 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Gadolinium-DTPA amphiphile nanoassemblies: agents for magnetic resonance imaging and neutron capture therapy†
Minoo J.
Moghaddam
*a,
Liliana
de Campo
ab,
Mioko
Hirabayashi
a,
Penny A.
Bean
a,
Lynne J.
Waddington
c,
Judith A.
Scoble
c,
Gregory
Coia
c and
Calum J.
Drummond
de
aCSIRO Materials Science and Engineering, PO BOX 52, North Ryde, NSW 1670, Australia. E-mail: minoo.moghaddam@csiro.au
bAustralian National University, Department of Applied Mathematics, Research School of Physical Science and Engineering, Canberra, 0200, Australia
cCSIRO Materials Science and Engineering, 343 Royal Parade, Parkville, VIC 3052, Australia
dCSIRO Materials Science and Engineering, Private Bag 10, Clayton South, VIC 3169, Australia
eSchool of Applied Sciences, College of Science, Engineering and Health, RMIT University, GPO Box 2476, Melbourne, VIC 3001, Australia
First published on 17th February 2014
Abstract
Engineering biocompatible and physiologically stable nanoscaled therapeutics and imaging agents with the ability to target tumor tissue is a key challenge for the advancement of cancer therapeutics and diagnostic imaging. Here, we present chelating amphiphiles with the capacity to form nanoassembled colloidal particles containing high payloads of gadolinium (Gd) ions. We present the in situ synthesis and complexation of Gd with colloidal nanoassemblies (NAs) based on diethylenetriamine pentaacetic acid (DTPA) amphiphiles. This method allows for facile simultaneous incorporation of several metal ions for applications in multimodal imaging and therapeutics. The diverse internally nanostructured NAs made from sole precursor amphiphiles and their Gd-complexes were investigated by synchrotron small angle X-ray scattering (SAXS) and cryo-TEM. Depending on the molecular structure of the amphiphiles, the structures of NAs range from micelles to liposomes, to colloidal particles of inverse hexagonal (hexosomes) and inverse bicontinuous cubic phases (cubosomes), to multilayered nanospheres. The in vitro contrast activity of these NAs exhibited high relaxivity values as T1-weighted magnetic resonance imaging (MRI) contrast enhancement agents. Further, an α-Flag antibody fragment (Fab′) was bioconjugated to the surface of the Gd-complexed NAs. The binding ability of these targeted NAs to a FLAG-tagged protein was confirmed by SDS-PAGE. The in vitro cytotoxicity against two cell lines showed that except for the negatively charged micellar Gd-DTPA amphiphile, liposomal and higher order internally nanostructured NAs had low cell toxicity. The efficient cellular uptake of Gd-NAs by melanoma cancer cells was also investigated.
1. Introduction
Self-assembly of amphiphiles into various highly ordered nanoscale particles has been the subject of intensive research and development over the last thirty years, particularly for the development of efficient drug delivery systems and biomedical imaging agents.1,2 NAs, either made from natural lipids or synthetic amphiphiles, have been explored as potential slow release matrices for a wide range of biologically active molecules and drugs.1–3 The characteristics of amphiphile-based colloidal particles as drug delivery systems can be excellent bio-compatibility and low toxicity, ease of synthesis and large scale production of the low molecular weight amphiphilic building blocks, the capacity to precisely control the composition of the NAs, and drug protection against biological degradation under physiological conditions. Here we focus on a series of Gd-chelate amphiphile NAs, possessing various supramolecular nanostructures, as prospective advanced MRI contrast enhancement agents (CEAs) and potentially also as neutron capture therapeutics.Among amphiphile self-assembly systems, liposomes are one of the most widely-used NAs and currently are the gold standard for delivery of drugs in preclinical and clinical studies, some of which are clinically approved for various anti-cancer and anti-fungal drugs.4,5 In more recent years, there has been a growing body of research on the application of highly ordered inverse liquid crystalline lipid-based NAs as drug delivery systems.2 Colloidal particles with nanostructures of two dimensional (2-D) and three dimensional (3-D) periodicity, namely “hexosomes” and “cubosomes”, have been increasingly exploited for drug delivery of poorly water-soluble hydrophobic or amphiphilic drugs.2,3,6–8 The key aspects of the internally higher ordered NAs are exemplified by their capacity to carry and encapsulate relatively high payloads of drugs, high thermodynamic stability, and depot systems for controlled drug release.2,9 In addition, similar to liposomal particles, the stabilizing surface-adsorbed polymer (steric barrier acting to prevent colloidal particle agglomeration) can enhance the bioavailability and pharmacokinetics of drugs and protect them against degradation.10–13 Non-lamellar (non-liposomal) highly ordered NAs have been recently investigated as medical imaging agents by our group and others.14–19
Gd-based agents still remain the most dominant reagents for improving the performance of MRI as a non-invasive imaging modality in the clinic and in clinical research.20,21 Unlike other imaging modalities such as computed tomography (CT), nuclear medicine and positron emission tomography (PET), CEAs for MRI contribute to the enhancement of image contrast of different soft tissues by lowering the spin–lattice (longitudinal) and spin–spin (transverse) relaxation times of the nearby water protons (T1 and T2 respectively). With the exception of some recent developments with ultra-small iron oxide nanoparticles,22T1-weighted CEAs are mainly constructed from paramagnetic Gd-chelates and typically reduce T1 to a greater extent than T2, resulting in brighter images.23,24 Further, Gd-based materials have been recently identified as suitable therapeutic agents for neutron capture therapy (NCT). This is due to the inherent neutron absorption and high thermal cross section of 157Gd amongst known stable elements in the periodic table. Gd provides a 65 fold increase in thermal neutron cross-section over the extensively explored Boron-10 in absorbing neutrons and in emission of radiation energy.25 Efficient targeted NAs bearing high payloads of Gd may therefore not only enhance the sensitivity of MRI, but also confer the potential of enhancing the success of Gd-NCT.26
In ongoing research in our laboratory, we have been designing chelating amphiphiles with the possibility of forming increasingly higher order 1, 2 and 3D periodic nanostructured liquid crystalline NAs, solely made from Gd-complexed amphiphiles, in order to increase the payload of paramagnetic metal ions by many orders of magnitude. It has been challenging to design and control the physicochemical properties of these NAs due to the large headgroups required for stable chelation of the metal ions, and the significant structural changes that they will undergo upon addition of metal ions. We have recently reported on DTPA-chelating amphiphiles with mono- and bis-oleyl (DTPA-MO and DTPA-BO) and phytanyl chains (DTPA-MP and DTPA-BP) and investigated their neat and bulk lyotropic liquid crystalline self-assembly, which gave us an insight into the complex mesophase behaviour of these chelating amphiphiles and their Gd complexes.27 The selection of oleyl and phytanyl chains was to increase the likelihood that the amphiphile chains were in the fluid state at room temperature or under physiological conditions, to allow for the formation of lyotropic liquid crystalline NAs.
In this report, our focus is on in situ synthesis of Gd-complexed DTPA amphiphilic NAs, their colloidal structures, relaxivity values in low magnetic field NMR, cell toxicity, biconjugation of targeting molecules to the surfaces of the NAs and their cellular uptake.
Gd-complexation with DTPA amphiphiles occurs very rapidly; therefore we performed complexation of Gd(III) subsequent to aqueous dispersion of precursor DTPA chelating amphiphiles. Post-complexation of the colloidal particles allowed us to finely tune the NAs internal nanostructures and investigate the structural changes as a function of molar ratio of Gd(III) to DTPA amphiphile. The post-complexation of metal ions also provides flexibility in simultaneous incorporation of several metal ions such as luminescent Eu(III) or radioisotope metal ions such as 111In or 99Tc for the application of these NAs to multimodal imaging. Synchrotron SAXS and Cryo-TEM were used to determine the structure and morphology of the NAs. We evaluated the relaxivity of the Gd-complexed NAs as CEAs in low field NMR and tested in vitro toxicity using two cell lines. Further, we present proof of concept for the facile preparation of multifunctional NAs and bio-conjugation of antibodies to the surface of the NAs that can be used as active targeting NAs. The cell internalization of these contrast agents was evaluated with Gd-NAs containing a fluorescent lipid probe.
Fine tuning of advanced stable nanostructured NAs bearing high payloads of Gd combined with efficient cellular uptake using selective targeting molecules is a critical component to enhance the sensitivity of MRI, whilst lowering the required dosage of Gd-complexes. Both of these features can be achieved by localizing high payloads of contrast agents via condensed and highly ordered nanostructured NAs, which can be targeted to diseased tissues such as cancer. In addition, these NAs may be useful as advanced neutron absorbing therapeutic agents for neutron capture therapy for tumours leading to sophisticated theranostic materials.
2. Materials and methods
2.1 Materials
The synthesis of the DTPA amphiphiles was performed as previously reported.27 The pluronic F127, poly(ethylene oxide)–poly(propylene oxide)–poly(ethylene oxide) block copolymers were purchased from BASF, Germany. All solvents were purchased from Merck Australia.Na-acetate and Gd-acetate, dicyclohexylcarbodiimide (DCCD), N-hydroxysuccinimide (HOSU), N-(2-aminoethyl) maleimide trifluoroacetate salt and xylenol orange were purchased from Sigma-Aldrich (Australia). Lissamine™ Rhodamine B 1,2-dihexadecanoyl-sn-glycero-3-phosphoethanolamine, triethyl-ammonium salt (rhodamine DHPE) was purchased from Molecular Probes (InVitrogen, now Life Technologies).
2.2 Synthesis of 2-amidoethyl maleimide-EDTA-BP
Ethylenenediaminetetracetic acid-bis phytanyl amphiphile (EDTA-BP) was synthesised as previously reported.14 HOSU (140 mg, 1.2 mmol) and DCCD (228 mg, 1.1 mmol) were added to EDTA-BP (852 mg, 1 mmol) solution in 10 mL of dichloromethane, and stirred at room temperature for 2 h. The formation of active ester was confirmed by ESI/MS.The white precipitate was filtered and N-(2-aminoethyl) maleimide trifluoroacetate salt (254 mg, 1 mmol) was added to the filtrate and stirred for 5 h at room temperature. The pH of the reaction mixture was adjusted to 8 by addition of N,N-diisopropylethylamine. The formation of the title compound was confirmed by ESI/MS. The solvent was evaporated to dryness and the oily residue was purified by flash chromatography using a silica column and a gradient of DCM/methanol as an eluting solvent. The pure fractions were collected, evaporated to dryness and freeze-dried. HPLC showed a single peak and negative ion mode ESI/MS showed a molecular ion [M − H] of m/z = 974.20.
2.3. Complexation of Gd(III) to DTPA amphiphile NAs in aqueous solution
Colloidal dispersions of DTPA-MO and -MP were prepared by addition of 100 mM Na-acetate buffer (100 mM, pH 7.6) to the bulk amphiphile phase, followed by homogenization of the samples using a PathTech X120 probe sonicator at 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm for 1 min with further sonication, using a 600 W ultrasonic bath cleaner for 10 min. The final concentration of the amphiphile dispersions was adjusted to 40 mM for physicochemical characterization of the particles and further complexation with Gd(III) ions. Gd complexes were made by addition of a 40 mM solution of Gd-acetate, containing an appropriate amount of pluronic steric stabilizer solution F127 (10% by weight of the complexed amphiphile) to yield particles containing a 1:1 molar ratio of Gd(III) to DTPA-MO or DTPA-MP. Amicon® Ultracel-3, 3K molecular cut-off centrifugal filters (Millipore, Australia) were used to remove any residual non-complexed Gd(III) ions for colloidal dispersion batches used for relaxivity measurement and cytotoxicity assay.
000 rpm for 1 min with further sonication, using a 600 W ultrasonic bath cleaner for 10 min. The final concentration of the amphiphile dispersions was adjusted to 40 mM for physicochemical characterization of the particles and further complexation with Gd(III) ions. Gd complexes were made by addition of a 40 mM solution of Gd-acetate, containing an appropriate amount of pluronic steric stabilizer solution F127 (10% by weight of the complexed amphiphile) to yield particles containing a 1:1 molar ratio of Gd(III) to DTPA-MO or DTPA-MP. Amicon® Ultracel-3, 3K molecular cut-off centrifugal filters (Millipore, Australia) were used to remove any residual non-complexed Gd(III) ions for colloidal dispersion batches used for relaxivity measurement and cytotoxicity assay.
The bis-conjugate dispersions were prepared according to the procedure noted above, except that both conjugates were initially solubilised in an appropriate amount of ethanol as a hydrotropic solvent, to prepare 200 mM solutions. The ethanolic solutions were added via a syringe to appropriate volumes of 100 mM Na-acetate solutions to prepare 40 mM dispersions of the precursor chelating amphiphiles, whilst undergoing harsh vortexing. Gd-complexed dispersed particles were prepared by drop-wise addition of appropriate solutions of Gd-acetate and pluronic solution to the DTPA-bis conjugate NAs at 0.5:1 Gd(III) to DTPA-BO or DTPA-BP molar equivalents. Further, we investigated the structural changes as a function of incremental changes in the concentration of chelated Gd(III) by SAXS, using dispersions of various molar ratios of Gd(III) to DTPA-BO or -BP ranging from 0.1:1 to 1:1.
NAs used for antibody conjugation were prepared as described above with the exception that 10% maleimido-EDTA-BP conjugate was incorporated within the bulk phase of the precursor chelating amphiphile prior to hydration in Na-acetate solution and complexation with a mixture of Gd(III) and Eu(III) at a 0.5:1 molar ratio of metal ion (Gd/EU:0.499/0.001) to DTPA-BP.
The excess Gd(III) + EU(III) were filtered through a Millipore Ultracel-3 membrane as noted before. The free Gd(III) ion in the NA dispersion was measured by an assay using xylenol orange.28
The particle sizes of the colloidal dispersions were measured using a Malvern Zetasizer Nano ZS (Malvern Instruments, Worcestershire, UK) and using an intensity-weighted mode. The sizes were averaged over three measurements.
2.4. Biconjugation of anti-FLAG antibody to the dispersed NAs
Anti-FLAG antibody29 was cleaved with pepsin and the resulting Fab′ fragment reduced with 2 mM tris (2-carboxyethyl) phosphine hydrochloride (TCEP-HCl) for 30 minutes on ice before being desalted into PBS on a PD10 column (GE Healthcare Australia). Dispersed particles of Gd/Eu-DTPA-BP (4 mL, 13.3 mM) containing 10% maleimide-EDTA-amphiphiles were incubated with the Fab′ fragment (3 mg) for one week at 4 °C before any remaining maleimide groups were blocked by reaction with 14 mM 2-mercaptoethanol for 3 h. Unconjugated Fab′ and 2-mercaptoethanol were removed by extensive dialysis against PBS using a 300 kDa molecular weight cut-off membrane (Spectrum Laboratories). The control dispersion without the Fab′ was prepared by reacting the maleimide-containing particles with 14 mM 2-mercaptoethanol for 3 h, followed by dialysis.2.5. Synchrotron small and wide angle X-ray scattering
All SAXS measurements on dispersions were conducted at the Australian Synchrotron Radiation Source (Melbourne, Australia). The diffraction patterns were transmission-corrected and background-subtracted. Sample-detector distance calibration was performed using silver behenate as the standard sample. A multi-capillary (24) sample holder with a Peltier temperature controller, designed and manufactured at the Australian synchrotron, was used as a semi-high-throughput sample holder for the analysis of the dispersions. The actual temperature of the sample during analysis was checked by inserting a thermocouple into a water-filled capillary in the sample holder. 2-D scattering images were radially averaged to conventional scattering plots using the program SAX15-ID or Scatterbrain. Scattering intensities I(q) were plotted as a function of the scattering vector q, where q = (4π/λ)sin(θ/2), in which λ is the wavelength and θ is the scattering angle.2.6. Cryogenic transmission electron microscopy (Cryo-TEM)
A laboratory-built humidity-controlled vitrification system was used to prepare the samples for Cryo-TEM. Humidity was kept close to 80% for all experiments and ambient temperature was ∼22 °C. A 4 μl aliquot of the sample was pipetted onto a 300-mesh copper grid coated with lacy formvar-carbon film (ProSciTech, Thuringowa, Queensland). After 30 s adsorption time, the grid was blotted manually using Whatman 541 filter paper, for between 2 and 10 s. The grid was then plunged into liquid ethane cooled by liquid nitrogen. Frozen grids were stored in liquid nitrogen until examined.The samples were examined using a Gatan 626 cryoholder (Gatan, Pleasanton, CA, USA) and a Tecnai 12 Transmission Electron Microscope (FEI, Eindhoven, The Netherlands) at an operating voltage of 120 kV, equipped with a FEI Eagle 4k × 4k CCD (FEI, Eindhoven, the Netherlands). Alternatively, a Tecnai TF30 cryo-TEM operating at 200 kV and equipped with a Gatan US 1000-2 K CCD camera and digital micrograph imaging software was used for imaging the samples. At all times low dose procedures were followed, using an electron dose of 8–10 electrons per Å2 for all imaging.
2.7. Assessment of T1 and T2 relaxivities
The proton longitudinal and transverse relaxation time (T1 and T2) for each sample were performed with a Maran Ultra 23.4 MHz (0.54 T) bench-top NMR (Oxford Instruments Molecular Bio-tools Ltd, Oxon, UK), equipped with a variable temperature probe.T 1 was measured with an inversion recovery sequence. The recycle delay time was set to five times the T1 value. In general, 20 points were taken for each T1 measurement. T2 was measured with a Carr–Purcell–Meiboom–Gill (CPMG) sequence and were fitted into multiple or distributed exponentials.
Measurements were conducted on four dilutions of each NA formulation from 20 mM to 2.5 mM, and the relaxivity was calculated from the slope of inverse relaxation time versus Gd(III) concentration.
2.8. Determination of Gd(III)
Inductively coupled plasma optical emission spectrometry (ICP-OES) was performed with a Varian Vista MPX (Varian, Palo Alto, CA) to determine the Gd(III) contents of each NA. Samples and vials were weighed and recorded. The contents were rinsed with water in plastic hotblock vials and 2 mL of 5% nitric acid was added. The samples and acids were heated until turbidity disappeared. The empty vials and lids were dried and cooled and reweighed and the sample weight determined by difference. Calibration standards were made in the same acid mixture and concentrations of Gd(III) within each NA formulation were determined and the relaxivity values were corrected.2.9. Pull-down assay
To 0.5 mL dispersed particles (conjugated or blocked) collected by centrifugation was added 15 μg sErbB4.497.Fc.FLAG30 or 15 μg bovine serum albumin (BSA) in a total volume of 50 μl. The mixtures were incubated for 30 minutes at room temperature with constant mixing before being centrifuged and the supernatant removed. The particles were washed with phosphate-buffered saline (PBS) and then re-suspended in 200 μl PBS. 12 μl aliquots of the unbound fraction or the suspension (containing bound protein) were analyzed by non-reduced sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE).2.10. Cell cytotoxicity assay
Cells were seeded at an optimum density to generate a confluent monolayer by completion of the experiment in Dulbecco's Modified Eagle Medium (DMEM) containing 10% fetal bovine serum (FBS), penicillin/streptomycin (Pen/Strep) + L-glutamine. Mouse fibroblast L929 cells or human breast cancer cells (MCF7) were seeded into 96-well tissue culture treated polystyrene (TCPS) wells at 2 × 105 cells per ml seeding density, 100 μl per well. Cells were incubated at 37 °C in a 5% CO2 humidified incubator for 24 h prior to use. At 24 h, samples and control solutions were prepared in tissue culture media containing 10% FBS. Media were removed from wells and replaced with 100 μl per well of sample or control solution as per the standard plate format.Samples were diluted to 200 μM in the tissue culture media and then serially diluted in doubling dilutions down to 0.78 μM concentration. Control samples included: 5% PBS in tissue culture media used as a negative control and 5% dimethyl sulfoxide (DMSO) in tissue culture media used as a positive killing control and media alone (no treatment). These were selected to show the working range of the assay.
After 20 hours, representative images of wells were recorded using the Olympus i80 Imaging System. At 24 h, media were removed and replaced with methylthiazol tetrazolium (MTT) at 0.5 mg mL−1 in fresh tissue culture medium, using 100 μl per well and incubated for a further 4 h. Media were removed from wells and replaced with 100 μl per well of DMSO and placed on an orbital shaker for 15 min to dissolve stain. Plates were read at T595/R655 wavelengths using a Spectramax 340PC plate reader.
2.11. Cell uptake assay using fluorescent probes
Human melanoma cells (SK-Mel 28) and human lung fibroblast cells (MRC-5) were seeded at 30000 cells per well in an eight-well chamber slide and incubated for 24 h in a humidified incubator with 5% CO2 at 37 °C h. NAs with red fluorescent-lipid dye were made from mixing the Gd-DTPA-MO/BO (1:1) with ∼1% of Lissamine™ rhodamine B 1,2-dihexadecanoyl-sn-glycero-3-phosphoethanolamine, triethylammonium salt (Rhodamine DHPE, Life Technologies), as noted in section 2.3. Cells were exposed to 100 μl aliquots of 100, 50 or 25 μM of NA dispersion in tissue culture media in the presence or absence of 10% FBS for 4 hours in an incubated environment. At the conclusion of the incubation period, media were removed from the wells and washed with PBS for three times, fixed with formaldehyde, washed again with warm PBS and then embedded with antifade 4′,6-diamidino-2-phenylindole (DAPI) gel mounting medium (Life Technologies). Fluorescent microscope images were obtained on a Nikon Eclipse 90i inverted microscope equipped with a Nikon camera with fluorescence and phase contrast. The filters used were a DAPI filter and a tetramethyl rhodamine isothiocyanate (TRITC) filter (excitation 540 nm/emission 605 nm).
3. Results and discussion
3.1. Synthesis and characterization of Gd-DTPA amphiphile NAs
Colloidal dispersions of Gd-DTPA amphiphile NAs were initially prepared by dispersion of pre-complexed Gd-DTPA-mono and -bis oleyl (Gd-DTPA-MO and Gd-DTPA-BO) and phytanyl chains (Gd-DTPA-MP and Gd-DTPA-BP), Fig. 1, which were prepared by our previously reported method.27 The Gd-DTPA-MO and -MP, containing one negative charge per headgroup, formed clear micellar and liposomal solutions (ESI, Fig. 1Sa†). The Gd-DTPA-BO and -BP dispersions formed liposomal and crystalline particles, as shown by their SAXS analysis (Fig. 1Sb†), and were not stable as dispersion. Precipitation of some crystalline NAs was obvious to the naked eye over a period of 24 h. Therefore, we focused our studies on an alternative in situ incremental addition of Gd-ions into the aqueous dispersion of DTPA chelating amphiphile NAs, and investigated the structural changes by synchrotron SAXS.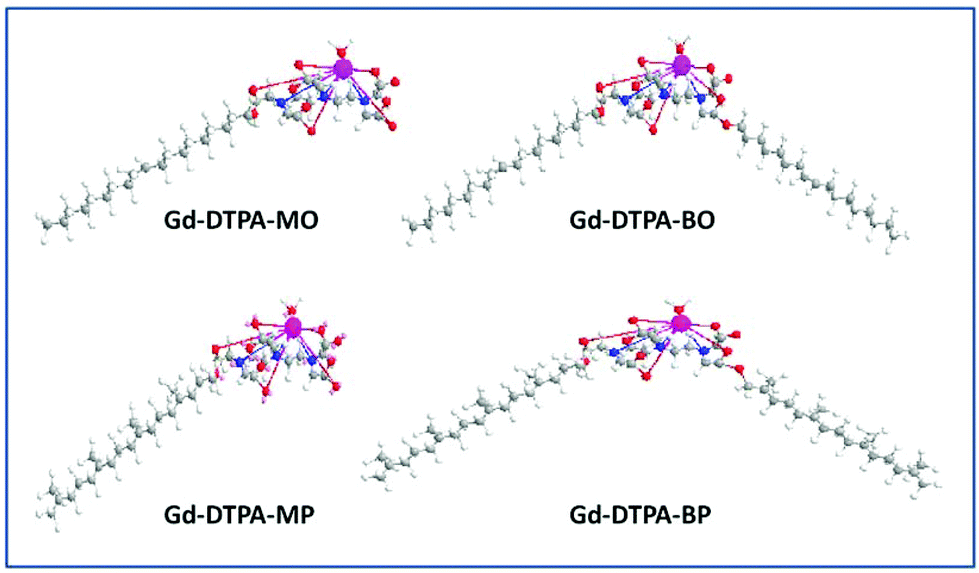 | ||
| Fig. 1 Molecular structure of Gd-mono and bis DTPA-oleyl and phytanyl amphiphiles. | ||
The Gd(III) complexation occurred very rapidly following the addition of Gd ions as was confirmed by ESI/MS. We prepared the Gd-DTPA-MO and Gd-DTPA-MP at a 1:1 molar ratio of Gd(III) to the respective chelating amphiphile NAs, whereas for bis conjugate NAs we found that raising the ratio of Gd(III) to the DTPA amphiphiles above 0.5:1 molar ratio resulted in partial precipitation. Except for the SAXS studies, the Gd-bis conjugates evaluated in this study for their relaxivity or cell toxicity were prepared at 0.5:1 molar equivalent of Gd(III) to DTPA-BO and DTPA-BP. Dispersions of 20–40 mM amphiphiles in 100 mM Na-acetate solutions were used for the analysis of synchrotron SAXS and subsequent complexation reactions with Gd(III) ions, unless otherwise noted.
The SAXS scattering patterns for DTPA-MO, DTPA-MP and their respective complexed dispersions at 25 °C are shown in Fig. 2a and 2b. The scattering curves of the starting samples, DTPA-MO and DTPA-MP, indicated the predominant formation of rather spherical micelles with a radius of gyration resulting from the Guinier approximation of 38 and 30 Å, respectively. Upon complexation of Gd, the forward scattering and thus the negative slope illustrated in the log–log plots significantly increased, indicating a change in the micellar shape from rather spherical to more elongated or flat. For Gd-DTPA-MO, the intensity slope at low q decreased with q−1, consistent with the formation of rod-shaped micelles, while the slope in Gd-DTPA-MP was steeper (q−1.25), suggesting that also flatter structures like vesicles were present, since a q−2 slope would indicate flat structures like lamellae. Detailed analysis and fit with model structures using SANSView (DANSE project, University of Tennessee) is also shown in the ESI, Fig. 2S.†
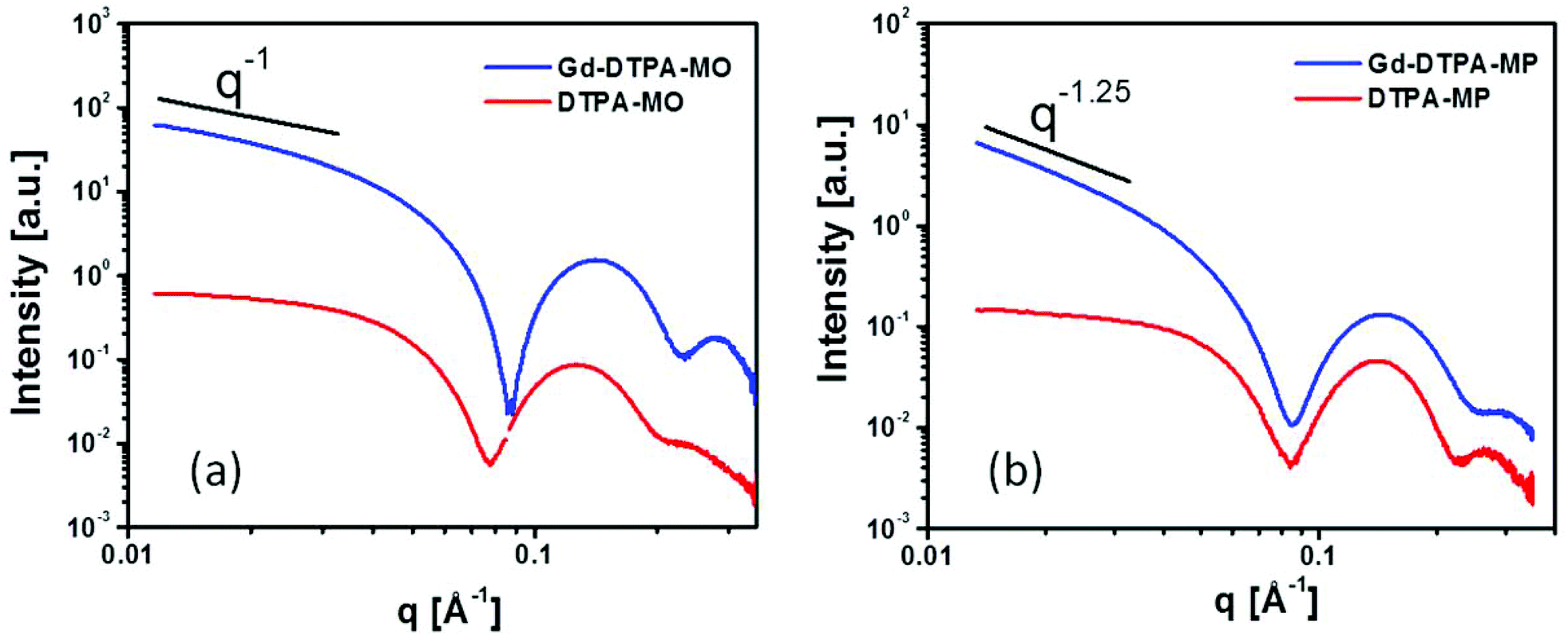 | ||
| Fig. 2 SAXS patterns of colloidal dispersion of DTPA-MO (a), DTPA-MP (b) before and after complexation with Gd(III) at a molar ratio of 1:1 to the amphiphile dispersion; upon complexation, the forward scattering significantly increases, suggesting a structural change from spherical to rod-like micelles and vesicular structures. | ||
The morphology and size distribution of these colloidal NAs were also examined by cryo-TEM and a particle size analyser. The DTPA-MO dispersion showed solid dot shaped spherical micelles with some coexisting small unilamellar liposomes (Fig. 3a). The complexed Gd-DTPA-MO (1:1) resulted in the formation of mostly spherical micelles with some co-existing cylindrical micelles (Fig. 3b).
 | ||
| Fig. 3 Cryo-TEM micrographs of (a) DTPA-MO, (b) Gd-DTPA-MO (1:1), (c) DTPA-MP, and (d) Gd-DTPA-MP (1:1) dispersed particles; scale bars are 200 nm in (a), (c), and (d) and 100 nm in (b). | ||
The DTPA-MP dispersion formed mostly spherical micelles co-existing with some unilamellar liposomes (Fig. 3c), which upon complexation with Gd(III) at a molar ratio of 1:1 transformed to more uniform and small unilamellar liposomal particles (Fig. 3d). These morphologies correspond closely with those obtained by SAXS analysis, whilst the size and polydispersity index (Pdi) were aligned with those values obtained by a particle sizer as listed in Table 1.
| Amphiphile dispersion | r 1 (m M−1 S−1) | r 2 (m M−1 S−1) | r 2/r1 | Size (nm) | Pdi |
|---|---|---|---|---|---|
| a The relaxivity values of Magnevist were from ref. 21. | |||||
| DTPA-MO | — | — | — | 3 and 173 (bimodal) | 0.57 |
| Gd-DTPA-MO | 29.5 | 33.2 | 1.13 | 8 | 0.25 |
| DTPA-MP | — | — | — | 3 and 200 (bimodal) | 0.22 |
| Gd-DTPA-MP | 16.8 | 19.8 | 1.17 | 53 | 0.27 |
| DTPA-BO | — | — | — | 255 | 0.10 |
| Gd-DTPA-BO | 11.4 | 13.4 | 1.17 | 213 | 0.23 |
| DTPA-BP | — | — | — | 218 | 0.34 |
| Gd-DTPA-BP | 10.9 | 13.6 | 1.24 | 212 | 0.25 |
| Magnevista | 3.7 | 5.6 | 1.51 | — | — |
The SAXS patterns of DTPA-BO and their Gd-complexed NAs with various molar ratios from 0.1:1 to 1:1 are shown in Fig. 4a. The non-complexed amphiphile dispersion exhibited lamellar crystalline structures with equidistant peaks in the ratio of 1:2:3, superimposed on a broad peak and with a strong increase at low q values, which is typically observed for liposomal particles. The equidistant peaks are representative of a lamellar crystal structure with a repeat distance of 47.60 ± 0.5 Å, which is consistent with what we observed for a DTPA-BO bulk phase in excess aqueous solution.27 The incremental addition of Gd(III) to the colloidal DTPA-BO NAs resulted in gradual disappearance of the lamellar crystal peaks to nearly all liposomal particles at the molar ratio of 0.6:1. Any incremental increase above this ratio resulted in a new lamellar spacing, which was significantly smaller than that of the non-complexed particles, corresponding to 40.5 ± 0.5 Å. The closer packing of the lamellar structures is conceivably due to either the Gd complexed headgroups having zero net charge compared with the negatively charged non-complexed NAs or due to the tilting of chains or both. This lamellar repeat distance is equivalent to the Gd-DTPA-BO bulk lamellar crystals.27 In addition, the scattering pattern had low intensity as the material started to precipitate out from the dispersion. The visual observation of the dispersed NAs by cryo-TEM confirmed the formation of unilamellar and predominantly multilamellar liposomal NAs for DTPA-BO dispersed particles and occasional solid lamellar crystalline particles, as shown in Fig. 5a. Gd-DTPA-BO at a ratio of 0.5:1 exhibited mostly unilamellar liposomes and occasional multilamellar liposomes as shown in Fig. 5b. The morphology of Gd-DTPA-BO at a ratio of 1:1 revealed the formation of multilayered nanospheres (Fig. 5c), consistent with the SAXS results, showing that these layers are built from crystalline Gd-DTPA-BO (Fig. 4a). The dispersion was not stable over a long time, likely due to the neutral charges of the crystalline nanospheres.
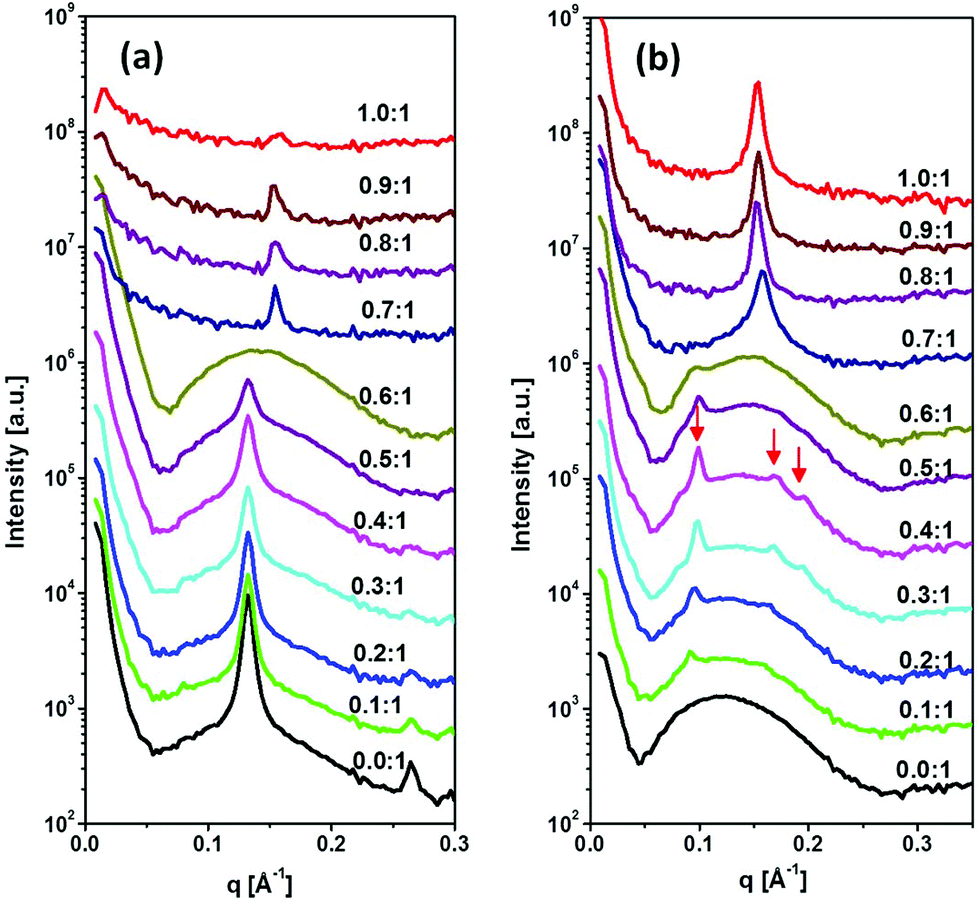 | ||
| Fig. 4 SAXS patterns of colloidal NAs before and after incremental addition of Gd(III) into DTPA-BO (a) and DTPA-BP (b). Red arrows show peaks in the ratio of 1:√3:√4, consistent with an inverse hexagonal structure (hexosomes). | ||
 | ||
| Fig. 5 Cryo-TEM micrographs of (a) DTPA-BO showing unilamellar and multilamellar liposomal NAs; (b) Gd-DTPA-BO (0.5:1) contained the structure of the liposomes upon complexation with Gd(III), albeit transformed to larger NAs; and (c) Gd-DTPA-BO (1:1) transformed to multilayered nanospheres; scale bars are 200 nm in (a) and (b) and 100 nm in (c). | ||
The SAXS diffraction patterns of DTPA-BP dispersed particles and their Gd-complexed nanoassemblies with an incremental increase in the molar ratio of Gd(III) to DTPA-BP from 0.1:1 to 1:1 are shown in Fig. 4b. The diffraction pattern of DTPA-BP is consistent with liposomal particles, exhibiting a broad peak at 0.11 Å−1. The incremental increase in Gd(III) concentration resulted in the emergence of a set of diffraction peaks in the ratio of 1:√3:√4, consistent with an inverse hexagonal structure (hexosomes). Upon dilution with water, a more prominent second set of diffraction peaks in the ratio of √2:√4:√6 appeared, consistent with an inverse bicontinuous cubic structure (cubosomes) with inherent Im3m symmetry (ESI, Fig. 3S†). The lattice parameters of the hexosomes and cubosomes at 0.5:1 molar ratio of Gd-DTPA-BP were 72.7 ± 0.5 Å and 128.3 ±0.5 Å, respectively. Both sets of scattering peaks were superimposed on a broad peak, indicative of the co-existence of liposomal particles. Any incremental increase of Gd(III) ions to DTPA-BP above 0.5:1 molar ratio resulted in the emergence of a new set of lamellar peaks at a higher reciprocal spacing at 0.158 Å−1, with a lamellar spacing of 39.7 ± 0.5 Å. The emergence of the closely packed lamellar structures is similar to our findings with Gd-DTPA-BO noted earlier. The lamellar d-spacing of these NAs was slightly increased to 41.5 ± 0.5 Å at the highest ratio of 1:1 Gd-DTPA-BP, consistent with our finding of the slightly hydrated crystals of Gd-DTPA-BP in excess water as previously reported.27
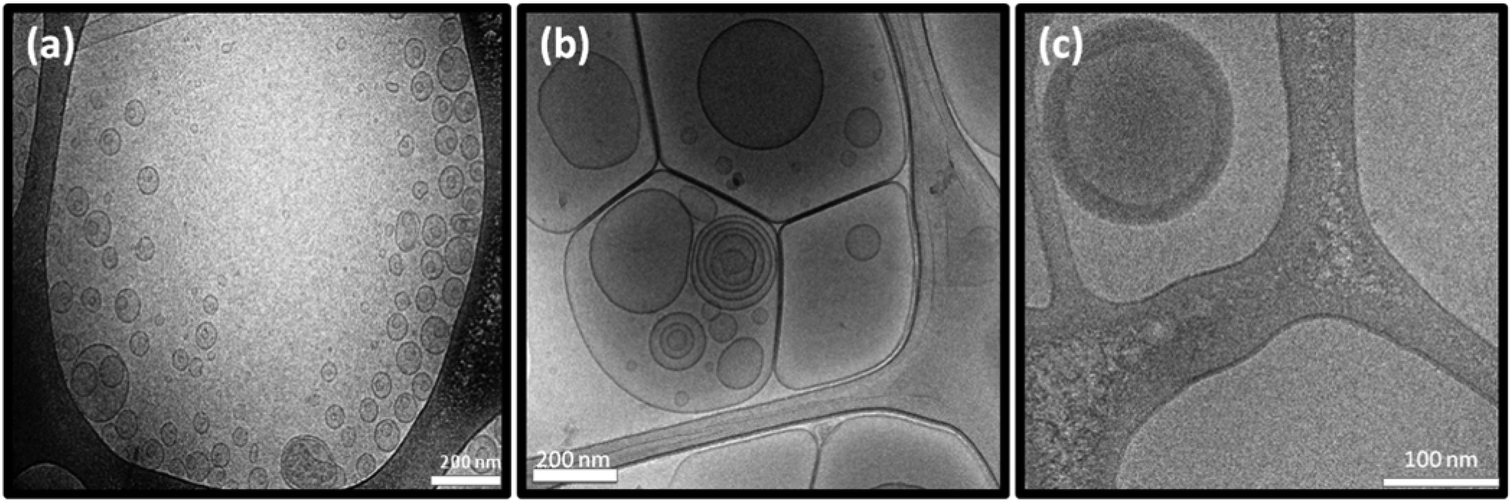
Cryo-TEM images of the DTPA-BP dispersion displayed mostly liposomal particles (Fig. 6a), consistent with our observation from SAXS analysis. At 0.5:1 molar ratio of Gd(III) to DTPA, the images showed mostly hexosomes co-existing with some liposomal particles (Fig. 6b). The fast Fourier transform (FFT) measurement by ImageJ (insets in Fig. 6b) showed internal hexagonal symmetry. This dispersion transformed to some cubosomes upon dilution of the dispersed colloidal NAs with water (Fig. 6c). The FFT by ImageJ also (insets in Fig. 6c) confirmed the internal cubic symmetry. It should be noted that this might be an attribute of a less ionic solution due to a drop in concentration of Na-acetate. These morphologies are consistent with those found by SAXS results on diluted dispersions (ESI, Fig. 3S†). Upon further addition of Gd(III) and at a 1:1 molar ratio, Gd-DTPA-BP NAs transformed to multilayered nanospheres (Fig. 6d), similar to our findings with Gd-DTPA-BO and consistent with SAXS analysis (Fig. 4b).
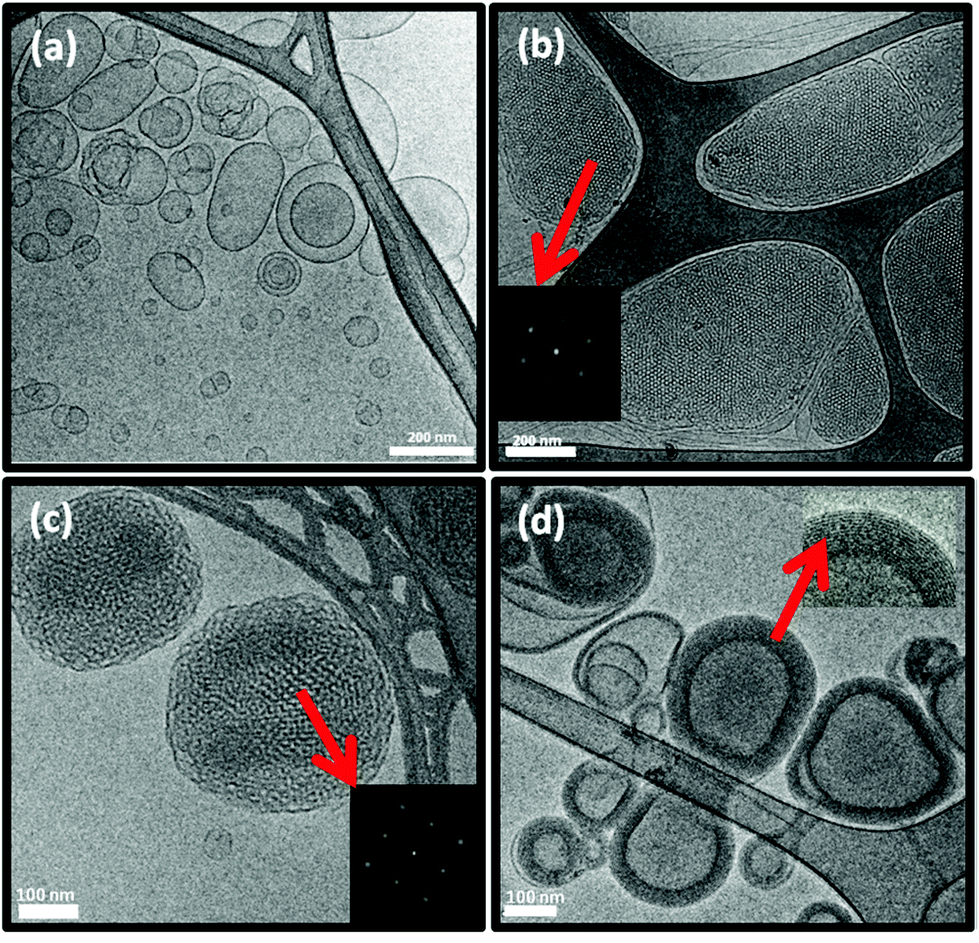 | ||
| Fig. 6 Cryo-TEM micrograph of (a) DTPA-BP showing liposomal particles; (b) and (c) Gd-DTPA-BP (0.5:1) showing mostly hexagonal and cubosomal particles co-existing with liposomal particles; (d) Gd-DTPA-BP (1:1) transformed to mutilayered nanosphere. Scale bars are 200 nm in (a) and (b) and 100 nm in (c) and (d). Insets in (b) and (c) show the hexagonal and cubic symmetries, whereas the inset in (d) shows the multiwalls of the slightly hydrated multilayered nanosphere. | ||
3.2. Relaxivities of the dispersed Gd complexed NAs
The normalized longitudinal and transverse relaxivity values (r1 and r2) of water protons in the solution containing dispersed particles of Gd-complexed DTPA chelating amphiphiles were examined at 37 °C using a 23.4 MHz low field NMR. Typical plots used to obtain longitudinal (r1) and transverse (r2) for [Gd-DTPA-BP·(H2O)] and the linearity of the relaxivity values versus concentration are exemplified in the ESI, Fig. 4S.† We note that all other NAs had a similar linear trend, suggesting that the micellar Gd-DTPA-MO has been measured above its critical micelle concentration. The free Gd concentration tested by xylenol orange and measuring the absorbance ratio at 573/433 nm proved to be less than 0.1% in all colloidal NA dispersions. The relaxivity values were then normalized to the Gd content determined by ICP-OES.Analogous to Magnevist [Gd-DTPA·(H2O)]−2, the Gd-DTPA amphiphiles, either possessing one or two oleyl and phytanyl chains, are likely to have one inner sphere water molecule coordinated to Gd(III). This has been shown by X-ray crystallography of Gd-DTPA derivatives, exemplified by bis ethylamide.31 Gd(III) is a nine coordinate metal ion with sites occupied by three amines nitrogen, one or two esters oxygen and four or three carboxylates oxygen in mono- or bis-amphiphiles respectively. The ninth coordination site, therefore, can be occupied by a water molecule in an aqueous solution.31Fig. 7 shows the normalized longitudinal and transverse relaxivity values of all Gd–NAs at 37 °C, compared with the values of magnevist [Gd-DTPA·(H2O)]−2 solution.32 These values are also tabulated in Table 1 with the size and polydispersity values for the different NAs.
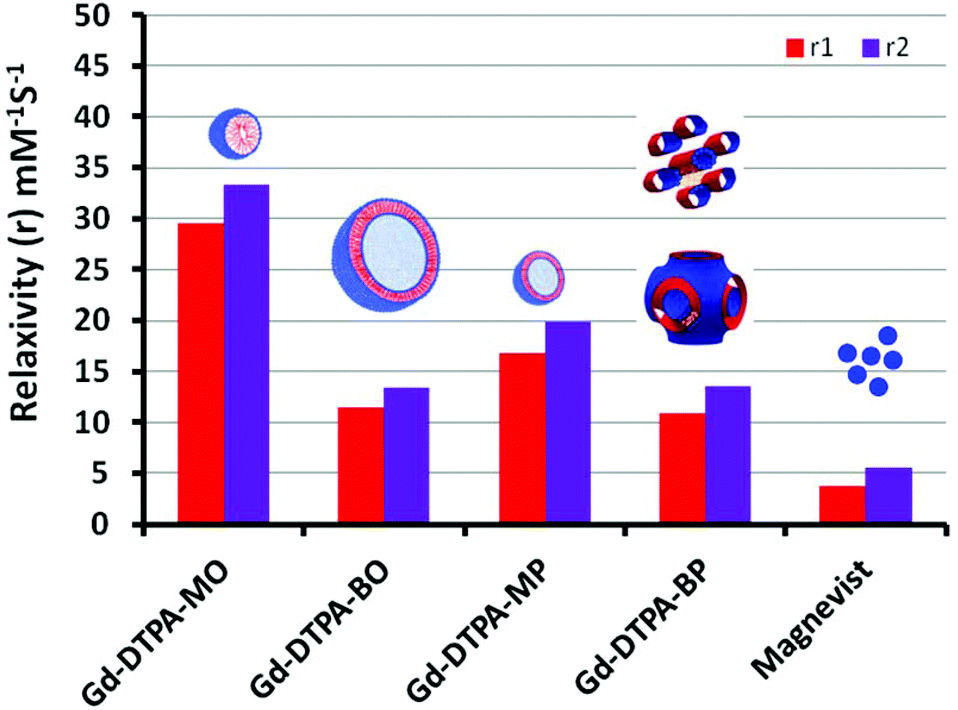 | ||
| Fig. 7 Relaxivity values of Gd-DTPA NAs measured at 0.54 T (23 MHZ) and 37 °C. | ||
All the Gd-NAs demonstrated ∼3–8 times the normalized longitudinal relaxivity of Magnevist at 23.4 MHz. The higher relaxivity values over the low molecular weight and non-self-assembled Gd-DTPA in Magnevist are most likely due to the slow rotational motion or tumbling rate of the Gd(III) within the self-assembled aggregates of various NAs. The relaxivity for Gd-DTPA-MO was the highest, followed by Gd-DTPA-MP, Gd-DTPA-BO and Gd-DTPA-BP. We note that due to the precipitation of [Gd-DTPA-BO·(H2O)]0 and [Gd-DTPA-BP·(H2O)]0 with neutral charges at 1:1 molar ratios, the relaxivity measurements shown here are based on complexed samples at 0.5:1 molar ratios of Gd to the respective amphiphiles. The ESI/MS obtained on all NAs revealed that Gd ions were coordinated to only one amphiphile molecule at all molar ratios. It is also notable that relaxivity measurements were conducted at 37 °C, at which temperature the SAXS scattering pattern of Gd-DTPA-BP NAs showed mostly hexosomal particles co-existing with some liposomal particles (data not shown). The results therefore imply that the hexosomal nanostructures with highly ordered water channels are not specifically advantageous to increase the normalized relaxivity values of the chelated Gd, compared to those in liposomal Gd-DTPA-BO. On the other hand, the improved relaxivity of Gd-DTPA-MO over MP NAs may suggest that spherical micelles may provide a better environment for relaxivity of Gd over those chelated to the bilayers of small liposomes of Gd-DTPA-MP, and that small liposomes of Gd-DTPA-MP may have an advantage over larger liposomes (Gd-DTPA-BO). Similar trends have been observed for small and large liposomal particles containing Gd-DTPA encapsulated in their interior cavity.33 We observed similar results for Mn-EDTA chelating amphiphiles where small unilamellar liposomes demonstrated higher normalized relaxivity values over NAs of larger particles.14,15
It is also noteworthy that all Gd-NAs examined here had r2/r1 values in the range of 1.13–1.24, representing T1-weighted (positive) CEAs. We note that despite the higher relaxivity per Gd of the spherical micelles and small unilamellar liposomes, they comprise much less Gd per NA, whereas the numbers of Gd in larger liposomes or hexosomes and cubosomes are likely to be ∼10- to 100-fold higher. Negatively charged contrast agents have shown some hemolytic properties due to their high osmolality.34 Further, the in vitro cytotoxicity of these NAs shown in the following sections demonstrated that Gd-bis conjugates with neutral charges are less toxic in comparison with their mono-counterparts with negative charges. At this stage, it is not clear whether this is due to negatively charged particles or due to smaller sizes of the mono conjugate NAs and a better uptake into cells and therefore higher toxicity.
3.3. Bioconjugation of antibody to the surfaces of the NAs and antigen recognition
Anti-FLAG Fab′ conjugated NAs and control NAs based on DTPA-BP and maleimide-EDTA-BP, containing 49% Gd and 1% Eu, were synthesised as described in the Materials section and shown schematically in Fig. 8. Eu(III) was incorporated into the NAs to provide luminescence property and evaluation by enzyme-linked immunosorbent assay (ELISA).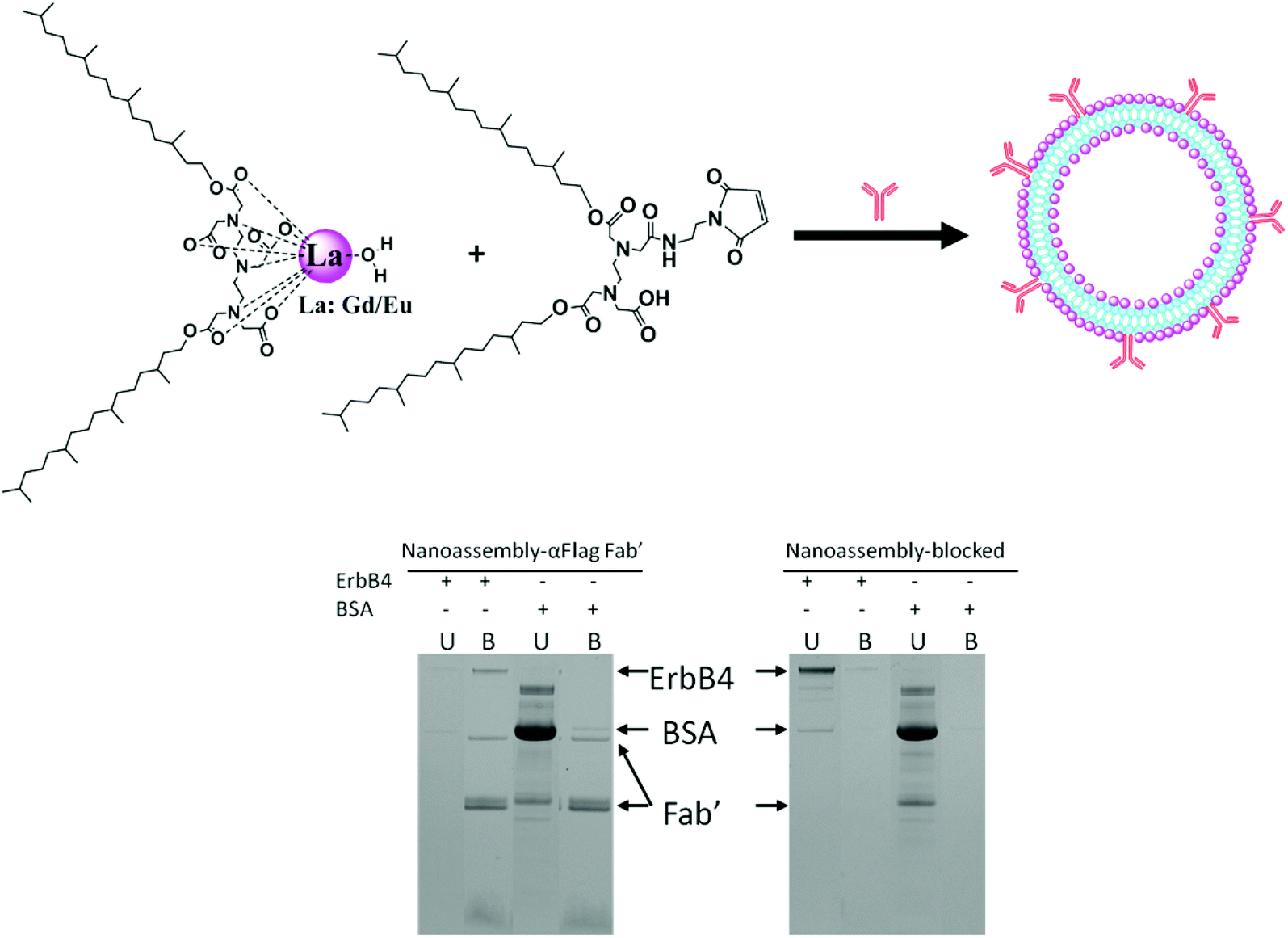 | ||
| Fig. 8 Formulation of NAs containing Gd/Eu chelated metal ions and maleimide-amphiphiles, and subsequent bioconjugation of anti-FLAG Fab′ antibody to NAs. SDS-PAGE demonstrates that FLAG-ErbB4 binds to the conjugated liposomal NAs in the left band (left gel) but not to the mercaptoethanol quenched NA in the left band (right gel), while BSA did not bind to any of the NA; U: unbound solution and B: bio-conjugated protein-liposome. | ||
Initially, 96 well plates, coated with FLAG-ErbB4 or the control BSA, were used to assess the binding recognition of the NAs bio-conjugated to anti-FLAG Fab′. We encountered problems with non-specific binding of the NAs due to the strong interaction of the liposomes with the surface of the 96 well plates, even in the absence of anti-FLAG Fab′. This led us to use an SDS-PAGE assay devoid of non-specific binding. The results are also shown in Fig. 8.
Gd–Eu NAs with the functional maleimide groups formed liposomal particles as shown by their cryo-TEM images in the ESI, Fig. 5S.† The dispersion was prepared without the use of stabilizing agents in order to expose the maleimide groups onto the bilayer membrane. Anti-FLAG Fab′ was bioconjugated to maleimide groups as described in the Experimental section. The control NAs containing mercaptoethanol-quenched maleimide groups were incubated with FLAG-ErbB4 or with BSA (control). Unbound protein (U) was removed and liposomes with bound protein (B) were washed and then dissolved in SDS. Bands corresponding to reduced and non-reduced anti-FLAG Fab′ were observed in the bioconjugated liposomes. FLAG-ErbB4 was found to bind to the conjugated liposomes, but not to the mercaptoethanol-quenched control liposomes, while BSA bound to neither the bioconjugated nor the quenched liposomes. This result indicated that anti-FLAG Fab′ was bound in the right conformation and was exposed onto the surfaces of the liposomes, available for specific recognition by FLAG-ErbB4. This result revealed that the maleimide groups allowed for effective bioconjugation of the antibody in the right orientation and spacing and enabled effective binding to antigen.
3.4. In vitro cytotoxicity of the NAs
The cytotoxicity testing of the four Gd NAs was based on the International Organisation for Standardization Guidelines as stated in ISO 10993-5 (Biological evaluation of medical devices). This was done against two types of cells: firstly, L929 cells (mouse fibroblast cell line), as a model cell line with high sensitivity, and secondly, MCF7 cells (human breast adenocarcinoma cell line), a possible target cell population, to determine whether a similar sensitivity level of cytotoxicity was seen. A level of 80% cell attachment was deemed acceptable and below that level was considered cytotoxic. The morphology of the L929 cells incubated with NAs after 24 h incubation is shown in Fig. 9-a, and the respective cell viability in Fig. 9-b. It should be noted that the concentrations are in respect to the Gd in the NAs. It can be seen that the cells are well spread and that there are no distinct morphological changes after 24 h incubation for 50 μM concentration of the Gd-DTPA-MP, Gd-DTPA-BO, Gd-DTPA-BP in Fig. 9-a2, -a3 and -a4. Gd-DTPA-MO showed relatively higher toxicity at 50 μM, but showed no shrinkage or change to a rounded morphology at 25 μM (Fig. 9-a1). The control samples 5% PBS or 5% DMSO cell morphology are shown for comparison in Fig. 9-a5 and -a6 respectively. For L929 cells, the cytotoxic concentration of Gd-DTPA-MO was above 12.5 μM, whereas this concentration was above 50 μM for Gd-DTPA-MP and Gd-DTPA-BO and 25 μM for GD-DTPA-BP (Fig. 9-b). The MCF7 cells showed more tolerance and higher viability to various Gd-NAs. The level of 80% attachment of the MCF7 cells against different Gd-NAs showed that the Gd-DTPA-MO was cytotoxic at concentrations above 25 μM, whereas Gd-DTPA-MP had some toxicity above 50 μM concentration (Fig. 9-c). Both Gd-DTPA-BO and Gd-DTPA-BP essentially did not show cytotoxicity at the relevant toxicological range tested here.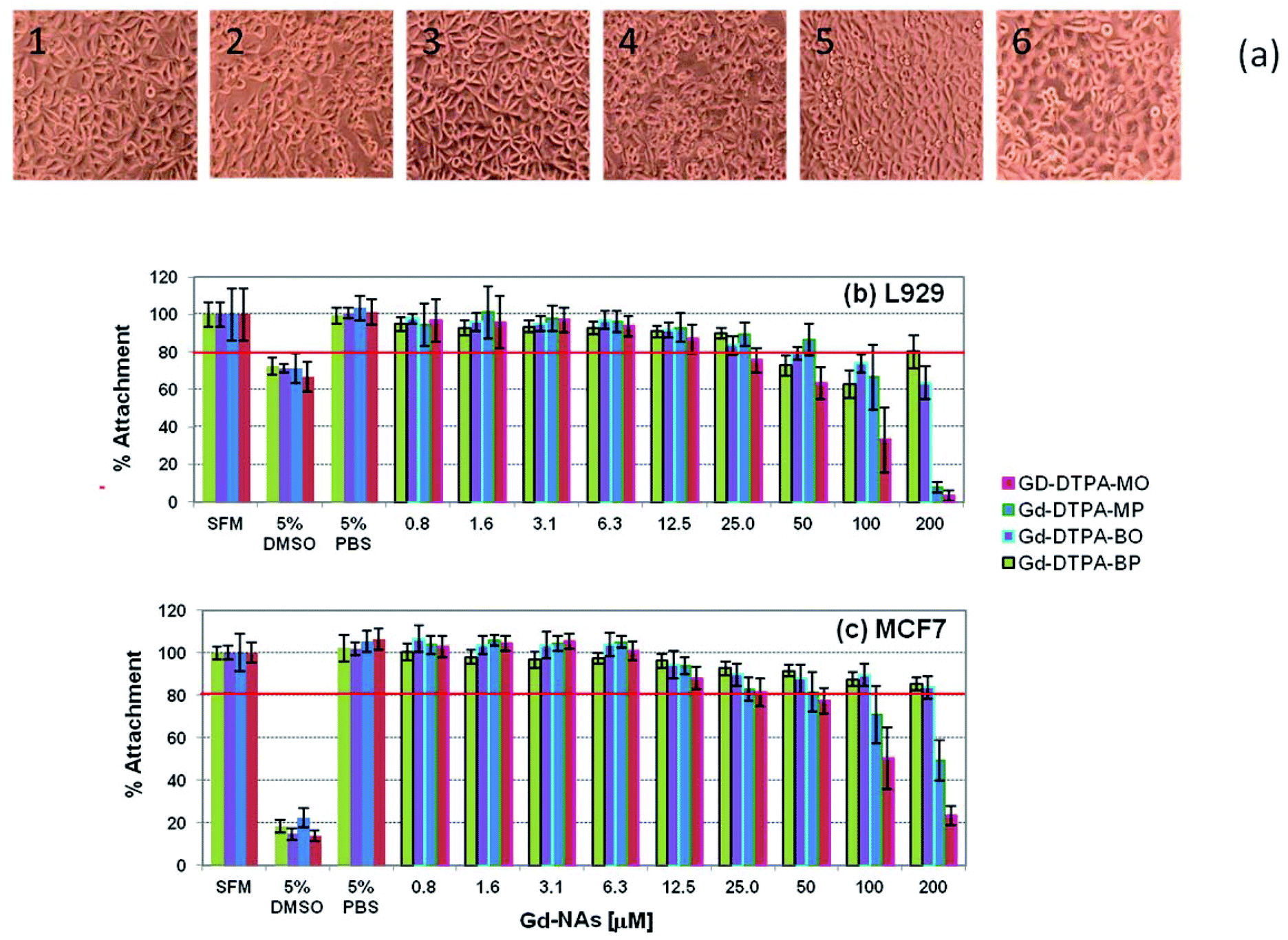 | ||
| Fig. 9 Morphology of the L929 cells after incubation with different Gd-NAs for 24 h; (a1) Gd-DTPA-MO (25 μM), (a2) Gd-DTPA-MP (50 μM), (a3) Gd-DTPA-BO (50 μM), (a4) Gd-DTPA-BP (50 μM), (a5) control PBS 5%, (a6) 5% DMSO; cell viability of L929 (b) and MCF7 (c) against different Gd NAs and the control samples at various concentrations. | ||
3.5. Cell uptake of fluorescent NAs and internalization into cells
To determine whether the Gd-NAs were able to enter the cells, we demonstrate here our results with a NA dispersion composed of Gd-DMO–DBO 1:1 (99%) and 1% rhodamine-labelled lipid. The experiment was conducted on both cancer cells and normal cells in order to determine whether a difference was present in the uptake of these NAs. The cell uptake was first assessed against a melanoma cancer cell line (SK-Mel 28), as shown in Fig. 10. The uptake was optimized by varying the concentration of the NA from 100, 50 to 25 μM. The 50 μM images were specifically chosen since it was non-toxic to the cells and we could observe a high concentration of the fluorescent NAs inside the cytoplasm. The images shown here were captured after 4 h incubation in serum free medium. The uptake of NAs by cells in 10% serum cell culture medium also showed considerable internalization of the NAs, but slightly less than serum free media (data not shown). We also examined the uptake of NAs by human lung fibroblasts, shown in the ESI, Fig. 6S.† The results showed that the Gd-NAs were also internalized into normal human fibroblast cells. It should be noted that the NAs used in this assay were not equipped with a targeting antibody and therefore are passively endocytosed into both normal and cancerous cells. This preliminary work underscores the importance of developing targeted NAs for selective cell uptake. Further work using cancer specific targeting molecules is ongoing and will be reported in the near future.
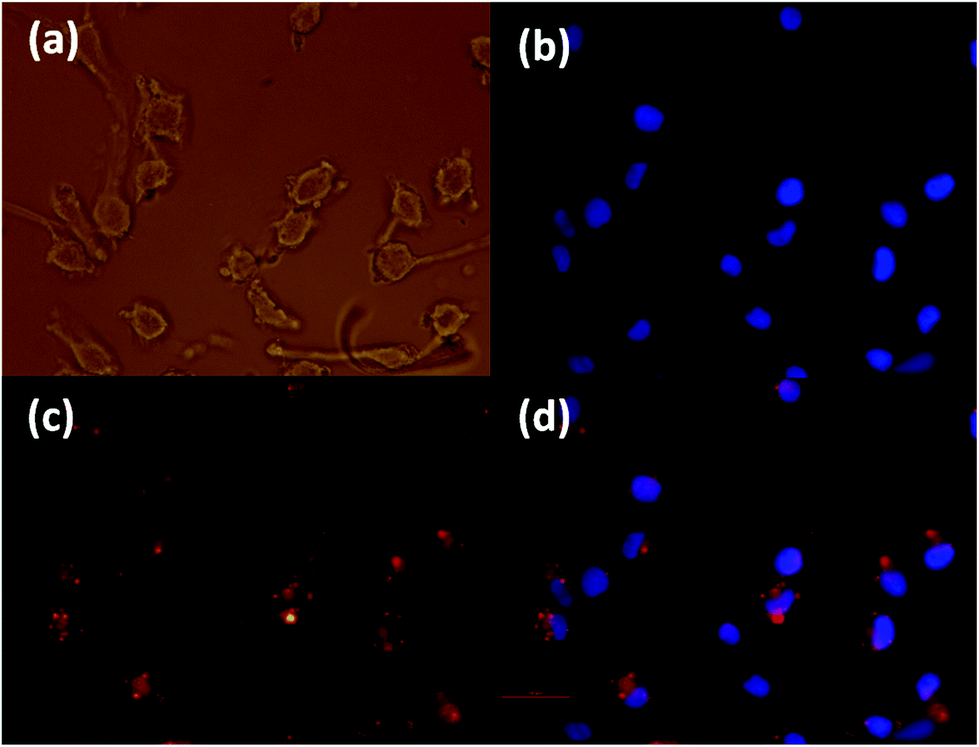 | ||
| Fig. 10 Fluorescence microscopy images (×40) of SK-Mel 28 (human melanoma cancer cells) uptake of Rhodamine-labeled (Gd-DTPA-MO/Gd-DTPA-BO) NA at 1:1 molar ratio after 4 h incubation with serum free medium; morphology of the cells (a), DAPI staining of the nuclei (b), Rhodamine-labeled Gd-NA internalized into cells’ cytoplasm (c), and images b and c overlapped (d). Scale bar is 50 μm. | ||
4. Conclusions
We have presented the in situ synthesis and formulation of colloidal Gd-DTPA NAs containing unsaturated oleyl and branched phytanyl chain(s) and their application as MRI CEAs. The in situ complexation markedly changed the NA structures of the precursor amphiphiles. Whilst Gd-DTPA-MO formed mostly micelles, the mono-phytanyl counterpart created mostly small unilamellar liposomal NAs. The Gd-bis conjugates formed various higher ordered nanostructures of liposomes, hexosomes and cubosomes at ratios of Gd to DTPA-amphiphiles less than 1 and transformed to multilayered nanospheres at 1:1 ratios.
This work clearly demonstrates that the judicious design of the nature and number of hydrophobic chains and the fine tuning of the Gd content can markedly alter the nanostructure and size of the NAs, both of which are important factors for the development of highly efficient contrast agents. All of these NAs exhibited 3- to 8-fold increased relaxivity over the low molecular weight commercial Gd-DTPA (Magnevist), suggesting that these liquid crystalline NAs have high prospectivity as novel CEAs for MRI.
The ease of complexation and preparation, the low toxicity, the possibility of concurrent incorporation of several metal ions (i.e. radioisotope metal ions and luminescent metal ions), high payloads of metal ions within the colloidal particles composed solely of metal ion chelated amphiphiles, combined with the ease of incorporation of targeting molecules into these NAs suggest the high promise of this type of amphiphile self-assembly material as advanced CEAs for cellular and molecular multimodal imaging systems, as well as nanomaterials for Gd-neutron capture therapy.
Acknowledgements
The authors acknowledge the Australian Synchrotron for awarding user time to conduct SAXS experimental work and Drs Nigel Kirby, Stephen Mudie and Adrian Hawley for their assistance in conducting the experimental work. We would like to thank Dr Eric Hanssen from the Electron Microscope Facility at the University of Melbourne, Bio21 Institute for access to the TF30 TEM. We thank Mr Chris Sheedy (CSIRO, Process Science and Engineering) for performing the ICP-OES analyses. CJD acknowledges the receipt of an Australian Research Council Federation Fellowship.References
- V. P. Torchilin, Nat. Rev. Drug Discov., 2005, 4, 145–160 CrossRef CAS PubMed.
- X. Mulet, B. J. Boyd and C. J. Drummond, J. Colloid Interface Sci., 2013, 393, 1–20 CrossRef CAS PubMed.
- C. J. Drummond and C. Fong, Curr. Opin. Colloid Interface Sci., 1999, 4, 449–456 CrossRef CAS.
- A. Samad, Y. Sultana and M. Aqil, Curr. Drug Delivery, 2007, 4, 297–305 CrossRef CAS.
- A. N. Lukyanov, T. A. Elbayoumi, A. R. Chakilam and V. P. Torchilin, J. Controlled Release, 2004, 100, 135–144 CrossRef CAS PubMed.
- J. Barauskas, M. Johnsson and F. Tiberg, Nano Lett., 2005, 5, 1615–1619 CrossRef CAS PubMed.
- B. J. Boyd, D. V. Whittaker, S. M. Khoo and G. Davey, Int. J. Pharm., 2006, 309, 218–226 CrossRef CAS PubMed.
- A. Yaghmur and O. Glatter, Adv. Colloid Interface Sci., 2009, 147–48, 333–342 CrossRef PubMed.
- S. M. Sagnella, X. Gong, M. J. Moghaddam, C. E. Conn, K. Kimpton, L. J. Waddington, I. Krodkiewska and C. J. Drummond, Nanoscale, 2011, 3, 919–924 RSC.
- K. Larsson, Curr. Opin. Colloid Interface Sci., 2000, 5, 64–69 CrossRef CAS.
- M. Johnsson, J. Barauskas, A. Norlin and F. Tiberg, J. Nanosci. Nanotechnol., 2006, 6, 3017–3024 CrossRef CAS PubMed.
- L. Lopes, D. Ferreira, D. de Paula, M. Garcia, J. Thomazini, M. Fantini and M. Bentley, Pharm. Res., 2006, 23, 1332–1342 CrossRef CAS PubMed.
- T. Kaasgaard and C. J. Drummond, Phys. Chem. Chem. Phys., 2006, 8, 4957–4975 RSC.
- M. J. Moghaddam, L. de Campo, L. J. Waddington and C. J. Drummond, Soft Matter, 2010, 6, 5915–5929 RSC.
- M. J. Moghaddam, L. de Campo, L. J. Waddington, A. Weerawardena, N. Kirby and C. J. Drummond, Soft Matter, 2011, 7, 10994–11005 RSC.
- G. Z. Liu, C. E. Conn and C. J. Drummond, J. Phys. Chem. B, 2009, 113, 15949–15959 CrossRef CAS PubMed.
- C. E. Conn, V. Panchagnula, A. Weerawardena, L. J. Waddington, D. F. Kennedy and C. J. Drummond, Langmuir, 2010, 26, 6240–6249 CrossRef CAS PubMed.
- B. W. Muir, D. P. Acharya, D. F. Kennedy, X. Mulet, R. A. Evans, S. M. Pereira, K. L. Wark, B. J. Boyd, T.-H. Nguyen, T. M. Hinton, L. J. Waddington, N. Kirby, D. K. Wright, H. X. Wang, G. F. Egan and B. A. Moffat, Biomaterials, 2012, 33, 2723–2733 CrossRef CAS PubMed.
- D. P. Acharya, B. A. Moffat, A. Polyzos, L. Waddington, G. Coia, D. K. Wright, H. X. Wang, G. F. Egan, B. W. Muir and P. G. Hartley, RSC Adv., 2012, 2, 6655–6662 RSC.
- M. Bottrill, L. K. Nicholas and N. J. Long, Chem. Soc. Rev., 2006, 35, 557–571 RSC.
- R. Uppal and P. Caravan, Fut. Med. Chem., 2010, 2, 451–470 CrossRef CAS PubMed.
- S. Laurent, D. Forge, M. Port, A. Roch, C. Robic, L. Vander Elst and R. N. Muller, Chem. Rev., 2008, 108, 2064–2110 CrossRef CAS PubMed.
- P. Caravan, J. J. Ellison, T. J. McMurry and R. B. Lauffer, Chem. Rev., 1999, 99, 2293 CrossRef CAS PubMed.
- S. Aime, S. G. Crich, E. Gianolio, G. B. Giovenzana, L. Tei and E. Terreno, Coord. Chem. Rev., 2006, 250, 1562–1579 CrossRef CAS PubMed.
- G. De Stasio, P. Casalbore, R. Pallini, B. Gilbert, F. Sanità, M. T. Ciotti, G. Rosi, A. Festinesi, L. M. Larocca, A. Rinelli, D. Perret, D. W. Mogk, P. Perfetti, M. P. Mehta and D. Mercanti, Cancer Res., 2001, 61, 4272–4277 CAS.
- P. Sharma, S. C. Brown, G. Walter, S. Santra, E. Scott, H. Ichikawa, Y. Fukumori and B. M. Moudgil, Adv. Powder Technol., 2007, 18, 663–698 CrossRef CAS PubMed.
- M. J. Moghaddam, L. de Campo, N. Kirby and C. J. Drummond, Phys. Chem. Chem. Phys., 2012, 14, 12854–12862 RSC.
- A. Barge, G. Cravotto, E. Gianolio and F. Fedeli, Contrast Media Mol. Imaging, 2006, 1, 184–188 CrossRef PubMed.
- B. L. Brizzard, R. G. Chubet and D. L. Vizard, Biotechniques, 1994, 16, 730–735 CAS.
- E. J. Koziolek, J. E. Donoghue, J. D. Bentley, G. Lovrecz, O. Dolezal, C. W. Ward, J. Rothacker, E. C. Nice, A. W. Burgess, M. Hafner, T. G. Johns and T. E. Adams, Growth Factors, 2012, 30, 310–319 CrossRef CAS PubMed.
- M. S. Konings, W. C. Dow, D. B. Love, K. N. Raymond, S. C. Quay and S. M. Rocklage, Inorg. Chem., 1990, 29, 1488–1491 CrossRef CAS.
- M. F. Wendland, M. Saeed, K. Lauerma, N. Derugin, J. Mintorovitch, F. M. Cavagna and C. B. Higgins, Magn. Reson. Med., 1997, 37, 448–456 CrossRef CAS.
- C. Tilcock, E. Unger, P. Cullis and P. Macdougall, Radiology, 1989, 171, 77–80 CAS.
- P. L. Anelli, L. Lattuada, V. Lorusso, M. Schneider, H. Tournier and F. Uggeri, Magn. Reson. Mater. Phys. Biol. Med., 2001, 12, 114–120 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c3bm60235d |
| This journal is © The Royal Society of Chemistry 2014 |