
Metal-free covalent organic frameworks containing precise heteroatoms for electrocatalytic oxygen reduction reaction†
Jiali
Li
a,
Ji
Jia
a,
Jinquan
Suo
a,
Cuiyan
Li
a,
Zhiwei
Wang
b,
Hui
Li
*a,
Valentin
Valtchev
 c,
Shilun
Qiu
a,
Xiaoming
Liu
*a and
Qianrong
Fang
*a
c,
Shilun
Qiu
a,
Xiaoming
Liu
*a and
Qianrong
Fang
*a
aCollege of Chemistry, Jilin University, Changchun 130012, China. E-mail: postlh@jlu.edu.cn; xm_liu@jlu.edu.cn; qrfang@jlu.edu.cn
bInstitute of Theoretical Chemistry, College of Chemistry, Jilin University, Changchun 130023, China
cQingdao Institute of Bioenergy and Bioprocess Technology, Chinese Academy of Sciences, Normandie Univ, 6 Marechal Juin, 14050 Caen, France
First published on 31st July 2023
Abstract
Covalent organic frameworks (COFs) have been used for electrocatalytic oxygen reduction reaction (ORR) due to their structural tunability, well-defined electroactive sites, and easy introduction of heteroatoms. Researchers have incorporated heteroatoms into COF architectures to enhance their performance by tuning the electronic environment of oxygen intermediates. However, only a few heteroatoms (O, S, N, P) have been introduced into the backbone of COFs, and the effects of different types of heteroatoms on the electronic structure of COFs have not been specifically investigated. Furthermore, the development of COF electrocatalysts with highly active ORR is still at an early stage. Herein, we report a series of metal-free benzotrithiophene-based COFs containing various heteroatoms (Se, S or O), BTT-COFs (named JUC-616, JUC-617, and JUC-618, respectively), and explore their ORR catalytic activity. Remarkably, JUC-616 involving precise Se atoms exhibits a half-wave potential of 0.78 V and a high turnover frequency (TOF) of 0.0062 s−1 at 0.75 V vs. the reversible hydrogen electrode (RHE), which is the best among the metal- and pyrolysis-free COF-based electrocatalysts reported so far. Thus, this work shows the promising potential of functionalized COFs with precise heteroatoms for electrocatalysis.
Introduction
The oxygen reduction reaction (ORR) has attracted a lot of attention over the last decade as it is a key process for sustainable and green energy storage and conversion technologies such as fuel cells1 and rechargeable metal–air batteries.2,3 Nevertheless, an efficient ORR process requires robust catalysts to overcome the inherently sluggish kinetics of oxygen activation and accelerate the reaction rate.4,5 Nowadays, platinum (Pt)-based nanomaterials are the most efficient ORR catalysts, but problems such as high price, poor durability, and small Pt reserves hinder their large-scale commercial applications.6,7 Therefore, it is of great significance to develop metal-free electrocatalysts with excellent ORR performance as an alternative to Pt-containing materials. At present, heteroatoms have been shown to be a promising strategy for preparing metal-free carbon-based electrocatalysts.8 By integrating different heteroatoms into different nanostructures of carbon, various metal-free heteroatom-doped carbon composites have been developed, ranging from single-atom doping (N, P, B, S, etc.) to multi-heteroatom doping (N/P/S or N/S/B, etc.).9 However, traditional heteroatom-doped carbon materials, such as heteroatom-doped carbon nanotubes, reduced graphene oxides, mesoporous carbon, etc., usually have random structures with high uncertainty,10 and thus the way in which dopants promote the ORR by tuning the electronic structure of effective defects is not well understood.Covalent organic frameworks (COFs) are a new class of crystalline porous polymeric materials linked by strong covalent bonds between nodes and linkers.11,12 Since the first report by Yaghi's group in 2005,13 COFs have attracted great scientific interest for applications in various fields such as catalysis,14–18 energy storage,19–21 optoelectronics,22–24etc.25–29 owing to their large acceptable surface area, high crystallinity and structural tunability. Compared with traditional heteroatom-doped materials, COFs retain their original skeleton and crystal structure, and have fast ion transport channels and well-defined electroactive sites. More importantly, they enable precise tuning of environmental features around active sites, such as electronic properties and coordination states. The large number of heteroatoms in COFs leads to a redistribution of charges, while some positively charged atoms are more favourable for the adsorption of O2, acting as reaction centres.30 In addition, the activity, selectivity and stability of catalysts depend on the scale, heteroatom content, doping location of active sites, and porous structure, and COFs allow precise control of these elements simultaneously due to their fixed backbone structures, which facilitates in-depth exploration of the relationship between catalyst structure and catalytic performance.31 Over the past few years, preliminary explorations have shown that two-dimensional (2D) COFs can serve as promising platforms for electrocatalytic reactions.32–34 Generally, a post-pyrolysis strategy is often employed to improve the catalytic performance of COFs for the ORR.35,36 However, the high-temperature treatment process usually inevitably leads to energy consumption and uncontrollable structural changes, resulting in poorly defined active sites, which limits a deep understanding of the ORR mechanism.37 To avoid the hassle of pyrolysis, some pyrolysis-free COFs were recently explored and showed precise skeleton structures and accurate active sites.38–41
In 2020, we proposed for the first time to introduce S element into the framework of non-pyrolyzable COFs to precisely modulate the performance of the ORR.42 So far, although there have been many studies on the catalytic performance of heteroatom-containing COFs for the ORR, the effect of doping various heteroatoms with different electronegativities on the electronic structure of COFs has rarely been reported, and the development of COF electrocatalysts with highly active ORR is still in its infancy.43 Multi-heteroatom doping is a more effective and versatile approach than single dopants,44 so it is crucial to study the effect of COFs containing multiple heteroatoms on the ORR performance. However, it is common to investigate heteroatoms such as O, S, N, and P in the framework of COFs, and further exploration of atoms with higher activity is needed to investigate more active catalysts. In 2021, Han's research group introduced Se atoms to prepare Se single-atom catalysts with excellent catalytic performance, but this study led to incomplete pyrolysis structures, which hindered the precise tuning of atoms.45 Se, a chalcogen element with a larger atom size and high polarizability than S or O, has been shown to increase the electrical transport properties of framework materials.46,47 In addition, Se has a large atomic radius, small ionization energy, and abundant d electrons, which can facilitate charge transfer and increase the catalyst affinity for (O) OH species.48 Combining the unique properties of COF materials such as high atomic utilization, pre-designed structure and easy introduction of various heteroatoms, doping Se into the COF architectures and studying its effect on ORR performance will provide a new direction for the application of COF materials in electrocatalysts.
Herein, we report a series of novel metal-free electrocatalysts with excellent ORR performance by precisely introducing heteroatoms (Se, S, or O) into periodic benzotrithiophene-based COFs (BTT-COFs) via Schiff-base condensation reaction. Moreover, all these COFs possess a uniform porous structure with high surface area and electron donor–acceptor (D–A) characteristics, which can facilitate mass transport, expose a large number of active sites, and improve their electrical conductivity for fast electron transport.49 The influence of different heteroatoms on the electronic structure of COF materials was investigated in detail by calculating the adsorbed-state differential charges and D-band differential charges of the three materials. Remarkably, the Se-containing COF showed excellent electrocatalytic performance with a half-wave potential of 0.78 V and a high turnover frequency (TOF) of 0.0062 s−1 at 0.75 V vs. the reversible hydrogen electrode (RHE), which is better than that of other metal- and pyrolysis-based COF electrocatalysts reported so far.
Results and discussion
Selecting benzo[1,2-b:3,4-b′:5,6-b′′]trithiophene-2,5,8-tricarbaldehyde (BTT) with electron-donating properties as a triangular knot50 and electron-accepting 4,4′-benzoselenadiazole-4,7-diyl-diaminobenzene (BSD), 4,4′-benzothiadiazole-4,7-diyl-diaminobenzene (BTD) or 4,4′-benzoxadiazole-4,7-diyl-diaminobenzene (BXD) as a linear linker,51 three isomorphic BTT-COFs with D–A structural features, JUC-616, JUC-617 and JUC-618, were successfully designed and synthesized by imine reversible condensation reaction under solvothermal conditions, giving brown to orange microcrystalline powders (Scheme 1, the corresponding synthesis details are given in the ESI†). They were insoluble in common organic solvents and water. Fourier transform infrared spectroscopy (ESI Fig. S1†) showed a peak located at ∼1613 cm−1 for all of the BTT-COFs, corresponding to the formation of the imine bond. Additionally, the signals of –NH2 nearly disappeared compared with their corresponding monomers, demonstrating the conversion of the amine to imine groups. The successful formation of the designed COF frameworks and the presence of imine linkage were confirmed by cross-polarization magic angle spinning (CP/MAS) 13C solid-state NMR (ESI, Fig. S2†). Furthermore, X-ray photoelectron spectroscopy (XPS) was performed to verify the molecular structures of BTT-COFs. For example, the survey spectrum showed that the JUC-616 was composed of carbon, nitrogen, sulfur, and selenium (ESI, Fig. S3a†). The XPS spectra of N 1s display two distinct peaks, each exhibiting similar C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N–C binding energies of 398.6, 398.6, and 398.7 eV for the three materials, respectively. This similarity arises from the identical chemical environment of N in the imine bond.52 However, the different electronegativities of O, S, and Se introduce variability in the binding energies of the second peak associated with N. Specifically, the Se–NC peak appears at 399.1 eV in JUC-616, the S–NC peak at 399.3 eV in JUC-617, and the O–NC peak at 400.5 eV in JUC-618.53–55 For the high-resolution S 2p spectrum of JUC-616, the binding energies at 165.3 and 164.1 eV were ascribed to C–S–C in the BTT unit.56 Additionally, the peaks at 57.8 and 57.0 eV were assigned to Se 3d3/2 and Se 3d5/2 in the benzoselenadiazole group.57 Similarly, structural characterization of JUC-617 and JUC-618 was carried out to demonstrate their chemical composition and structure (ESI, Fig. S4†). Thermogravimetric analysis (TGA) revealed good thermal durability of the three COFs up to 400 °C in a N2 atmosphere, with less than 10% weight loss of the materials (ESI, Fig. S5†).
N–C binding energies of 398.6, 398.6, and 398.7 eV for the three materials, respectively. This similarity arises from the identical chemical environment of N in the imine bond.52 However, the different electronegativities of O, S, and Se introduce variability in the binding energies of the second peak associated with N. Specifically, the Se–NC peak appears at 399.1 eV in JUC-616, the S–NC peak at 399.3 eV in JUC-617, and the O–NC peak at 400.5 eV in JUC-618.53–55 For the high-resolution S 2p spectrum of JUC-616, the binding energies at 165.3 and 164.1 eV were ascribed to C–S–C in the BTT unit.56 Additionally, the peaks at 57.8 and 57.0 eV were assigned to Se 3d3/2 and Se 3d5/2 in the benzoselenadiazole group.57 Similarly, structural characterization of JUC-617 and JUC-618 was carried out to demonstrate their chemical composition and structure (ESI, Fig. S4†). Thermogravimetric analysis (TGA) revealed good thermal durability of the three COFs up to 400 °C in a N2 atmosphere, with less than 10% weight loss of the materials (ESI, Fig. S5†).
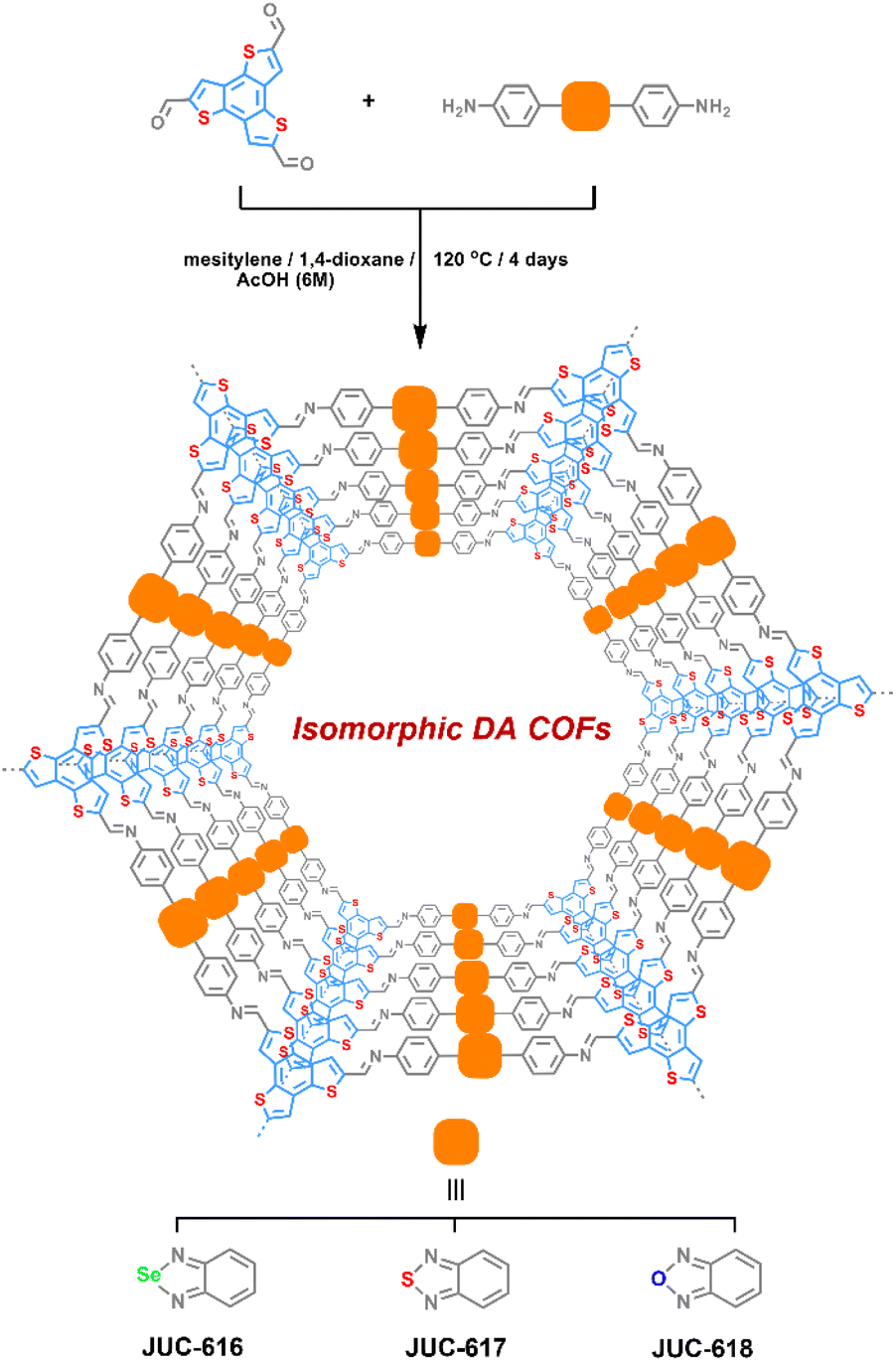 | ||
| Scheme 1 Synthesis and chemical structures of isomorphic BTT-COFs (JUC-616, JUC-617, and JUC-618). | ||
The crystalline structures of the three COFs were evaluated by powder X-ray diffraction (PXRD) analysis combined with theoretical simulations. As displayed in Fig. 1a–c, the experimental PXRD patterns of JUC-616, JUC-617, and JUC-618 showed a strong diffraction peak at 2.29°, which corresponds to the (100) reflection, implying the formation of long-range ordered structures. And other relatively broad peaks at about 3.96, 4.59, 6.05, 7.91, and 25.04° can be assigned to the (110), (200), (210), (220), and (001) planes, respectively. Notably, due to their isomorphic structures, the diffraction peak of the same plane of the three COFs appeared at almost the same position.58 Structural simulations using Materials Studio showed that all synthesized COFs are based on AA stacking of the 2D topology of hcg in space group P![[6 with combining macron]](https://www.rsc.org/images/entities/char_0036_0304.gif) (no. 174, ESI, Tables S1–S3†). The PXRDs of the three COFs based on AB stacking were also calculated, showing a mismatch with the experimental PXRD patterns (ESI, Fig. S6†). Full profile pattern matching (Pawley) refinements were performed from their PXRD patterns. The unit cell parameters were also obtained (a = b = 44.5786 Å and c = 3.5617 Å for JUC-616, a = b = 44.5873 Å and c = 3.5439 Å for JUC-617, a = b = 44.6085 Å and c = 3.5302 Å for JUC-618, and α = β = 90° and γ = 120° for all of the COFs). These results yielded unit cell parameters that closely match with the observed values with an excellent agreement factor (a = b = 44.5787 Å, c = 3.5618 Å, α = β = 90°, and γ = 120° for all of the COFs, Rwp = 3.23% and Rp = 2.45% for JUC-616, Rwp = 3.09% and Rp = 2.19% for JUC-617, and Rwp = 1.57% and Rp = 1.17% for JUC-618, respectively).
(no. 174, ESI, Tables S1–S3†). The PXRDs of the three COFs based on AB stacking were also calculated, showing a mismatch with the experimental PXRD patterns (ESI, Fig. S6†). Full profile pattern matching (Pawley) refinements were performed from their PXRD patterns. The unit cell parameters were also obtained (a = b = 44.5786 Å and c = 3.5617 Å for JUC-616, a = b = 44.5873 Å and c = 3.5439 Å for JUC-617, a = b = 44.6085 Å and c = 3.5302 Å for JUC-618, and α = β = 90° and γ = 120° for all of the COFs). These results yielded unit cell parameters that closely match with the observed values with an excellent agreement factor (a = b = 44.5787 Å, c = 3.5618 Å, α = β = 90°, and γ = 120° for all of the COFs, Rwp = 3.23% and Rp = 2.45% for JUC-616, Rwp = 3.09% and Rp = 2.19% for JUC-617, and Rwp = 1.57% and Rp = 1.17% for JUC-618, respectively).
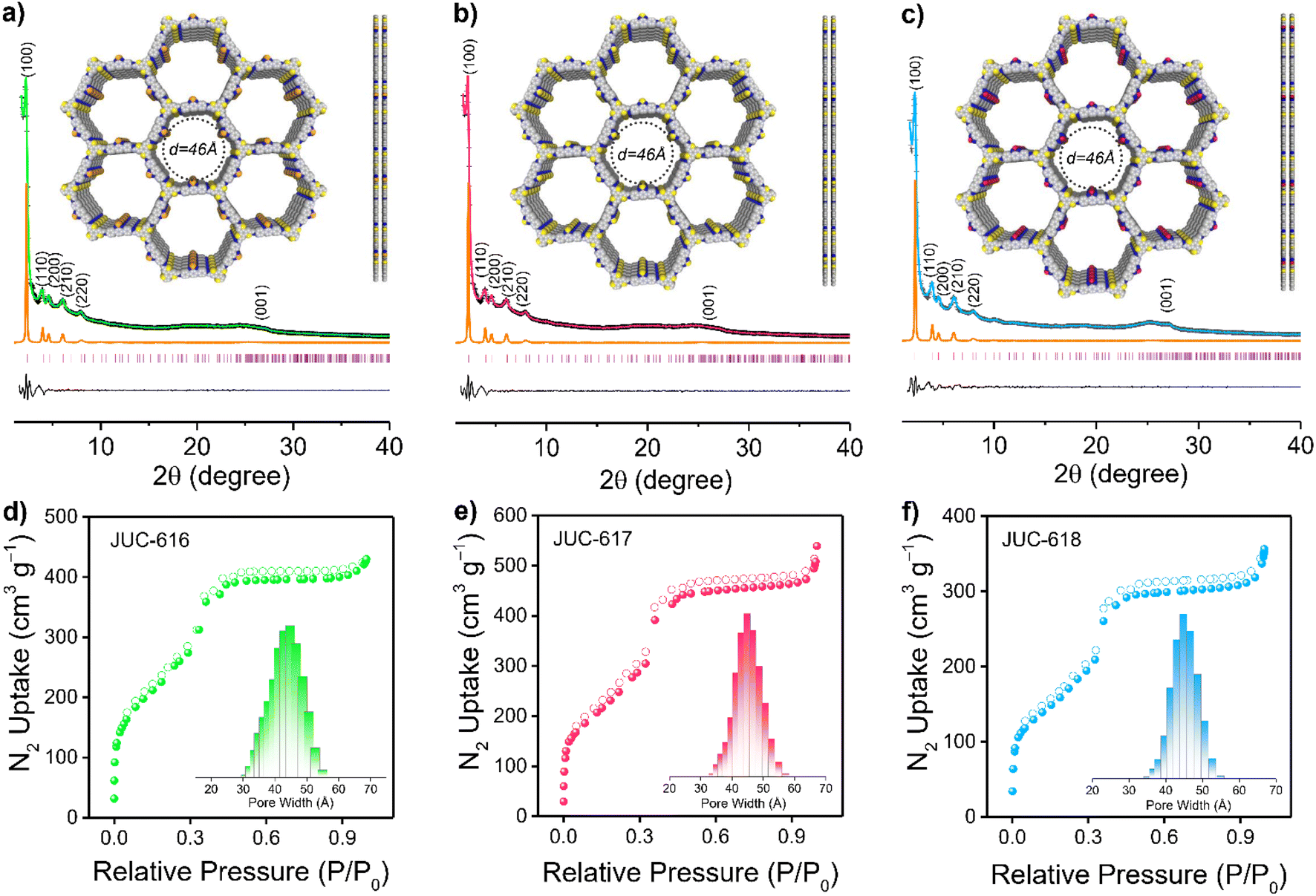 | ||
| Fig. 1 Powder XRD patterns of JUC-616 (a), JUC-617 (b) and JUC-618 (c). The observed XRD patterns (+), Pawley refined (pink), simulation (orange), the difference between the observed and refined profiles (black) and Bragg position (green). Insets: Crystal structures with AA stacking modes. Nitrogen adsorption (filled circles)–desorption (open circles) isotherms of JUC-616 (d), JUC-617 (e) and JUC-618 (f). Insets: Pore size estimated using non-local density functional theory. | ||
The morphology of the BTT-COFs was studied by field-emission scanning electron microscopy (FE-SEM), which exhibits needle-like or spherical nanoclusters aggregated into coral-like masses (ESI, Fig. S7†). In addition, high-resolution transmission electron microscopy (HR-TEM) images revealed long-range channels (ESI, Fig. S8†), illustrating further their high crystallinity. The lattice fringes of the BTT-COFs were calculated using software to be 0.38 nm counter to the (001) crystal plane, which is close to the simulated layer distances (0.35 nm). Furthermore, the nitrogen adsorption–desorption isotherms of the as-synthesized COFs were performed at 77 K to estimate their porosity. As illustrated in Fig. 1d–f, the sorption curves of JUC-616, JUC-617, and JUC-618 matched the type IV reversible isotherms, which are characteristic of mesoporous materials. The Brunauer–Emmett–Teller (BET) surface area was evaluated to be 893 m2 g−1 for JUC-616, 943 m2 g−1 for JUC-617, and 630 m2 g−1 for JUC-618, respectively. Their total pore volumes were calculated to be 0.66, 0.78, and 0.53 cm3 g−1 at P/P0 = 0.99. Additionally, the pore size distribution calculated using non-local density functional theory (NLDFT) shows that the three isomorphic COFs exhibit the same homogeneous pores with a pore size of 45 Å (Fig. 1d–f, insets), which matched the theoretical value of 46 Å based on the AA stacking mode (Fig. 1a–c, insets).
The UV-Vis diffuse reflectance spectrum (UV-DRS) of BTT-COFs displayed a broad absorption band with a tail extending to 1000 nm (ESI, Fig. S9†), which corresponds to their D–A structural characteristic. According to the Tauc plot, the optical bandgaps (Eg) of JUC-616, JUC-617, and JUC-618 were evaluated to be 1.98, 2.06, and 2.08 eV, respectively (ESI, Fig. S10†). Compared to JUC-617 and JUC-618, JUC-616 had a smaller Eg, which was more conducive to electron transmission.59,60 Based on the electrochemical Mott–Schottky spectra, we estimated the conduction band minimum (CBM) of JUC-616 to be −1.10 V vs. NHE, which is more negative than the −1.04 V for JUC-617 and −0.84 V for JUC-618 (ESI, Fig. S11†), implying a stronger electron reduction capacity of JUC-616.61
The electrocatalytic ORR performance of the obtained COFs was then evaluated at room temperature by a three-electrode system in an O2-saturated 0.1 M KOH electrolyte at a scan rate of 1600 rpm. As depicted in Fig. 2a, the linear sweep voltammetry (LSV) curves of the samples showed that JUC-616 exhibited better ORR performance than the isomorphic JUC-617 and JUC-618, which clearly indicated the importance of introducing Se atoms into the skeleton to enhance the ORR activity. The electrocatalytic ORR activity of the COFs was verified by cyclic voltammetry (CV), and compared with JUC-617 (0.73 V) and JUC-618 (0.70 V), JUC-616 exhibited the highest electrochemical ORR activity with an oxygen reduction peak at 0.74 V vs. RHE (ESI, Fig. S12†). The half-wave potential of JUC-616 was 0.78 V vs. RHE, which was more positive than for JUC-617 (0.73 V vs. RHE) and JUC-618 (0.70 V vs. RHE). Furthermore, the limiting current densities of the three isomorphic COFs were close to 6.0 mA cm−2, indicating that their mass transfer is almost identical due to their similar pore structures, surface areas, and morphologies. Surprisingly, we noticed that the ORR performance of JUC-616 was outstanding among the state-of-the-art metal-free COFs reported without a pyrolysis process (ESI, Table S4†). As shown in Fig. 2b, the Tafel slope of JUC-616 was determined to be 52.91 mV dec−1, which is smaller than that of JUC-617 (75.40 mV dec−1) and JUC-618 (78.96 mV dec−1), thus revealing the faster ORR kinetics of Se-containing JUC-616 (ESI, Fig. S13†). Obviously, the changing trend of the obtained Tafel slope value is consistent with the result of the LSV curve. Additionally, electrochemical impedance spectroscopy (EIS) measurements were performed on the obtained COFs to evaluate their conductive properties. Accordingly, the charge transfer resistances of JUC-616, JUC-617, and JUC-618 were 769, 1600, and 1941 Ω, respectively (Fig. 2c), which indicated that JUC-616 had a faster electron transfer capability. The electron transfer number (n) of JUC-616, JUC-617, and JUC-618, calculated from Koutecky–Levich (K–L) plots, was 3–3.8 in the potential range of 0.2–0.6 V vs. RHE (Fig. 2d and ESI Fig. S14 and S15 and Table S5†), suggesting a four-electron pathway in the ORR process.62 Similar to the K–L equation, the electron transfer number (n) of JUC-616, JUC-617, and JUC-618 from the rotating ring disk experiments, was 3–3.7 in the potential range of 0.0–0.7 V vs. RHE (ESI, Fig. S16†).
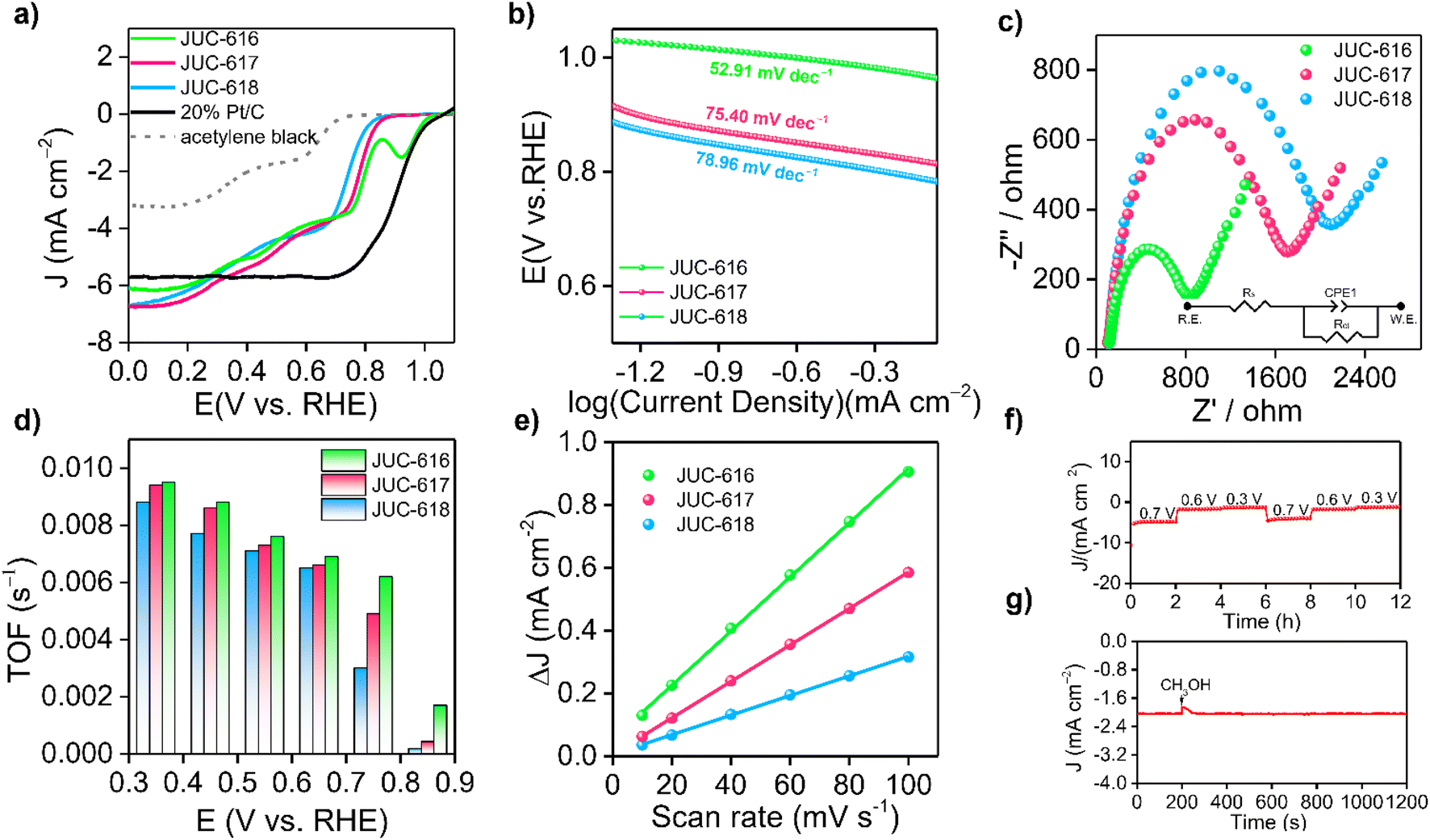 | ||
| Fig. 2 (a) LSV curves of the three isomorphic COFs in O2-saturated 0.1 M KOH electrolyte at a scan rate of 1600 rpm. (b) Tafel plots of the three COFs. (c) The impedance of COFs at open circuit voltage. Inset: Equivalent circuit. (d) TOFs and mass activities of COFs. (e) Cdl values of COFs. (f) Multi-step amperometry curve of JUC-616 at applied voltages of 0.3, 0.6 and 0.7 V. (g) Current–time curve of JUC-616 at 0.7 V vs. RHE. | ||
Next, the origin of the high performance of JUC-616 was also discussed from the experimental point of view. The linear relationship between electrochemically active surface area (ECSA) and double layer capacitance (Cdl) is well known.63 Therefore, Cdl measurements were performed by the CV method (ESI, Fig. S17†). As shown in Fig. 2e, the Cdl for JUC-616 is 8.62 mF cm−2, which is larger than the 5.80 mF cm−2 for JUC-617, 3.12 mF cm−2 for JUC-618, and 7.6 mF cm−2 for JUC-528 with a bithiophene-sulfur structure,42 indicating that the Se-containing COF can provide more efficient active sites. The intrinsic activity of the materials was investigated by calculating the conversion frequency (TOF) at various potentials. As shown in Fig. 2d, JUC-616 has the largest TOF value at different potentials, which promotes the adsorption and desorption of O2 and products on the catalyst surface.64 These obtained results clearly demonstrate that Se-containing JUC-616 shows the highest electrocatalytic ORR activity among the three isomorphic COFs. In addition, as shown in Fig. 2f, multi-step chronoamperometry measurements were also conducted at potentials of 0.3, 0.6, and 0.7 V, and the experimental data revealed that the current density of JUC-616 remained almost constant at each applied potential throughout the experimental cycle, indicating that JUC-616 has high electrochemical stability in alkaline solution.65 A negligible loss for the current density was found when adding methanol into the electrolyte at 0.7 V vs. RHE (Fig. 2g), proving the good methanol tolerance of JUC-616. Comparative analysis of the three materials before and after electrocatalysis revealed that both their structure and morphology remained essentially unaltered. This observation serves as compelling evidence of the high stability exhibited by these materials throughout the electrocatalytic process (ESI, Fig. S18 and S19†).
To further explore the catalytically active sites and mechanism of Se-doped COF, theoretical calculations were carried out using the density functional theory (DFT) method. In order to more intuitively visualize the effects of O, S, and Se on the ORR process, we performed theoretical calculations using O, S, and Se as the active sites. The calculation of the Mulliken charge of BTT-COFs identified the active sites as O, S and Se on the diazole, which are more prone to nucleophilic reactions favoring the ORR (ESI, Fig. S20 and Table S6†). In addition, by calculating the differential charge of the adsorbed state, it is shown that Se and S have a strong adsorption on OH*, proving that S and Se are the active sites. Electron transfer from COFs to the OH* intermediate during the ORR can also be visualized in the charge difference map. As shown in Fig. 3a, the yellow and cyan isosurfaces represent electron-rich and depleted regions, respectively. Obviously, there is no bonding or charge transfer between OH* and JUC-618, while the Se and S atoms of BSD and BTD units in JUC-616 and JUC-617 lose electrons and OH* gains electrons. In contrast, the charge transfer between OH* and JUC-616 and JUC-617 is large. Density of states calculations were performed for the O, S, and Se atoms in the three materials, and the DOS of their PZ orbitals were calculated (Fig. 3c and ESI Fig. S21†), revealing the binding energy of the PZ band center to the reaction intermediate. Compared with JUC-617 (−3.44 eV) and JUC-618 (−3.81 eV), JUC-616 has a higher PZ band center and is more capable of binding to reaction intermediates (−3.06 eV), thus exhibiting higher activity, similar to the trend of activity predicted by “d band theory”.66,67 Additionally, three adsorption intermediate models were constructed, as shown in Fig. 3a and S16 in the ESI,† and for four-electron transfer, we have calculated the free energies and overpotentials of the ORR on BTT-COFs and determined the rate-limiting step. The free energy diagram of the ORR (Fig. 3c) indicates that the rate-limiting step of the ORR is the transformation of OH* to OH− by all COFs in the last step of the ORR. According to the ΔG diagrams in Fig. 3d, the highest energy barrier of the three isomorphic COFs for electrocatalytic ORR occurs in the process from OH* to OH−, where Se-doped JUC-616 shows a lower energy barrier of 0.30 eV than JUC-617 (0.47 eV) and JUC-618 (0.55 eV). Additionally, JUC-616 has the lowest overpotential of 0.37 eV (ESI, Table S7†), indicating that it is more favorable for the ORR.
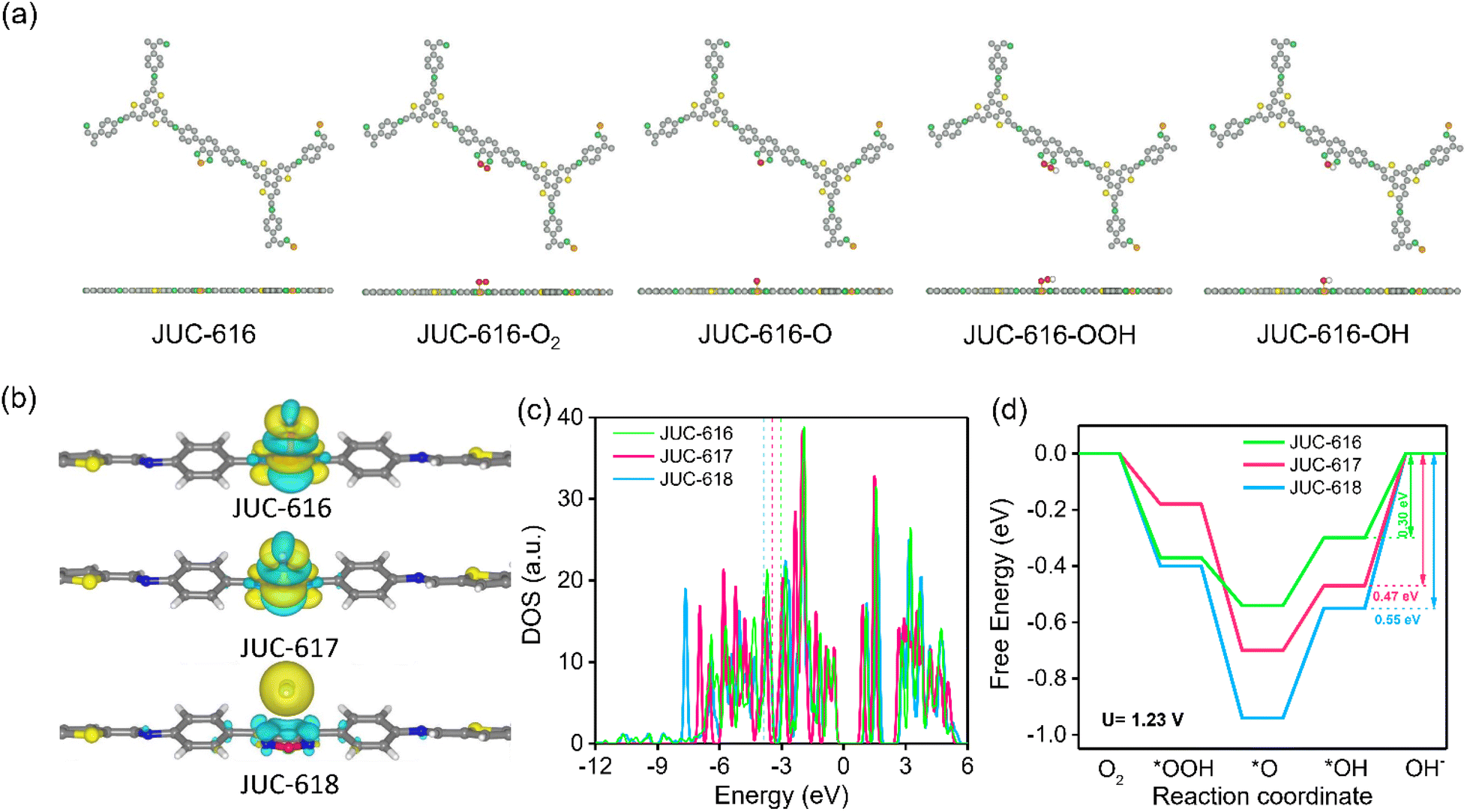 | ||
| Fig. 3 (a) Models for the adsorption of different ORR intermediates by JUC-616. The grey, green, yellow, red, orange and white spheres represent C, N, S, O, Se and H atoms, respectively. (b) The charge distribution of JUC-616. The curry, silvery white, green and yellow spheres represent C, N, Se and S atoms, respectively. Free energy diagrams of Se, S, and O atoms in JUC-616, JUC-617 and JUC-618 at the applied potentials of 0 V (c) and 1.23 V (d), respectively. | ||
Conclusions
In summary, a series of metal-free BTT-based COFs were designed and synthesized via the reversible condensation of imines under solvothermal conditions. They have high crystallinity, intrinsic porosity, and electron donating and accepting properties. As a metal- and pyrolysis-free electrocatalyst, JUC-616 exhibited excellent ORR activity with a half-wave potential of 0.78 V (vs. RHE) and a Tafel slope of 52.91 mV dec−1, which are better than those of previously reported COF materials. Theoretical and experimental results demonstrated that the Se atoms in the skeleton of COFs could serve as highly active sites for electrocatalytic ORR. We further highlight that this work is the first report on improving the ORR activity of COFs via a Se-doping strategy, which facilitates the rational design and application of COF-based electrocatalysts. Therefore, this work develops a general strategy to enrich the structural diversity of COF materials and promotes their potential applications.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Key R&D Program of China (2022YFB3704900 and 2021YFF0500500), National Natural Science Foundation of China (22025504, 21621001, and 22105082), the SINOPEC Research Institute of Petroleum Processing, “111” project (BP0719036 and B17020), China Postdoctoral Science Foundation (2020TQ0118 and 2020M681034), and the program for JLU Science and Technology Innovative Research Team.References
- I. Staffell, D. Scamman, A. Velazquez Abad, P. Balcombe, P. E. Dodds, P. Ekins, N. Shah and K. R. Ward, Energy Environ. Sci., 2019, 12, 463–491 RSC.
- C.-Y. Su, H. Cheng, W. Li, Z.-Q. Liu, N. Li, Z. Hou, F.-Q. Bai, H.-X. Zhang and T.-Y. Ma, Adv. Energy Mater., 2017, 7, 1602420 CrossRef.
- H. Wang, C. Tang, B. Wang, B. Li and Q. Zhang, Adv. Mater., 2017, 29, 1702327 CrossRef PubMed.
- Z. W. Seh, J. Kibsgaard, C. F. Dickens, I. Chorkendorff, J. K. Nørskov and T. F. Jaramillo, Science, 2017, 355, eaad4998 CrossRef PubMed.
- A. Kulkarni, S. Siahrostami, A. Patel and J. K. Nørskov, Chem. Rev., 2018, 118, 2302–2312 CrossRef CAS PubMed.
- A. Chen and P. Holt-Hindle, Chem. Rev., 2010, 110, 3767–3804 CrossRef CAS PubMed.
- M. K. Debe, Nature, 2012, 486, 43–51 CrossRef CAS PubMed.
- Z. Zhao, B. Wang, Z. You, Q. Zhang, W. Song and X. Long, Small, 2023, 19, 2207298 CrossRef CAS PubMed.
- X. Feng, Y. Bai, M. Liu, Y. Li, H. Yang, X. Wang and C. Wu, Energy Environ. Sci., 2021, 14, 2036–2089 RSC.
- K. Chalapat, N. Chekurov, H. Jiang, J. Li, B. Parviz and G. S. Paraoanu, Adv. Mater., 2013, 25, 1 CrossRef.
- P. J. Waller, F. Gándara and O. M. Yaghi, Acc. Chem. Res., 2015, 48, 3053–3063 CrossRef CAS PubMed.
- Y. Song, Q. Sun, B. Aguila and S. Ma, Adv. Sci., 2019, 6, 1801410 CrossRef PubMed.
- A. P. Côté, A. I. Benin, N. W. Ockwig, M. O'keeffe, A. J. Matzger and O. M. Yaghi, Science, 2005, 310, 1166–1170 CrossRef PubMed.
- Z. Zhang, J. Jia, Y. Zhi, S. Ma and X. Liu, Chem. Soc. Rev., 2022, 51, 2444–2490 RSC.
- Z. Li, J. Wang, S. Ma, Z. Zhang, Y. Zhi, F. Zhang, H. Xia, G. Henkelman and X. Liu, Appl. Catal., B, 2022, 310, 121335 CrossRef CAS.
- F. Chen, X. Guan, H. Li, J. Ding, L. Zhu, B. Tang, V. Valtchev, Y. Yan, S. Qiu and Q. Fang, Angew. Chem., Int. Ed., 2021, 60, 22230–22235 CrossRef CAS PubMed.
- Q. Fang, S. Gu, J. Zheng, Z. Zhuang, S. Qiu and Y. Yan, Angew. Chem., Int. Ed., 2014, 53, 2878–2882 CrossRef CAS PubMed.
- F. Tan, Y. Zheng, Z. Zhou, H. Wang, X. Dong, J. Yang, Z. Ou, H. Qi, W. Liu, Z. Zheng and X. Chen, CCS Chem., 2022, 4, 3751–3761 CrossRef CAS.
- Y. Zhang, J. Duan, D. Ma, P. Li, S. Li, H. Li, J. Zhou, X. Ma, X. Feng and B. Wang, Angew. Chem., Int. Ed., 2017, 56, 16313–16317 CrossRef CAS PubMed.
- S. Wang, Q. Wang, P. Shao, Y. Han, X. Gao, L. Ma, S. Yuan, X. Ma, J. Zhou, X. Feng and B. Wang, J. Am. Chem. Soc., 2017, 139, 4258–4261 CrossRef CAS PubMed.
- Y. Du, H. Yang, J. M. Whiteley, S. Wan, Y. Jin, S. H. Lee and W. Zhang, Angew. Chem., Int. Ed., 2016, 55, 1737–1741 CrossRef CAS PubMed.
- E. Jin, M. Asada, Q. Xu, S. Dalapati, M. A. Addicoat, M. A. Brady, H. Xu, T. Nakamura, T. Heine, Q. Chen and D. Jiang, Science, 2017, 357, 673–676 CrossRef CAS PubMed.
- H. Ding, J. Li, G. Xie, G. Lin, R. Chen, Z. Peng, C. Yang, B. Wang, J. Sun and C. Wang, Nat. Commun., 2018, 9, 5234 CrossRef CAS PubMed.
- A. C. Jakowetz, T. F. Hinrichsen, L. Ascherl, T. Sick, M. Calik, F. Auras, D. D. Medina, R. H. Friend, A. Rao and T. Bein, J. Am. Chem. Soc., 2019, 141, 11565–11571 CrossRef CAS PubMed.
- K. Geng, T. He, R. Liu, S. Dalapati, K. T. Tan, Z. Li, S. Tao, Y. Gong, Q. Jiang and D. Jiang, Chem. Rev., 2020, 120, 8814–8933 CrossRef CAS PubMed.
- X. Guan, F. Chen, Q. Fang and S. Qiu, Chem. Soc. Rev., 2020, 49, 1357–1384 RSC.
- W. Xian, P. Zhang, C. Zhu, X. Zuo, S. Ma and Q. Sun, CCS Chem., 2021, 3, 2464–2472 CrossRef CAS.
- M. Hao, Z. Chen, X. Liu, X. Liu, J. Zhang, H. Yang, G. I. N. Waterhouse, X. Wang and S. Ma, CCS Chem., 2022, 4, 2294–2307 CrossRef CAS.
- Y. Liu, J. Ren, Y. Wang, X. Zhu, X. Guan, Z. Wang, Y. Zhou, L. Zhu, S. Qiu, S. Xiao and Q. Fang, CCS Chem., 2022, 1–13 Search PubMed.
- Y. Wang, M. Jiao, W. Song and Z. Wu, Carbon, 2017, 114, 393–401 CrossRef CAS.
- M. Liu, S. Liu, C. Cui, Q. Miao, Y. He, X. Li, Q. Xu and G. Zeng, Angew. Chem., Int. Ed., 2022, 61, e202213522 CAS.
- Y. Yusran, Q. Fang and V. Valtchev, Adv. Mater., 2020, 32, 2002038 CrossRef CAS PubMed.
- X. Zhao, P. Pachfule and A. Thomas, Chem. Soc. Rev., 2021, 50, 6871–6913 RSC.
- D. Yang, Y. Tao, X. Ding and B. Han, Chem. Soc. Rev., 2022, 51, 761–791 RSC.
- Z. Xiang, D. Cao, L. Huang, J. Shui, M. Wang and L. Dai, Adv. Mater., 2014, 26, 3315–3320 CrossRef CAS PubMed.
- Q. Xu, Y. Tang, X. Zhang, Y. Oshima, Q. Chen and D. Jiang, Adv. Mater., 2018, 30, 1706330 CrossRef PubMed.
- S. Tao and D. Jiang, CCS Chem., 2021, 3, 2003–2024 CrossRef CAS.
- S. Royuela, E. Martínez-Periñán, M. P. Arrieta, J. I. Martínez, M. M. Ramos, F. Zamora, E. Lorenzo and J. L. Segura, Chem. Commun., 2020, 56, 1267–1270 RSC.
- L. Zhai, S. Yang, X. Yang, W. Ye, J. Wang, W. Chen, Y. Guo, L. Mi, Z. Wu, C. Soutis, Q. Xu and Z. Jiang, Chem. Mater., 2020, 32, 9747–9752 CrossRef CAS.
- S. Chang, C. Li, H. Li, L. Zhu and Q. Fang, Chem. Res. Chin. Univ., 2022, 38, 396–401 CrossRef CAS.
- J. Yue, Y. Wang, X. Wu, P. Yang, Y. Ma, X. Liu and B. Tang, Chem. Commun., 2021, 57, 12619–12622 RSC.
- D. Li, C. Li, L. Zhang, H. Li, L. Zhu, D. Yang, Q. Fang, S. Qiu and X. Yao, J. Am. Chem. Soc., 2020, 142, 8104–8108 CrossRef CAS PubMed.
- C. Yang, S. Tao, N. Huang, X. Zhang, J. Duan, R. Makiura and S. Maenosono, ACS Appl. Nano Mater., 2020, 3, 5481–5488 CrossRef CAS.
- J. P. Paraknowitsch, J. Zhang, D. Su, A. Thomas and M. Antonietti, Adv. Mater., 2010, 22, 87–92 CrossRef CAS PubMed.
- H. Hu, J. Wang, B. Cui, X. Zheng, J. Lin, Y. Deng and X. Han, Angew. Chem., Int. Ed., 2022, 61, e202114441 CAS.
- A. Abouimrane, D. Dambournet, K. W. Chapman, P. J. Chupas, W. Weng and K. Amine, J. Am. Chem. Soc., 2012, 134, 4505–4508 CrossRef CAS PubMed.
- B. Zhang, J. Zhang, F. Zhang, L. Zheng, G. Mo, B. Han and G. Yang, Adv. Funct. Mater., 2020, 30, 1906194 CrossRef CAS.
- H. Liang, L. Jia, F. Chen, S. Jing and P. Tsiakaras, Appl. Catal., B, 2022, 317, 121698 CrossRef CAS.
- X. Cui, L. Gao, R. Ma, Z. Wei, C. Lu, Z. Li and Y. Yang, J. Mater. Chem. A, 2021, 9, 20985–21004 RSC.
- Z. Li, Z. Zhang, R. Nie, C. Li, Q. Sun, W. Shi, W. Chu, Y. Long, H. Li and X. Liu, Adv. Funct. Mater., 2022, 32, 2112553 CrossRef CAS.
- Z. Li, S. Han, C. Li, P. Shao, H. Xia, H. Li, X. Chen, X. Feng and X. Liu, J. Mater. Chem. A, 2020, 8, 8706–8715 RSC.
- D. Yan, Z. Wang, P. Cheng, Y. Chen and Z. Zhang, Angew. Chem., 2021, 60, 6055–6060 CrossRef CAS PubMed.
- N. Zheng, Y. Feng, Y. Zhang, R. Li, C. Bian, L. Bao, S. Du, H. Dong, Y. Shen and W. Feng, ACS Appl. Mater. Interfaces, 2019, 11, 24360–24366 CrossRef CAS PubMed.
- L. Guo, Y. Niu, S. Razzaque, B. Tan and S. Jin, ACS Catal., 2019, 9, 9438–9445 CrossRef CAS.
- S.-X. L. Luo, R. Y. Liu, S. Lee and T. M. Swager, J. Am. Chem. Soc., 2021, 143, 10441–10453 CrossRef CAS PubMed.
- W.-R. Cui, Y.-J. Li, Q.-Q. Jiang, Q. Wu, R.-P. Liang, Q.-X. Luo, L. Zhang, J. Liu and J.-D. Qiu, Cell Rep. Phys. Sci., 2022, 3, 100630 CrossRef CAS.
- T. Zhang, T. Cai, W. Xing, T. Li, B. Liang, H. Hu, L. Zhao, X. Li and Z. Yan, Energy Storage Mater., 2021, 41, 667–676 CrossRef.
- R. Chen, Y. Wang, Y. Ma, A. Mal, X. Gao, L. Gao, L. Qiao, X. Li, L. Wu and C. Wang, Nat. Commun., 2021, 12, 1354 CrossRef CAS PubMed.
- B. He, G. Li, J. Li, J. Wang, H. Tong, Y. Fan, W. Wang, S. Sun and F. Dang, Adv. Energy Mater., 2021, 11, 2003263 CrossRef CAS.
- S. Bai, T. Li, H. Wang, L. Tan, Y. Zhao and Y. Song, Chem. Eng. J., 2021, 419, 129390 CrossRef CAS.
- T. Tian, X. Jin, N. Guo, H. Li, Y. Han and Y. Yuan, Appl. Catal., B, 2022, 308, 121227 CrossRef CAS.
- J. Zhang, L. Qu, G. Shi, J. Liu, J. Chen and L. Dai, Angew. Chem., Int. Ed., 2016, 55, 2230–2234 CrossRef CAS PubMed.
- W. Huang, J. Li, X. Liao, R. Lu, C. Ling, X. Liu, J. Meng, L. Qu, M. Lin, X. Hong, X. Zhou, S. Liu, Y. Zhao, L. Zhou and L. Mai, Adv. Mater., 2022, 34, 2200270 CrossRef CAS PubMed.
- W. Moschkowitsch, O. Lori and L. Elbaz, ACS Catal., 2022, 12, 1082–1089 CrossRef CAS.
- H. Zhang, P. Li, S. Chen, F. Xie and D. J. Riley, Adv. Funct. Mater., 2021, 31, 2106835 CrossRef CAS.
- J. K. Nørskov, F. Abild-Pedersen, F. Studt and T. Bligaard, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 937–943 CrossRef PubMed.
- S. Zhou, W. Pei, Y. Zhao, X. Yang, N. Liu and J. Zhao, npj Comput. Mater., 2021, 7, 186 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Materials and characterization, SEM images, TEM images, FT-IR spectra, TGA curves, PXRD patterns, and unit cell parameters. See DOI: https://doi.org/10.1039/d3ta03534d |
| This journal is © The Royal Society of Chemistry 2023 |