cohen, V. Dwarki and R. N. Zuckermann, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 1517 CrossRef CAS.
- M. L. Hebert, D. S. Shah, P. Blake, J. P. Turner and S. L. Servoss, Org. Biomol. Chem., 2013, 11, 4459 CAS.
- S. Kimura, T. Kidchob and Y. Imanishi, Polym. Adv. Technol., 2001, 12, 85 CrossRef CAS; A. Makino, E. Hara, I. Hara, R. Yamahara, K. Kurihara, E. Ozeki, F. Yamamoto and S. Kimura, J. Controlled Release, 2012, 161, 821 CrossRef PubMed.
- A. R. Statz, R. J. Meagher, A. E. Barron and P. B. Messersmith, J. Am. Chem. Soc., 2005, 127, 7972 CrossRef CAS PubMed.
- A. R. Statz, J. P. Park, N. P. Chongsiriwatana, A. E. Barron and P. B. Messersmith, Biofouling, 2008, 24, 439 CrossRef CAS PubMed.
- K. H. A. Lau, C. Ren, S. H. Park, I. Szleifer and P. B. Messersmith, Langmuir, 2012, 28, 2288 CrossRef CAS PubMed.
- A. R. Statz, J. H. Kuang, C. L. Ren, A. E. Barron, I. Szleifer and P. B. Messersmith, Biointerphases, 2009, 4, FA22 CrossRef CAS PubMed.
- J. Satulovsky, M. A. Carignano and I. Szleifer, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 9037 CrossRef CAS PubMed.
- S. H. Lin, B. Zhang, M. J. Skoumal, B. Ramunno, X. P. Li, C. Wesdemiotis, L. Y. Liu and L. Jia, Biomacromolecules, 2011, 12, 2573 CrossRef CAS PubMed.
- H. O. Ham, S. H. Park, J. W. Kurutz, I. G. Szleifer and P. B. Messersmith, J. Am. Chem. Soc., 2013, 135, 13015 CrossRef CAS PubMed.
- J. M. Holub, H. J. Jang and K. Kirshenbaum, Org. Biomol. Chem., 2006, 4, 1497 CAS.
- H. Yuasa, H. Honma, H. Hashimoto, M. Tsunooka and K. Kojima-Aikawa, Bioorg. Med. Chem. Lett., 2007, 17, 5274 CrossRef CAS PubMed.
- M. M. Lee, J. L. Childs-Disney, A. Pushechnikov, J. M. French, K. Sobczak, C. A. Thornton and M. D. Disney, J. Am. Chem. Soc., 2009, 131, 17464 CrossRef CAS PubMed; M. D. Disney, M. M. Lee, A. Pushechnikov and J. L. Childs-Disney, ChemBioChem, 2010, 11, 375 CrossRef.
- J. W. Robinson and H. Schlaad, Chem. Commun., 2012, 48, 7835 RSC.
- S. B. L. Vollrath, C. H. Hu, S. Brase and K. Kirshenbaum, Chem. Commun., 2013, 49, 2317 RSC.
- S. H. Lahasky, W. K. Serem, L. Guo, J. C. Garno and D. H. Zhang, Macromolecules, 2011, 44, 9063 CrossRef CAS.
- D. G. Udugamasooriya, S. P. Dineen, R. A. Brekken and T. Kodadek, J. Am. Chem. Soc., 2008, 130, 5744 CrossRef CAS PubMed.
- C.-L. Chen, J. Qi, R. N. Zuckermann and J. J. DeYoreo, J. Am. Chem. Soc., 2011, 133, 5214 CrossRef CAS PubMed.
- M. L. Huang, D. Ehre, Q. Jiang, C. H. Hu, K. Kirshenbaum and M. D. Ward, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 19922 CrossRef CAS PubMed.
- C. A. Olsen, ChemBioChem, 2010, 11, 152 CrossRef CAS PubMed.
- Y.-U. Kwon and T. Kodadek, J. Am. Chem. Soc., 2007, 129, 1508 CrossRef CAS PubMed; N. C. Tan, P. Yu, Y. U. Kwon and T. Kodadek, Bioorg. Med. Chem., 2008, 16, 5853 CrossRef PubMed.
- N. P. Chongsiriwatana, J. A. Patch, A. M. Czyzewski, M. T. Dohm, A. Ivankin, D. Gidalevitz, R. N. Zuckermann and A. E. Barron, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 2794 CrossRef CAS PubMed.
- O. E. Vercillo, C. K. Z. Andrade and L. A. Wessjohann, Org. Lett., 2008, 10, 205 CrossRef CAS PubMed.
- I. Dijkgraaf, J. A. W. Kruijtzer, C. Frielink, A. C. Soede, H. W. Hilbers, W. J. G. Oyen, F. H. M. Corstens, R. M. J. Liskamp and O. C. Boerman, Nucl. Med. Biol., 2006, 33, 953 CrossRef CAS PubMed.
- T. Hara, S. R. Durell, M. C. Myers and D. H. Appella, J. Am. Chem. Soc., 2006, 128, 1995 CrossRef CAS PubMed.
- S. H. Lahasky, X. K. Hu and D. H. Zhang, ACS Macro Lett., 2012, 1, 580 CrossRef CAS.
- G. Maayan and L. K. Liu, Biopolymers, 2011, 96, 679 CrossRef CAS PubMed.
- D. B. Robinson, G. M. Buffleben, M. E. Langham and R. N. Zuckermann, Biopolymers, 2011, 96, 669 CrossRef CAS.
- P. S. Farmer and E. J. Ariëns, Trends Pharmacol. Sci., 1982, 3, 362 CrossRef CAS.
Footnote |
| † Although the term “peptoids” was originally conceived as any non-peptidic chemical analog that mimics the biological action of peptides,65 and has been used to describe a number of different peptidomimetic polyamides, most reports of peptoids, e.g. those indexed in PubMed, refer to N-substituted glycines. |
| This journal is © The Royal Society of Chemistry 2014 |