Silver nanoparticle loaded PLGA composite nanoparticles for improving therapeutic efficacy of recombinant IFNγ by targeting the cell surface†
Nidhi
Chaubey
a,
Amaresh Kumar
Sahoo
b,
Arun
Chattopadhyay
bc and
Siddhartha Sankar
Ghosh
*ab
aDepartment of Biotechnology, Indian Institute of Technology Guwahati, Guwahati, Assam 781039, India. E-mail: sghosh@iitg.ernet.in
bCentre for Nanotechnology, Indian Institute of Technology Guwahati, Guwahati, Assam 781039, India
cDepartment of Chemistry, Indian Institute of Technology Guwahati, Guwahati, Assam 781039, India
First published on 11th March 2014
Abstract
The field of medical science has advanced significantly with the discoveries of new drugs and the development of sophisticated biomedical tools; still cancer therapy remains one of the major hurdles currently. Herein, we report a new approach, which exhibits complementary anti-cancer effects of recombinant IFNγ protein and silver nanoparticles (Ag NPs) when loaded together in PLGA composite NPs (GST IFNγ–Ag PLGA NPs). IFNγ acts as an antiviral and tumoricidal agent. To augment therapeutic efficacy, IFNγ was cloned, purified as GST tagged IFNγ recombinant protein, and immobilized on the composite NPs preloaded with Ag NPs. The NPs were characterized using UV-vis spectroscopy, transmission electron microscopy (TEM), field emission scanning electron microscopy (FESEM) and dynamic light scattering (DLS) analysis. Finally, the composite NPs were delivered into two different human cancer cell types, HeLa (cervical cancer) and MCF-7 (breast cancer) cells. Our results demonstrated that the recombinant IFNγ could block the cell cycle at the G1 phase and its anticancer activity could be potentiated in the presence of Ag NPs. The interaction between the recombinant IFNγ with its cell surface receptors facilitated the delivery of the composite NPs, and thus the combination of the duos ultimately led to induction of apoptosis in the cancer cells.
Introduction
In the last few decades, an ample growth has occurred in the field of cancer therapy, due to the advent of new molecular entities and the unearthing of crucial cell signalling pathways, which provide new opportunities in both therapeutic and basic research frontiers. However, suitable drug molecule(s) or novel therapeutic tactics that aim to replace conventional chemo/radio therapy have not been translated to the therapeutic level, at least not in clinical practice so far. The preferential delivery of therapeutic agents at tumour sites while avoiding their undesired accumulation at other sites i.e. “spatial drug delivery” is a matter of pivotal importance to curb the amount and subsequent side effects of drugs is now under consideration.1–3 In this scenario, nanoparticle mediated drug delivery offers promising opportunities, as it exhibits high efficacy due to the favourable size and tunable physicochemical properties. Moreover, conjugation of specific ligand(s) on the surface of the NPs (nanoparticles) provides a significant level of selectivity by active targeting.4–7 However, the major difficulty faced by any chemotherapy drug to access a tumour site is the intricate cellular organization of the surrounding cells. In such cases, NP mediated drug delivery increases blood circulation half-life and bioavailability of the drugs within a therapeutic frame and consequently, avoids immediate dose and frequency escalation thus mediating a controlled release of drug.8,9Furthermore, it would be beneficial to apply recombinant therapeutic agents of human origin, which may reduce the likelihood of host antagonistic responses. In this context, protein therapeutics has gained an edge over other options as their rate of industrial translation is high. For example, recombinant insulin isolated from E. coli has replaced the whole market from animal source,10 and the recombinant IFNα-2a is now used in cancer therapy and the treatment of autoimmune diseases.11 Another such molecule is IFNγ, a naturally occurring cytokine which is produced and secreted by immune system cells e.g., white blood cells and natural killer cells. IFNγ interacts with its cell surface receptors and induces signal transduction through the JAK-STAT pathway,12 which subsequently elicits multiple responses, such as, the prevention of viral replication, cell growth inhibition and cell differentiation.13–16 IFNγ is a FDA approved cytokine,17 which is involved in the activation of T cells and macrophages, exerting tumoricidal and antimicrobial activity, having a huge impact on host defence systems.18 Moreover, recent studies portray the important role of IFNγ in cancer immune surveillance.19–21
However, therapeutic applications of IFNγ are limited due to its shorter life time and rapid clearance from the body, and furthermore, higher doses and repetitive use cause severe side effects.8,22 Therefore, prolonging the sustainability of IFNγ in blood would be a viable option for exploiting maximum therapeutic efficacy by its possible integration with nanoparticles. Recent studies from several laboratories including ours, demonstrated that Ag NPs have great potential as antibacterial as well as anti-cell proliferative agents. Essentially, Ag NPs lead to apoptosis in mammalian cells.23–25
Keeping in this view, we have cloned, expressed and purified the GST fused IFNγ protein. It is to be mentioned here that the GST tag minimises the steps of protein purification, provides stability and facilitates the single step purification of the recombinant protein.26,27
Recombinant IFNγ protein was expressed in inclusion bodies,28 which was further purified and delivered to cancer cells by freshly synthesized Ag NP coated poly(lactic-co-glycolic acid) (PLGA) composite NPs. PLGA is a biocompatible and biodegradable polymer like poly(ester amide)s, comprising monomers of lactic and glycolic acids.29–33 We explored the complementary effects of the composite NPs, where the recombinant IFNγ was shown to block the cell cycle and the Ag NPs had induced apoptosis by cell surface receptor mediated targeting with the recombinant IFNγ as ligand. This combination therapy is a novel approach, where the additive effects of the recombinant IFNγ and Ag NPs at low concentrations may provide a new platform to enhance the therapeutic efficacy of IFNγ stabilized composite nanoparticles. Scheme 1 provides a brief overview of the current work encompassing cloning, purification, functional aspects of recombinant protein and delivery with Ag NPs for therapeutic application.
 | ||
| Scheme 1 Schematic representation of the development and functional aspects of composite nanoparticles (GST IFNγ–Ag PLGA NPs). | ||
Materials and methods
Cloning, expression, purification and activity assay of recombinant IFNγ
Cell culture
Cell lines were obtained from NCCS Pune, India. The cells were periodically subcultured in DMEM medium supplemented with 10% FBS, 20 U mL−1 penicillin and 50 mg mL−1 streptomycin in 5% CO2 humidity at 37 °C.Cloning of human IFNγ
The IFNγ gene was cloned from human blood cDNA. Gene specific forward primer (5′ATGTGGCTGCAGAGCCTGCTG3′) and reverse primer (5′TCACTCCTGGACTGGCTCCCA3′) were designed for PCR and the cloning of IFNγ in pGEMT Easy Vector. Afterwards the IFNγ gene was subcloned into N terminal GST tag pGEX4T2 Expression Vector (Amersham).Expression of IFNγ in E. coli Rosetta gami strain
The pGEX4T2 IFNγ construct was transformed into E. coli Rosetta gami expression host. A starter mini culture was developed by inoculating a single colony in LB medium supplemented with 100 μg mL−1 ampicillin for 14 h at 37 °C. This culture was further used as inoculum (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :250 dilution) for scale up in 500 mL LB media with ampicillin. At 0.4–0.6 OD590, IPTG was added to the growing culture for expression of Lac operon regulated recombinant protein and allowed to grow for another 12 h for induction of recombinant GST tagged IFNγ protein. Finally the grown culture was centrifuged at 12000 g for 15 minutes to harvest the bacterial pellet and stored at −20 °C for further application.
:250 dilution) for scale up in 500 mL LB media with ampicillin. At 0.4–0.6 OD590, IPTG was added to the growing culture for expression of Lac operon regulated recombinant protein and allowed to grow for another 12 h for induction of recombinant GST tagged IFNγ protein. Finally the grown culture was centrifuged at 12000 g for 15 minutes to harvest the bacterial pellet and stored at −20 °C for further application.
Solubilisation, refolding and purification of GST IFNγ from inclusion bodies
GST IFNγ overexpressing bacterial cells were suspended in ice chilled lysis buffer (50 mM Tris pH 8.0 with 1% of sarkosyl, 1 mM PMSF, 1 mM EDTA) and sonicated at 20 kpsi for 10 minutes for cell lysis.34,35 The presence of GST IFNγ protein was confirmed by 12% SDS-PAGE analysis of the cell lysate. After cell lysis, 20 mM CHAPS and 2% Triton ×100 was added in the lysed cell solution and kept continuously stirring for 30 minutes to form micelles of sarkosyl. This step opens up the covering of sarkosyl from GST IFNγ and makes the protein free. CHAPS and Triton ×100 are neutral detergent molecules, which help in the micelle formation of sarkosyl itself and thus the solubilised protein becomes free with its cationic surroundings made by sarkosyl. This solution was separated from the debris through centrifugation followed by dialysis in PBS (pH 7.4). This protein mixture was passed through a glutathione agarose 4B matrix (Sigma). The recombinant GST IFNγ protein was eluted with 10 mM reduced glutathione and checked by silver staining in 12% SDS-PAGE.Western blot analysis
Purified GST IFNγ was confirmed by anti-GST Western blot. Mouse anti-GST (Sigma) and mouse anti-human IFNγ antibodies were used as primary antibodies (BD Pharmingen, USA). Horse radish peroxidase conjugated goat anti-mouse polyclonal IgG was used as secondary antibody (BD Pharmingen, USA).MALDI analysis
Purified GST IFNγ protein was characterized with MALDI-TOF (4800 Plus Applied Biosystem) before loading on nanoparticles. Recombinant protein was eluted by in gel trypsin digestion (Sigma Kit) for 12 h and spotted on α-cyano-4-hydroxycinnamic acid matrix of MALDI plate. The samples were analyzed by MALDI-TOF.36,37Measurement of reactive oxygen species (ROS) generated by IFNγ on RAW 264.7 cells
To determine the activity of GST IFNγ purified protein in ROS generation, we added 5 μM dichloro fluorescein (DCFDA, Sigma) on RAW 264.7 mouse macrophages after 24 h of treatment with recombinant GST IFNγ protein. The samples were analysed by flow cytometry (FacsCalibur, BD Biosciences, NJ, USA) at an excitation wavelength of 488 nm and emission wavelength 530 nm. The fluorescence data were recorded with the CellQuest program (BD Biosciences) for 15000 cells in each sample.
Synthesis of PLGA and composite (Ag PLGA NPs) NPs
The PLGA NPs were synthesized by using the emulsification solvent evaporation method.29–31 In brief, 10 mg of PLGA was dissolved in 1.5 mL of chloroform. Then, the PLGA solution was added dropwise into 2 mL of 0.5% PVA (5 mg mL−1) solution under continuous sonication at 40 kpsi for 10 minutes. During sonication the temperature of the reaction was maintained at 90 °C which facilitated the removal of organic solvent. The NPs were collected by centrifugation at 6000 rpm for 5 minutes. The collected NPs were extensively washed with water for the complete removal of organic solvent, which is toxic to mammalian cells, and finally dissolved in deionized water. For the preparation of fluorescent NPs, 10 μL of fluorescein (1 mg mL−1) was added into the PVA solution before the addition of PLGA. For the synthesis of the composite NPs of Ag NPs and PLGA NPs, firstly, Ag NPs were synthesised with the reaction of 200 μL of 10 mM Ag NO3 and 1 mL of sodium borohydride (10 mM) in 2 mL of 0.5% PVA (5 mg mL−1) where PVA acted as the stabilizing agent of the Ag NPs. Then, the preformed PVA stabilized Ag NPs were used as emulsifying agent for the synthesis of the composite NPs (Ag PLGA NPs) by maintaining the same reaction conditions as described above.Hydrodynamic diameter and zeta potential measurement
Hydrodynamic diameter and zeta potential of PLGA and composite NPs (Ag PLGA NPs) were analysed by Malvern Zeta Sizer Nano ZS in PBS buffer. The same experiment was also performed after incubation with GST IFNγ protein.Surface morphology study
The surface morphology of the NPs was studied with field emission scanning electron microscopy (FESEM) and transmission electron microscopy (TEM). For FESEM analysis, 20 μL of the sample was drop casted on a glass slide which was wrapped with aluminium foil and sputter-coated with gold film using a sputter coater (SC7620 “Mini”, Polaron Sputter Coater, Quorum Technologies, New Haven, England) and analysed by FESEM. For the TEM analysis, 7 μL of the sample was added on the carbon coated copper grid, dried and analysed under TEM (TEM; JEM 2100; Jeol, Peabody, MA, USA) which was operating at an accelerating voltage of 200 keV.Determination of IFNγ protein binding efficiency with NPs
Binding of GST IFNγ protein with the PLGA and composite NPs was confirmed by fluorescence spectroscopy study. For that, different amounts of protein (100–300 ng) were incubated with a fixed amount of NPs (1 μg) for 1 h at 37 °C, followed by centrifugation (6000 rpm, 5 minutes) to remove the unbound protein. The fluorescence intensity of the fluorescein isothiocyanate (FITC) at 520 nm (when excited at 480 nm) for both the NPs such as PLGA and composite NPs were gradually decreased with increasing concentration of the protein. The protein binding was also validated by UV-vis spectroscopy study. For that, both the NPs i.e. PLGA and composite NPs (20 μg) were incubated with different amounts of the recombinant IFNγ (500–3000 ng) for 1 h at 37 °C, followed by centrifugation at 6000 rpm for 2 minutes. The NPs were collected by discarding the supernatant and measuring the absorbance at 280 nm to find out the amount of protein immobilized on the surface of the NPs.Protein release assay
To study the protein release, 5 μg of GST-IFNγ was incubated with 200 μg of Ag PLGA NPs at 37 °C for 72 h in PBS (pH 7.4). The samples were taken out periodically and centrifuged to pellet down the nanoparticles. The supernatant was collected and the absorbance was measured at 280 nm by UV-vis spectroscopy, which corresponds to the amount of released protein.Fourier transform infrared (FTIR) spectroscopy analysis
To perform FTIR analysis, the samples were first lyophilized to remove water. Then the dried samples were mixed with KBr to make the pellets and analysed by Perkin-Elmer spectra in the range 4000–400 cm−1.Fluorescence microscopy study
To perform the microscopy imaging, cells were grown in a 6-well plate (1 × 105 cells per well) for 12 h. The cells were treated with the PLGA NPs, composite NPs (Ag PLGA NPs) and GST IFNγ protein immobilized NPs for 3 h. After that, the cells were thoroughly washed with PBS to remove the residual NPs and observed by epifluorescence microscopy (20×, Nikon ECLIPSE, TS100, Tokyo, Japan). As FITC was incorporated with the NPs, treated cells showed green emission with excitation with blue light as compared to the control cells.Protease protection assay
For protease protection assay, 500 ng of free GST IFNγ as well as the GST IFNγ loaded Ag PLGA NPs (20 μg) were incubated with protease (Qiagen) at 37 °C for 30 minutes. After that, samples were analyzed using 12% SDS-PAGE analysis.Acridine orange/ethidium bromide (AO/EtBr) double staining
For the double staining, 5 × 103 HeLa cells and 3 × 103 MCF-7 cells were seeded in 96 well plates and grown overnight. Cells were treated with PLGA, composite NPs (GST IFNγ–Ag PLGA NPs), GST IFNγ protein and protein immobilized NPs for 24 h. Then a mixture of acridine orange (20 μg mL−1) and ethidium bromide (20 μg mL−1) was applied to the cells for 5 minutes. The cells were washed with PBS to remove the free dye and observed using epifluorescence microscopy at 20× (Nikon ECLIPSE, TS100, Tokyo, Japan).Cell viability assay
The cell viability after treatment with the NPs and the protein immobilized NPs were performed by MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) assay. 5 × 103 HeLa cells and 3 × 103 MCF-7 cells were seeded in 96 well plates and grown overnight. Then, varying amounts of PLGA, composite NPs (Ag PLGA NPs), GST protein, GST IFNγ protein and protein immobilized Ag PLGA NPs were added separately to the cells. MTT assay was performed after 48 h. The colourless MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) was converted into blue colour formazan by respiring mitochondria of the viable cells. Water insoluble formazan was solubilized by adding DMSO, and the colour product measured (absorbance at 550 nm) was directly correlated with the number of viable cells.Cell cycle analysis
Cell cycle analysis was performed by FACS, for that cells were grown in a 6 well plate (1 × 105 cells per well) for 12 h followed by the treatment with the GST IFNγ protein, composite NPs (Ag PLGA NPs) and GST IFNγ protein immobilized NPs for 48 h. The cells were trypsinized and collected by centrifugation at 650 rcf, 6 minutes at 4 °C. The cells were then fixed with 70% ethanol at −20 °C for 1 h. The 100 μg mL−1 RNase was added into the fixed cells and incubated for 30 minutes at 37 °C. Next, 40 μg mL−1 propidium iodide (PI) was added with 0.01% Triton X-100 into the cells and samples were analysed by Florescence Activated Cells Sorter (FacsCalibur, BD Biosciences, NJ, USA). The fluorescence data were recorded with the Cell Quest program (BD Biosciences) for 10000 cells in each sample.
Caspase 3 assay
HeLa cells were seeded (1 × 105 cells per well) and treated at IC50 of the Ag PLGA NPs in serum media, (1.36 μg mL−1), for 18 h. Afterwards, cells were trypsinized and fixed with 0.1% of formaldehyde in PBS for 15 min at room temperature (25 °C). Further, cells were pelleted down by centrifugation at 300 g and incubated with 1 mL of membrane permealization solution (0.5% of Tween 20 in PBS) for another 15 min in the dark followed by washing with double volume of PBS. Cells were treated with PE conjugated Rabbit Anti-Active-Caspase-3 (Catalog no. 550821, BD Biosciences) for 30 min in the dark. Finally, the samples were analysed in FACS.Results and discussion
Cloning, expression and characterization of human IFNγ
A 501 bp IFNγ gene was PCR amplified from human blood cDNA and initially cloned into the pGEMT Easy Cloning Vector. The clone was confirmed by Eco RI restriction digestion (Fig. S1†) and further subcloned into pGEX4T2 Bacterial Expression Vector to generate recombinant pGEX4T2-IFNγ plasmid (Fig. 1A). The clone was further confirmed by restriction digestion with Xho I and Bam HI enzymes followed by sequencing. The construct was transformed into E. coli Rosetta gami and the recombinant IFNγ expression was induced with IPTG (Fig. 1B). However, the recombinant protein mainly appeared in inclusion bodies (Lane 5, Fig. 1B), which was solubilised using 1% sarkosyl, 20 mM CHAPS and 2% Triton X-100. The recombinant IFNγ was purified to a single band of 44 kDa using glutathione agarose affinity chromatography (Lane 2, Fig. 1C).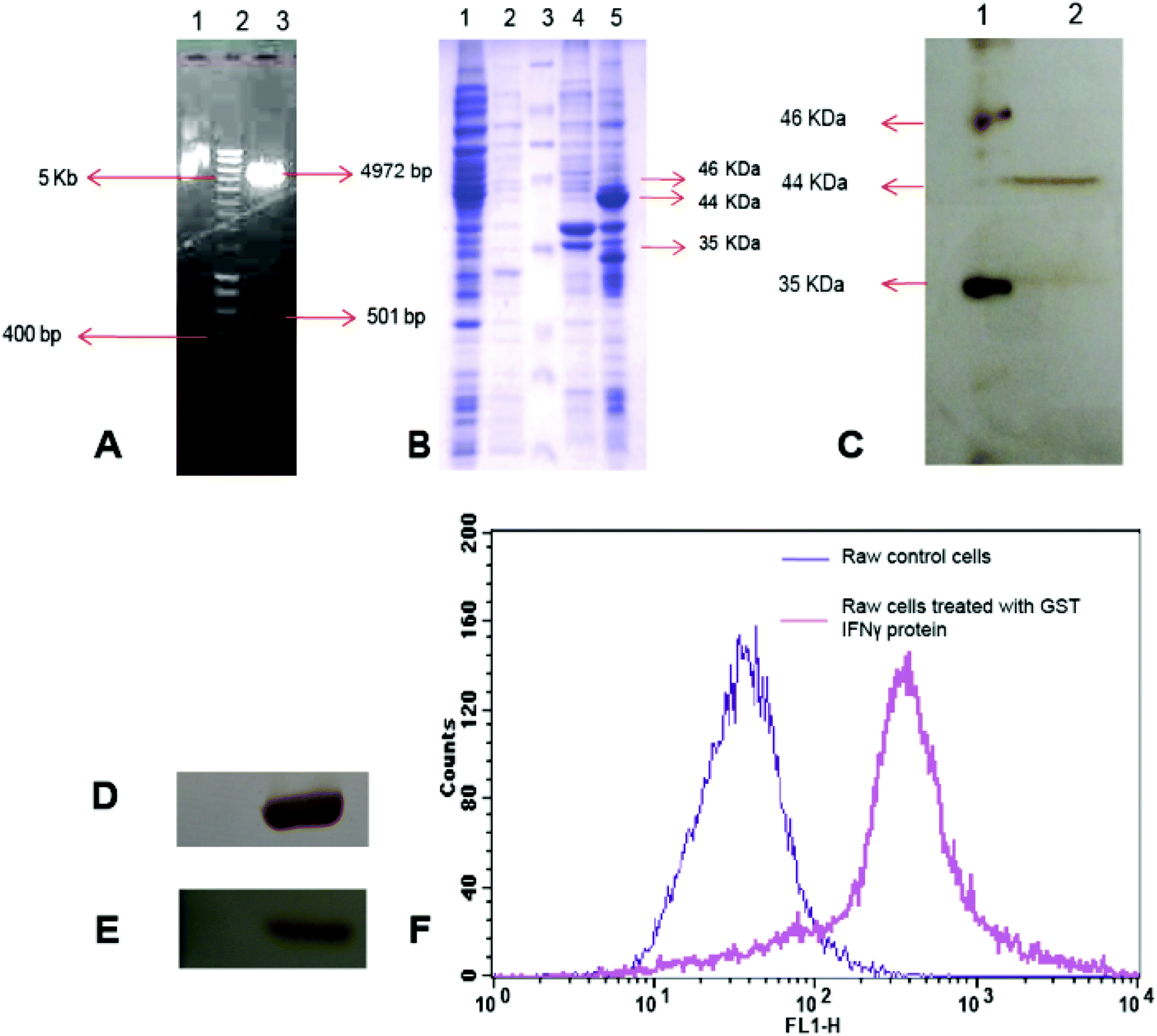 | ||
| Fig. 1 (A) Agarose gel electrophoresis of pGEX4T2-IFNγ plasmid, lane 1: undigested plasmid, lane 2: hyper ladder (NEB), lane 3: pGEX4T2-IFNγ plasmid digested with Bam HI and Xho I showing 4972 bp vector backbone 501 bp IFNγ insert (B) SDS-PAGE of E. coli Rosseta gami cell lysates. Lane 1: supernatant of pGEX4T2 transformed IPTG induced cell lysates, lane 2: cell pellet of pGEX4T2 backbone transformed IPTG induced cells, lane 3: protein marker, lane 4: supernatant of pGEX4T2-IFNγ transformed cell lysate after IPTG induction, lane 5: inclusion bodies of pGEX4T2-IFNγ transformed cells after IPTG induction (C) SDS-PAGE of purified GST IFNγ using GST glutathione affinity chromatography, lane 1: protein marker and lane 2: purified GST IFNγ protein, (D, E) are Western blots with anti GST and anti IFNγ antibodies, respectively (F) FACS analysis of recombinant IFNγ mediated reactive oxygen species (ROS) generation in macrophage (RAW 264.7) cells. The FL1-H corresponds to the green emission of the DCF. | ||
Western blotting also showed a single band of 44 kDa for GST tagged IFNγ either with anti-GST antibody (Fig. 1D) or with anti-IFNγ antibody (Fig. 1E), separately. Furthermore, MALDI-TOF analysis of trypsin digested recombinant IFNγ protein depicted significant similarity with human IFNγ protein by Mascot search in NCBI BLAST showing 39% sequence coverage with a score value of 89 (Fig. S2 and S3†).
Activity study of the recombinant IFNγ
Macrophage cells are known to produce a high amount of reactive oxygen species (ROS) in response to IFNγ.38 Thus, measurement of ROS in macrophage cells, after treatment with IFNγ is a well-established bioassay for functional activity of IFNγ.39We have treated mouse macrophage cells (RAW 264.7) with purified recombinant GST IFNγ protein for 24 h, and measured ROS by FACS analysis,40 using 2,7-dichlorofluoresceindiacetate (DCFDA) staining method. The non-fluorescent DCFDA freely diffuses into the cells through the plasma membrane, and converts to green fluorescent DCF due to intracellular oxidation by ROS. Thus, the amount of fluorescence intensity of DCF produced is directly correlated with the amount of ROS production inside the treated cells.
The FACS results demonstrated that cells treated with recombinant IFNγ protein generated ROS, which was evident from a prominent shift of fluorescence intensity in the FL-1H channel corresponds to the green emission of DCF compared to the untreated cells (Fig. 1F).
This result revealed that recombinant IFNγ purified obtained from a bacterial expression host could retain functional activity. The anti-cell proliferative activity of the recombinant IFNγ was further tested on MCF-7 and HeLa cells using a MTT assay. The results showed a maximum of 20–25% cell death in a wide concentration range (2.5–3000 ng mL−1) of the recombinant protein (Fig. S4†). The MTT assay for activity of GST and PLGA was also examined, which showed no significant effect of GST and PLGA on cell viability (Fig. S4†). Cell cycle analysis using flow cytometry revealed cell cycle arrest at the G1 phase after treatment with 25 ng mL−1 recombinant IFNγ, whereas the other phases were unaffected (Fig. S5†). The low cell death could be due to the short half-life of IFNγ and there was also no signature of apoptosis after treatment. Thus, it was clear from our results that although the recombinant IFNγ could block the cell cycle, it was not able to kill cancer cells effectively. Therefore, we further immobilized the recombinant IFNγ on the surface of Ag NPs coated PLGA NPs. This was being performed with the hope that the attachment of the recombinant IFNγ with polymer nanoparticles (NPs) would not only impart stability, but also increase its anti-cancer effect due to Ag NPs. However, the Ag NPs, a known anti-proliferative agent is required at high concentration to kill cancer cells. Moreover, the composite NPs could be targeted easily on the cells with the help of the interaction between IFNγ and its cell surface receptors. Hence, this combination therapy module could potentiate the anti-proliferative effect of the recombinant IFNγ in the presence of a low amount of Ag NPs.
Nanoparticle synthesis, characterization and binding confirmation with GST IFNγ protein
The composite nanoparticles (Ag PLGA NPs) were synthesised in two steps; first Ag NPs were synthesised by reaction of sodium borohydride and Ag NO3 in the presence of PVA, which acted as a stabilizing agent for Ag NPs. The formation of Ag NPs was confirmed by its characteristic surface plasmon peak at 415 nm in UV-Vis spectroscopy (Fig. S6†). The Ag NPs–PVA composite was used to stabilize the PLGA NPs by using emulsification solvent evaporation method. Usually, PVA, a biodegradable polymer, is used as an emulsifying agent during the synthesis of PLGA NPs. However, in the present case, the preformed Ag NPs–PVA composite was used for the same. Thus, we were able to deposit Ag NPs on the surface of the PLGA NPs. In addition, we have incorporated FITC during synthesis of the NPs to make these NPs fluorescent. The PLGA NPs were also synthesized without Ag NPs to perform the control experiments. The formation of NPs was confirmed by TEM analysis (Fig. 2). The TEM images of PLGA and the composite NPs (Ag PLGA NPs) showed that both NPs were spherical in shape and average particle sizes were 266.40 ± 29.08 nm and 259.28 ± 34.31 nm, respectively (Fig. 2A and C). Interestingly, the Ag NPs were clearly visible to be embedded in the PLGA NPs in the case of the composite NPs. The average Ag NP size was found to be 7.04 ± 3.2 nm. The selected area electron diffraction (SAED) analysis of the same sample showed corresponding Scherrer ring patterns, confirming the presence of the metallic silver (Fig. 2C). The FESEM analysis of PLGA and composite NPs showed average particle sizes around 238.23 ± 60.1 nm (Fig. 2B) and 246.45 ± 48.57, respectively (Fig. S7†), which are in close compliance with the TEM results. In addition, we performed DLS analysis to determine the hydrodynamic diameter and the zeta potential of the composite NPs. The hydrodynamic diameter was 356 nm, and the zeta potential value was −26 mV (Fig. S8†). | ||
| Fig. 2 (A) and (B) are the TEM and FESEM images of PLGA NPs, respectively (C) TEM image with inset SAED of the Ag NPs (D) Magnified TEM image of a single composite NP (Ag PLGA NPs) showing Ag NPs on the surface of the PLGA NPs. | ||
To immobilize the purified GST IFNγ protein on the surface of the PLGA and composite NPs, different amounts of recombinant IFNγ were incubated with a fixed amount of NPs for 1 h at 37 °C, followed by centrifugation to remove the unbound protein. The protein bound NPs collected by centrifugation revealed that the fluorescence intensity of the NPs gradually decreased with the addition of protein up to 25–30 ng protein μg−1 of NPs; after that it became saturated (Fig. S9†). The protein immobilized NPs were further probed for quantitative estimation by UV-vis spectroscopy. Absorption at 280 nm, which corresponds to the amount of bound protein, revealed that maximum 25 ng protein was bound with 1 μg composite NPs (Ag PLGA NPs) (Fig. 3A and 4B). It should be mentioned here that the recombinant GST IFNγ protein has a PI value of 8.76 (based on ExPASy PI Calculator) and thus, it is positively charged at physiological pH (i.e. 7.4). On the other hand, the zeta potential of the NPs showed that the synthesized NPs were negatively charged; thereby creating a plausibility of electrostatic interaction between the duos. The interaction was also supported by zeta potential and FTIR analysis. The zeta potential value of the NPs decreased to −3.83 mV from −26 mV and also the hydrodynamic diameter increased from 356 nm to 375 nm after incubation with the recombinant IFNγ, possibly due to interaction with the protein. The FTIR spectra of the composite NP alone and after its interaction with the recombinant protein showed that most of the peaks were unaltered.
 | ||
| Fig. 3 UV-Vis study of the binding of GST IFNγ protein with (A) PLGA only (B) composite NPs (Ag PLGA NPs). The values are represented as the mean ± SD of three individual experiments (C) and (D) are the FTIR spectra of the composite NPs and the recombinant IFNγ immobilized GST IFNγ Ag PLGA composite NPs, respectively. | ||
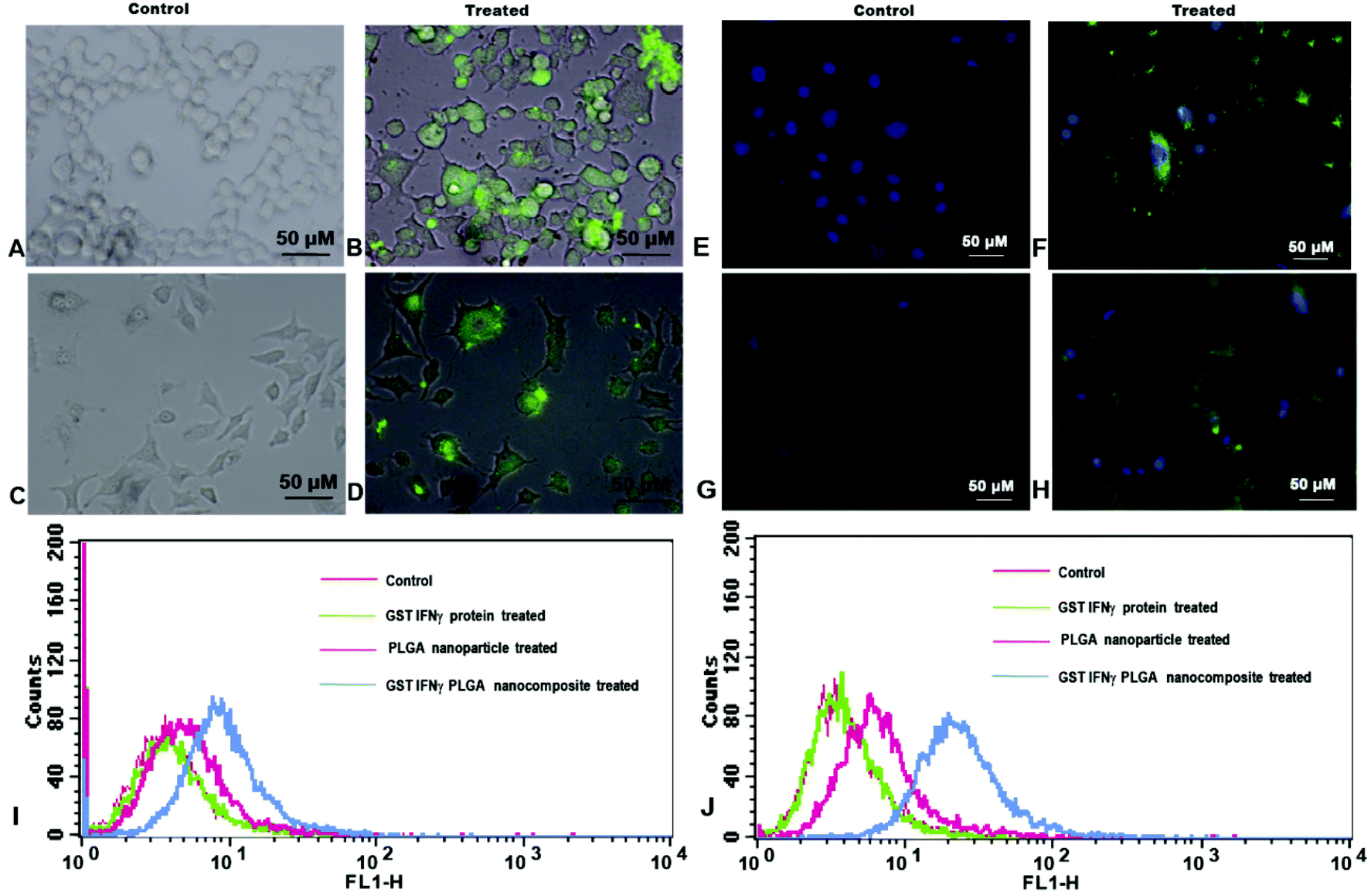 | ||
| Fig. 4 Fluorescent microscopy images of (A, B) HeLa control and GST IFNγ loaded Ag PLGA composite nanoparticle treated cells respectively (C, D) MCF-7 control and GST IFNγ loaded Ag PLGA composite nanoparticle treated cells, respectively (E, F) DAPI stained images of HeLa control and treated cells (G, H) DAPI stained MCF-7 control and treated cells respectively (I, J) GST IFNγ Ag PLGA cell surface binding study by FACS on HeLa and MCF-7 cells, respectively. | ||
However, the peaks at 1760 cm−1 and 1162 cm−1 corresponding to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and C–O stretching vibrations were more prominent in the case of only PLGA NPs.41 One additional peak appeared for protein immobilized NPs at 1651 cm−1 corresponding to amide bond I (HNCO) bond stretching, indicating the presence of protein on the surface of the NPs (Fig. 3C and D). In addition, the TEM image showed that the composite NPs didn't agglomerate or lose their structural integrity as the diameter and shape of the NPs remained similar after immobilization of the recombinant protein (Fig. S10†). Protein release data showed a maximum of 34% release of GST IFNγ from NPs in 72 h at physiological pH, while the rest was bound with the nanoparticles (Fig. S11†). The sustained release of the protein in physiological conditions possibly reduces the frequency of its doses. More importantly, we have performed the protease protection assay, which showed lesser protein degradation of GST IFNγ protein loaded on Ag PLGA NPs compared with free GST IFNγ protein, which confirms more stability of NPs conjugated protein than free protein (Fig. S12†).
O and C–O stretching vibrations were more prominent in the case of only PLGA NPs.41 One additional peak appeared for protein immobilized NPs at 1651 cm−1 corresponding to amide bond I (HNCO) bond stretching, indicating the presence of protein on the surface of the NPs (Fig. 3C and D). In addition, the TEM image showed that the composite NPs didn't agglomerate or lose their structural integrity as the diameter and shape of the NPs remained similar after immobilization of the recombinant protein (Fig. S10†). Protein release data showed a maximum of 34% release of GST IFNγ from NPs in 72 h at physiological pH, while the rest was bound with the nanoparticles (Fig. S11†). The sustained release of the protein in physiological conditions possibly reduces the frequency of its doses. More importantly, we have performed the protease protection assay, which showed lesser protein degradation of GST IFNγ protein loaded on Ag PLGA NPs compared with free GST IFNγ protein, which confirms more stability of NPs conjugated protein than free protein (Fig. S12†).
Cell surface interaction study
To study the interaction of protein immobilized NPs with the cells, HeLa and MCF-7 cells were incubated with FITC loaded GST IFNγ Ag PLGA NPs for 1 h and analysed with a fluorescence microscope as well as FACS. Fluorescence microscopy images showed green fluorescence emission at the surface of the treated cells when excited with blue light (Fig. 4B and 4D). This could be due to binding of IFNγ with its surface receptor which allowed the nanoparticles to attach on the cell surface. Under similar conditions, DAPI, a well-known nuclear dye stained the nuclei blue. Interestingly, the merged images showed both green and blue fluorescence for GST IFNγ Ag PLGA NPs treated cells (Fig. 4F and 4H) whereas the Ag PLGA NPs treated cells showed only blue fluorescence due to DAPI staining (Fig. 4E and 4G). Thus, the microscopy images illustrated the evidence of NPs interaction with the cell surface through IFNγ cell surface receptor. The finding was substantiated by FACS analysis of the cells treated with FITC loaded GST IFNγ Ag PLGA NPs. FACS results revealed a prominent shift of fluorescence intensity in the FL-1H channel corresponding to the green emission of FITC loaded NPs attached on cell surface, which was not seen in the control experiment (Fig. 4I and 4J). This is possibly due to binding of IFNγ with its cell surface receptor leading to the change in fluorescence intensity as observed during FACS analysis.42Cell viability
We have studied the impact of individual PLGA NPs and Ag PLGA NPs on HeLa and MCF-7 cells before studying the effect of the composite nanoparticles. The cells treated with various amounts of PLGA NPs and the Ag PLGA NPs were estimated for viability estimation by MTT assay. Our results showed that PLGA NPs did not produce cell mortality; however cell viability gradually decreased with increasing concentration of Ag PLGA NPs for both the cell lines. The IC50 of the Ag PLGA NPs, where the 50% cells were killed, was found to be 1.48 μg mL−1 and 1.36 μg mL−1 for the MCF-7 and HeLa cells, respectively (Fig. 5A and B). Finally, we performed a MTT assay to assess the cell viability in the presence of the protein immobilized composite NPs (Ag PLGA NPs). The protein was immobilized on the NPs at 37 °C for 1 h followed by centrifugation to remove the free protein (25 μg protein per mg Ag PLGA NPs) and the cells were treated with the protein immobilized NPs to check the combined effect. Cell viability data showed that protein immobilized NPs had killed the cancer cells more effectively than Ag PLGA NPs or GST IFNγ protein alone for both the cell lines of HeLa and MCF 7. | ||
| Fig. 5 Cell viability analysis of (A) HeLa (B) MCF-7 cells. The values are represented as the mean ± SD of three individual experiments with p < 0.005. | ||
At IC50 the GST IFNγ Ag PLGA composite GST IFNγ loaded PLGA nanoparticles comprises 0.95 μg mL−1 Ag NPs and 0.05 μg mL−1 IFNγ protein (Fig. 5). The amount of silver required here to attain the IC50 was much less than the amount required for Ag PLGA NPs alone.
Mode of cell death
We have stained the GST IFNγ PLGA nanoparticle treated cells and PLGA treated control cells with acridine orange (AO)/ethidium bromide (EtBr) and observed them under a fluorescence microscope. The live cells allowed permeation of AO, which stained the nuclei green, whereas the dead cells emitted red fluorescence due to uptake of EtBr. Thus dual staining facilitated an easier view of dead and live cell populations. The cells treated with the IC50 dose of protein immobilized NPs (Fig. 6B and 6D) showed more red cells compared to the control untreated cells. Moreover, careful observation depicted chromatin condensation and fragmentation in the case of NP treated cells, which is the distinctive feature of apoptotic cell death.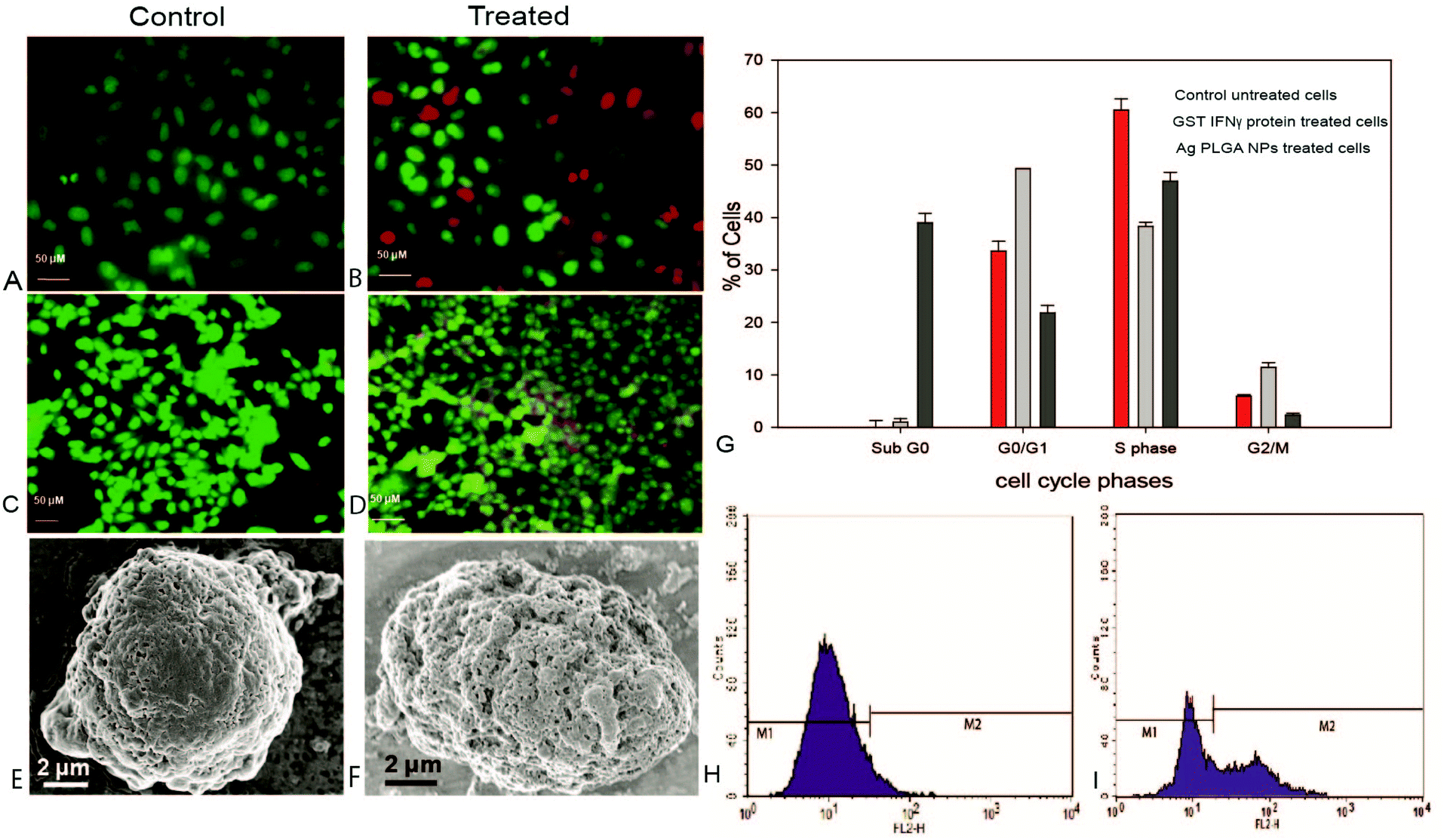 | ||
| Fig. 6 (A) AO/EtBr dual staining of control HeLa cells. (B) HeLa cells treated with GST IFNγ PLGA nanoparticles. (C) Control MCF-7 cells (D) MCF-7 cells treated with GST IFNγ PLGA nanoparticles (E) FESEM images of the untreated HeLa cells (F) HeLa cells treated with GST IFNγ Ag PLGA composite nanoparticles (G) cell cycle analysis of HeLa cells (H) Caspase 3 analysis of untreated HeLa cells and (I) HeLa cells treated with Ag PLGA NPs. | ||
The observation was substantiated with FESEM analysis, which revealed indentation and irregular membrane structure of the composite NPs treated cells compared to the control cell population. This was in agreement with initiation of apoptosis, which ultimately formed apoptotic bodies (Fig. 6F). Further, to understand the mode of cell death, the HeLa cells were treated with an IC50 dose of protein and composite NPs. Cell cycle analysis for HeLa cells revealed that recombinant IFNγ had caused cell cycle arrest mostly in the G1 phase. Treatment with recombinant IFNγ alone did not show any sub G0 population, whereas a significant (40%) sub G0 population was observed due to apoptosis after composite NPs treatment at IC50 of 1.36 μg mL−1 (Fig. 6G).
Further, to confirm the apoptotic mode of cell death, caspase-3 assay was performed in the HeLa cells. Caspase is the key protease, synthesized in an inactive form in cells, which is converted to its active form during the initial phase of apoptosis by self-proteolysis or by cleavage with other proteases. In untreated (control) HeLa cells, 98.99% of cells were found to be non-apoptotic (Fig. 6H), whereas around 45% of cells were apoptotic in caspase 3 assay, after 16 h treatment with Ag PLGA NPs (Fig. 6I). Thus, the overall results demonstrated that the protein immobilized Ag PLGA NPs were more effective in order to cause the apoptosis mediated cell death than their individual components, possibly to complementary effects.
Conclusion
In brief, we have generated recombinant human IFNγ clone, and purified the cytokine as GST tagged IFNγ. Our initial experiments showed that the recombinant IFNγ could block the cell cycle mostly at the G1 phase in two different cancer cells, HeLa and MCF-7 cells but could not cause the cell death. Hence, the recombinant IFNγ was immobilized on the composite PLGA NPs together with Ag NPs, where PLGA NPs provided stability to the protein and the Ag NPs have potentiated anticancer activity of the recombinant IFNγ. The combination of IFNγ and Ag NPs eventually led to induction of apoptosis to the cancer cells. Thus, the composite NPs mediated delivery of this clinically important cytokine opens up a new regimen in cancer therapy, which could minimize the repetitive doses and reduce the amount of Ag NPs for induction of apoptosis.Acknowledgements
The authors acknowledge the financial support of the Department of Biotechnology (no. BT/49/NE/TBP/2010 and BT/01/NE/PS/08). Assistance from the Central Instruments Facility (CIF) and the Centre for Nanotechnology, IIT Guwahati for TEM, FACS, DLS, SEM and FESEM are gratefully acknowledged.Notes and references
- T. Lammers, F. Kiessling, W. E. Hennink and G. Storm, J. Controlled Release, 2012, 161, 175–187 CrossRef CAS PubMed.
- Y. H. Bae and K. Park, J. Controlled Release, 2011, 153, 198–205 CrossRef CAS PubMed.
- F. Danhier, B. Ucakar, N. Magotteaux, M. E. Brewster and V. Préat, Int. J. Pharm., 2010, 392, 20–28 CrossRef CAS PubMed.
- R. Dinarvand, P. Cesar de Morais and A. D'Emanuele, J. Drug Delivery, 2012, 2012, 1–2 CrossRef PubMed.
- O. C. Farokhzad and R. Langer, ACS Nano, 2009, 3, 16–20 CrossRef CAS PubMed.
- K. Cho, X. Wang, S. Nie, Z. Chen and D. M. Shin, Clin. Cancer Res., 2008, 14, 1310–1316 CrossRef CAS PubMed.
- D. Peer, J. M. Karp, S. Hong, O. C. Farokhzad, R. Margalit and R. Langer, Nat. Nanotechnol., 2007, 2, 751–760 CrossRef CAS PubMed.
- S. Segura, S. Espuelas, M. J. Renedo and J. M. Irache, Drug Dev. Ind. Pharm., 2005, 31, 271–280 CrossRef CAS PubMed.
- N. Vij, T. Min, R. Marasigan, C. N. Belcher, S. Mazur, H. Ding, K.-T. Yong and I. Roy, J. Nanobiotechnol., 2010, 8, 22 CrossRef PubMed.
- Z. Vajo, J. Fawcett and W. C. Duckworth, Endocr. Rev., 2001, 22, 706–717 CrossRef CAS PubMed.
- L. M. Minasian, R. J. Motzer, L. Gluck, M. Mazumdar, V. Vlamis and S. E. Krown, J. Clin. Oncol., 1993, 11, 1368–1375 CAS.
- F. Lange, K. Rateitschak, B. Fitzner, R. Pöhland, O. Wolkenhauer and R. Jaster, Mol. Cancer, 2011, 10, 13 CrossRef CAS PubMed.
- A. Billiau and P. Matthys, Cytokine Growth Factor Rev., 2009, 20, 97–113 CrossRef CAS PubMed.
- C. E. Samuel, Clin. Microbiol. Rev., 2001, 14, 778–809 CrossRef CAS PubMed.
- F. Lange, K. Rateitschak, B. Fitzner, R. Pöhland, O. Wolkenhauer and R. Jaster, Mol. Cancer, 2011, 10, 13 CrossRef CAS PubMed.
- A. Kano, Y. Watanabe, N. Takeda, S. Aizawa and T. Akaike, J. Biochem., 1997, 121, 677–683 CrossRef CAS.
- S. Roy-Ghanta and J. S. Orange, Clin. Rev. Allergy Immunol., 2009, 38, 39–53 Search PubMed.
- J. Girdlestone and M. Wing, Eur. J. Immunol., 1996, 26, 704–709 CrossRef CAS PubMed.
- H. Ikeda, L. J. Old and R. D. Schreiber, Cytokine Growth Factor Rev., 2002, 13, 95–109 CrossRef CAS.
- G. P. Dunn, L. J. Old and R. D. Schreiber, Immunity, 2004, 21, 137–148 CrossRef CAS PubMed.
- G. Brandacher, C. Winkler, K. Schroecksnadel, R. Margreiter and D. Fuchs, Curr. Drug Metab., 2006, 7, 599–612 CrossRef CAS.
- Z. Dong, C. Zhang, H. Wei, R. Sun and Z. Tian, Can. J. Physiol. Pharmacol., 2005, 83, 1045–1053 CrossRef CAS PubMed.
- A. K. Sahoo, M. P. Sk, S. S. Ghosh and A. Chattopadhyay, Nanoscale, 2011, 3, 4226 RSC.
- P. Gopinath, S. K. Gogoi, P. Sanpui, A. Paul, A. Chattopadhyay and S. S. Ghosh, Colloids Surf., B, 2010, 77, 240–245 CrossRef CAS PubMed.
- P. Gopinath, S. K. Gogoi, A. Chattopadhyay and S. S. Ghosh, Nanotechnology, 2008, 19, 075104 CrossRef CAS PubMed.
- D. R. Smyth, M. K. Mrozkiewicz, W. J. McGrath, P. Listwan and B. Kobe, Protein Sci., 2003, 12, 1313–1322 CrossRef CAS PubMed.
- S. Ghosh, S. Rasheedi, S. S. Rahim, S. Banerjee, R. K. Choudhary, P. Chakhaiyar, N. Z. Ehtesham, S. Mukhopadhyay and S. E. Hasnain, BioTechniques, 2004, 37, 418 CAS , 420, 422–423.
- S. Petrov, G. Nacheva and I. Ivanov, Protein Expression Purif., 2010, 73, 70–73 CrossRef CAS PubMed.
- R. Dinarvand, N. Sepehri, S. Manoochehri, H. Rouhani and F. Atyabi, Int. J. Nanomed., 2011, 6, 877–895 CrossRef CAS PubMed.
- F. Danhier, E. Ansorena, J. M. Silva, R. Coco, A. Le Breton and V. Préat, J. Controlled Release, 2012, 161, 505–522 CrossRef CAS PubMed.
- H. Murakami, M. Kobayashi, H. Takeuchi and Y. Kawashima, Int. J. Pharm., 1999, 187, 143–152 CrossRef CAS.
- Z. Gu, A. Biswas, M. Zhao and Y. Tang, Chem. Soc. Rev., 2011, 40, 3638–3655 RSC.
- J. Wu and C.-C. Chu, J. Mater. Chem. B, 2013, 1, 353–360 RSC.
- D.-W. Park, S.-S. Kim, M.-K. Nam, G.-Y. Kim, J.-H. Kim and H.-S. Rhim, BMB Rep., 2011, 44, 279–284 CrossRef CAS PubMed.
- H. Tao, W. Liu, B. Simmons, H. Harris, T. Cox and M. Massiah, BioTechniques, 2010, 48, 61–64 CrossRef CAS PubMed.
- A. L. Yergey, J. R. Coorssen, P. S. Backlund Jr., P. S. Blank, G. A. Humphrey, J. Zimmerberg, J. M. Campbell and M. L. Vestal, J. Am. Soc. Mass Spectrom., 2002, 13, 784–791 CrossRef CAS.
- J. Havlis, H. Thomas, M. Sebela and A. Shevchenko, Anal. Chem., 2003, 75, 1300–1306 CrossRef CAS.
- C. Amatore, S. Arbault and A. C. W. Koh, Anal. Chem., 2010, 82, 1411–1419 CrossRef CAS PubMed.
- E. Eruslanov and S. Kusmartsev, in Advanced Protocols in Oxidative Stress II, ed. D. Armstrong, Humana Press, Totowa, NJ, 2010, vol. 594, pp. 57–72 Search PubMed.
- D. Yang, S. G. Elner, Z.-M. Bian, G. O. Till, H. R. Petty and V. M. Elner, Exp. Eye Res., 2007, 85, 462–472 CrossRef CAS PubMed.
- M. Khemani, M. Sharon and M. Sharon, ISRN Nanotechnol., 2012, 2012, 1–9 CrossRef PubMed.
- K. Schroder, P. J. Hertzog, T. Ravasi and D. A. Hume, J. Leukocyte Biol., 2004, 75, 163–189 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c3bm60251f |
| This journal is © The Royal Society of Chemistry 2014 |