DOI:
10.1039/C3AY41626G
(Paper)
Anal. Methods, 2014,
6, 223-228
Enantiomeric separation of sitagliptin by a validated chiral liquid chromatographic method
Received
19th September 2013
, Accepted 27th October 2013
First published on 22nd November 2013
1. Introduction
Sitagliptin ((2R)-4-oxo-4-[3-(trifluoromethyl)-5,6-dihydro[1,2,4]triazolo[4,3-a]pyrazin-7(8H)-yl]-1-(2,4,5-trifluorophenyl)butan-2-amine) (Fig. 1) is a new orally active, potent and selective DPP-IV inhibitor for the treatment of type 2 diabetes mellitus. It enhances the levels of active glucagon-like peptides, other incretins and facilitates glucose-dependent insulin secretion.1,2 It also improves glycemic control and regulates the growth of insulin producing β-cells in pancreatic islets. Many clinical findings suggest that a diabetic individual is more likely to develop hypertension, which subsequently predisposes the patient to cardiovascular diseases and many other complications.3 Moreover, hypertension along with diabetes impairs the renal function, thus resulting in the development of diabetic retinopathy.4 Therefore, an effective control of blood pressure is primarily required for a hypertensive diabetic patient in order to prevent and delay the subsequent manifestations.
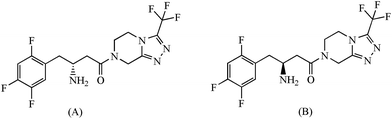 |
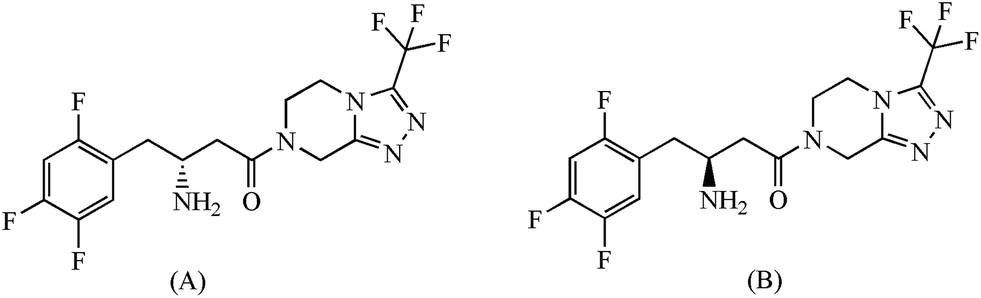
| | Fig. 1 Chemical structures of (A) sitagliptin and (B) (S)-enantiomer of sitagliptin. | |
A thorough literature search has revealed that several high performance liquid chromatography (HPLC), liquid chromatography-mass spectroscopy (LC-MS), gas chromatography-mass spectroscopy (GC-MS), high performance thin layer chromatography (HPTLC), capillary zone electrophoresis (CZE) and ultraviolet-visible spectrometry methods have been reported for determination of sitagliptin phosphate in tablet dosage form and in combination with other drugs in pharmaceutical formulation, and plasma.5–17 During the synthesis of sitagliptin there is a possibility of carrying an undesired (S)-enantiomer of enantiopure sitagliptin, therefore enantioselective analytical methods are necessary to ensure its therapeutic efficacy and safety. Therefore, it is a challenge to develop a sensitive and high-throughput assay for quantitation of sitagliptin enantiomers. High performance liquid chromatography (HPLC) is playing a more and more important role for the resolution of drug enantiomers in the field of pharmaceutical industry.18 However, the development of methods for the quantitative analysis of chiral compounds and for the assessment of enantiomeric purity is extremely challenging, because the same physical and chemical properties of the two enantiomers make discriminating and separating them very difficult. Recently, many chromatographic methods have been reported describing the use of chiral stationary phases in conjunction with HPLC, as ways to separate and thereby individually quantitate the enantiomers of an enantiomeric pair.19,20 B. V. Rami Reddy et al. reported a chiral HPLC method with less accuracy, less sensitivity and more run time for the enantiomeric separation of sitagliptin and more results are shown in Table 1.
Table 1 Comparison of the present method to the reported method
| Parameter |
Reported method |
Present method |
| Operating mode |
NP-HPLC |
RP-HPLC |
| Detector |
PDA |
PDA |
| Mobile phase |
n-Heptane–ethanol–diethyl amine (35![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :65:0.1, v/v/v) :65:0.1, v/v/v) |
KH2PO4–methanol–acetonitrile (60:30:10, v/v/v) |
| Column |
Chiralpak AD-H (250 mm × 4.6 mm, i.d., 5 μm) |
Chiralcel OD-RH (150 mm × 4.6 mm, i.d., 5 μm) |
| Flow rate |
1.0 mL min−1 |
0.5 mL min−1 |
|
λ
max
|
265 |
262 |
| Run time |
30 min |
10 min |
| Linearity range |
400–2250 ng mL−1 |
30–300 ng mL−1 |
| Correlation coefficient (r2) |
0.995 |
0.9996 |
| Limit of detection (LOD) |
150 ng mL−1 |
8 ng mL−1 |
| Limit of quantification (LOQ) |
400 ng mL−1 |
30 ng mL−1 |
| Reference |
21
|
— |
This paper describes a chiral HPLC method for the enantiomeric separation of sitagliptin enantiomers using the modified cellulose based chiral stationary phase, Chiralcel OD-RH. The aim of this work was to optimize the chromatographic conditions in terms of temperature, pH value of buffer solution and mobile phase composition in order to separate and identify the enantiomers of sitagliptin. The developed method was validated according to International Conference on Harmonization (ICH) guidelines22 for the quantitative determination of the (S)-enantiomer in sitagliptin.
2. Experimental
2.1. Chemicals
Sitagliptin and its (S)-enantiomer (Fig. 1) were kindly provided by Aurobindo Pharma Pvt. Ltd. (Hyderabad, India). HPLC grade methanol and acetonitrile were purchased from S.D. Fine-Chem. (Mumbai, India). Potassium di-hydrogen phosphate dihydrate, ortho-phosphoric acid and sodium hydroxide were purchased from Merck (Mumbai, India); glass-distilled and de-ionized water from Nanopure (Barnsted, USA) were used.
2.2. Instrumentation and chromatographic conditions
The HPLC system consisting of a quaternary LC-20AD pump, a SPD-M20A diode array detector, a SIL-20AC auto sampler, a DGU-20A5 degasser and a CBM-20A communication bus module (all from Shimadzu, Kyoto, Japan) was used. pH measurements were carried out by using an Elico, model LI 120, pH meter equipped with a combined glass–calomel electrode. It was calibrated using standard buffers of pH 4.0, 7.0 and 9.2. The chromatographic and the integrated data were recorded using an HP-Vectra (Hewlett Packard, Waldron, Germany) computer system using LC-Solution data acquiring software (Shimadzu, Kyoto, Japan). Before delivering the mobile phase into the system, it was filtered through 0.45 μm PTFE filters and degassed. The chromatographic conditions were optimized using a chiral stationary phase, Chiralcel OD-RH column (150 mm × 4.6 mm, 5 μm, Daicel, Japan). The isocratic mobile phase composition was a mixture of 3 M potassium di-hydrogen phosphate buffer (pH 4.0)–methanol–acetonitrile (60:30:10, v/v/v), which was pumped at a flow rate of 0.5 mL min−1. The temperature of the column was maintained at 25 °C, and the eluent was monitored at a wavelength in the range of 252 to 268 nm. The injection volume was 20 μL.
2.3. Sample preparation
Stock solutions of sitagliptin (1.0 mg mL−1) and (S)-enantiomer (1.0 mg mL−1) were prepared by dissolving the appropriate amount of the substances in the mobile phase. The analyte concentration of sitagliptin was fixed to be 10 μg mL−1. Sitagliptin solutions spiked with low levels of the (S)-enantiomer were prepared by transferring a calculated amount of undesired enantiomer stock solution with a pipette into a calculated amount of sitagliptin stock solution, and then the solution was added to volume with the mobile phase and mixed well.
3. Results and discussion
3.1. Optimization of chromatographic conditions
A racemic mixture solution of sitagliptin and its (S)-enantiomer (5 μg mL−1 each) prepared in the mobile phase was used in the method establishment. To develop a rugged and suitable HPLC method for the separation of the two enantiomers, different stationary phases and mobile phases were employed. Preliminary column screening involved protein-based chiral HPLC columns, namely Chiral AGP (150 mm × 4.0 mm, 5 μm), Chiral HSA (150 mm × 4.0 mm, 5 μm), and Chiral CBH (150 mm × 4.0 mm, 5 μm) of Chromtech and then Chiralpak AD-RH (150 mm × 4.6 mm, 5 μm) and Chiralcel OJ-RH (150 mm × 4.6 mm, 5 μm) of Daicel were also employed (for all these columns the mobile phase of 1 M potassium di-hydrogen phosphate buffer (pH 3.0)–methanol (70:30, v/v) was used). Only Chiralpak AD-RH [amylose tris-(3,5-dimethylphenylcarbamate)] and Chiralcel OD-RH [cellulose tris-(3,5-dimethylphenylcarbamate)] columns provided selectivity between the sitagliptin peak and the undesired enantiomer peak using a mobile phase consisting of 1 M potassium di-hydrogen phosphate buffer (pH 3.0)–methanol (70:30, v/v), but the retention times of sitagliptin and (S)-enantiomer were both longer than 20 min and the peaks were broad. We continued to select the best mobile phases that would give optimum resolution and selectivity for the two enantiomers. To shorten the retention times of the two enantiomers, the right amount of acetonitrile was added to the mobile phase. Then the pH value and concentration of the phosphate buffer solution in the mobile phase on Chiralcel OD-RH and Chiralpak AD-RH columns were optimized to enhance the chromatographic efficiency and resolution between the enantiomers (Tables 2 and 3). Good separation was achieved on Chiralcel OD-RH and Chiralpak AD-RH columns using 3 M potassium di-hydrogen phosphate buffer (pH 4.0)–methanol–acetonitrile (60:30:10, v/v/v) as the mobile phase. The peaks of the two enantiomers became broad and the resolution fell obviously with high pH value of phosphate buffer. Low pH (pH 4.0 compared with pH 5.0) and higher concentration of phosphate buffer gave more chromatographic efficiency and resolution. However, for long working life of the columns, we chose 3 M potassium di-hydrogen phosphate buffer (pH 4.0) as the final aqueous phase. Due to the better chromatographic results obtained on the Chiralcel OD-RH column, further method optimization and quantification of the (S)-enantiomer were carried out on this column. Based on the data obtained from method development and optimization activities, the Chiralcel OD-RH (150 mm × 4.6 mm, 5 μm) column with the mobile phase of 3 M potassium di-hydrogen phosphate buffer (pH 4.0)–methanol–acetonitrile (60:30:10, v/v/v) was selected for the final method. The flow rate of the experimental method was 0.5 mL min−1 with an injection volume of 20 μL. The column temperature was 25 °C, and the detection wavelength was in the range of 252 to 268 nm. Under these conditions, the two enantiomers were separated well and the peak of the (S)-enantiomer eluted before the peak of sitagliptin. In the optimized method, the typical retention times of the (S)-enantiomer and sitagliptin were approximately 3.610 and 5.364 min, respectively. Baseline separation of sitagliptin and (S)-enantiomer was obtained with a total run time of 10 min. The chromatographic separation of sitagliptin enantiomers under the optimized conditions using a photo diode array (PDA) detector on Chiralcel OD-RH and Chiralpak AD-RH columns is shown in Fig. 2. The system suitability test results of the chiral HPLC method on Chiralcel OD-RH and Chiralpak AD-RH columns are presented in Table 4.
Table 2 The selectivity of sitagliptin enantiomers on a Chiralcel OD-RH column with different pH values of buffer salt solutions in modifiersa
| Potassium di-hydrogen phosphate buffer concentration |
pH |
K
′1
|
K
′2
|
Rs |
α
|
|
K
′1: retention factor of the (S)-enantiomer; K′2: retention factor of sitagliptin; Rs: resolution; α: selectivity.
|
| 1 M |
3.0 |
3.82 |
4.33 |
1.51 |
1.12 |
| 3.5 |
3.36 |
3.91 |
2.34 |
1.10 |
| 4.0 |
3.35 |
3.68 |
2.85 |
1.11 |
| 5.0 |
5.51 |
5.93 |
1.17 |
1.08 |
| 2 M |
3.0 |
3.91 |
4.14 |
2.01 |
1.11 |
| 3.5 |
3.55 |
3.71 |
2.63 |
1.14 |
| 4.0 |
3.28 |
3.58 |
3.17 |
1.15 |
| 5.0 |
5.69 |
5.58 |
1.29 |
1.09 |
| 3 M |
3.0 |
3.63 |
4.25 |
2.65 |
1.13 |
| 3.5 |
3.47 |
3.91 |
3.09 |
1.20 |
| 4.0 |
2.99 |
3.57 |
3.43 |
1.19 |
| 5.0 |
4.74 |
5.39 |
1.93 |
1.11 |
Table 3 The selectivity of sitagliptin enantiomers on a Chiralpak AD-RH column with different pH values of buffer salt solutions in modifiersa
| Potassium di-hydrogen phosphate buffer concentration |
pH |
K
′1
|
K
′2
|
Rs |
α
|
|
K
′1: retention factor of the (S)-enantiomer; K′2: retention factor of sitagliptin; Rs: resolution; α: selectivity.
|
| 1 M |
3.0 |
3.99 |
4.33 |
1.67 |
1.18 |
| 3.5 |
3.52 |
3.92 |
2.07 |
1.10 |
| 4.0 |
3.37 |
3.69 |
2.13 |
1.11 |
| 5.0 |
5.51 |
5.93 |
1.17 |
1.08 |
| 2 M |
3.0 |
3.70 |
4.61 |
1.91 |
1.11 |
| 3.5 |
3.62 |
3.91 |
2.01 |
1.13 |
| 4.0 |
3.19 |
3.66 |
2.47 |
1.12 |
| 5.0 |
5.27 |
5.84 |
1.53 |
1.06 |
| 3 M |
3.0 |
3.59 |
4.11 |
2.20 |
1.17 |
| 3.5 |
3.41 |
3.86 |
2.80 |
1.10 |
| 4.0 |
2.99 |
3.55 |
2.96 |
1.18 |
| 5.0 |
4.73 |
5.29 |
1.98 |
1.09 |
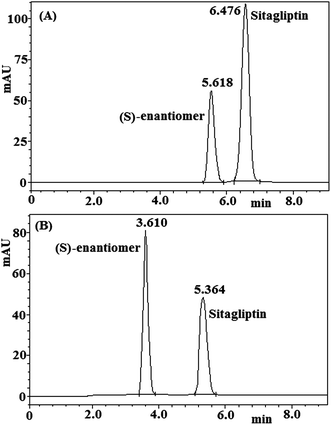 |
| | Fig. 2 Enantiomeric resolution of racemic sitagliptin on (A) a Chiralpak AD-RH column and (B) a Chiralcel OD-RH column. | |
Table 4 System suitability test results using Chiralcel OD-RH and Chiralpak AD-RH columnsa
| Column name |
Compound (n = 3) |
K′ |
Rs |
N
|
T
|
α
|
|
n = 3 determinations; K′: retention factor; Rs: resolution; N: the number of theoretical plates; T: tailing factor; α: enantioselectivity.
|
| Chiralcel OD-RH |
(S)-Enantiomer |
3.08 |
— |
13587 |
1.11 |
— |
| Sitagliptin |
3.63 |
3.36 |
13864 |
1.09 |
1.21 |
| Chiralpak AD-RH |
(S)-Enantiomer |
2.57 |
— |
5781 |
1.02 |
— |
| Sitagliptin |
3.19 |
2.59 |
9984 |
0.81 |
1.20 |
3.2. Validation of the method
The described method was validated in terms of linearity, limit of detection (LOD), limit of quantification (LOQ), recovery, stability, precision and accuracy according to International Conference on Harmonization (ICH) guidelines.22
3.2.1. Method reproducibility.
Method reproducibility was determined by measuring repeatability and intermediate precision of retention times and peak areas for each enantiomer. The repeatability and intermediate precision were expressed as relative standard deviation (R.S.D.). For this study, sitagliptin (10 μg mL−1) spiked with the (S)-enantiomer (0.5%, 50 ng mL−1) was analyzed in six replicates each day over a period of three consecutive days to establish repeatability. The R.S.D. values were less than 0.5% for the retention times of the enantiomers, 1.0% for the sitagliptin peak area and 5.0% for the (S)-enantiomer peak area (Table 5). In the intermediate precision study, results showed that R.S.D. values were in the same order of magnitude as those obtained for repeatability studies (Table 5). All these values indicated that the method was precise.
Table 5 Validation results of the developed chiral LC method
| Validation parameter |
Results |
|
Repeatability (n = 6, % R.S.D.)
|
| Retention time (S-enantiomer) |
0.3 |
| Retention time (sitagliptin) |
0.4 |
| Area (S-enantiomer) |
4.7 |
| Area (sitagliptin) |
0.9 |
| |
|
Repeatability (n = 18, % R.S.D.)
|
| Retention time (S-enantiomer) |
0.7 |
| Retention time (sitagliptin) |
0.5 |
| Area (S-enantiomer) |
4.8 |
| Area (sitagliptin) |
1.0 |
| |
|
LOD–LOQ (S-enantiomer)
|
| Limit of detection (ng mL−1) |
8 |
| Limit of quantification (ng mL−1) |
30 |
| Precision at LOQ (% R.S.D.) |
4.9 |
| |
|
Linearity (S-enantiomer)
|
| Calibration range (ng mL−1) |
30–300 |
| Calibration points (n) |
6 |
| Correlation coefficient (r2) |
0.9996 |
| Slope (% R.S.D.) |
3.7 |
| Intercept (% R.S.D.) |
7.8 |
3.2.2. Limit of detection and limit of quantification of (S)-enantiomer.
The limit of detection (LOD), defined as the lowest concentration of the analyte that can be clearly detected above the baseline signal, is estimated at 3 times the signal to noise ratio. The limit of quantitation (LOQ), defined as the lowest concentration of the analyte that can be quantified with suitable precision and accuracy, is estimated at 10 times the signal to noise ratio. LOD and LOQ were achieved by injecting a series of dilute solutions of the (S)-enantiomer. The precision of the developed chiral method for the (S)-enantiomer was checked by analyzing six test solutions of the (S)-enantiomer prepared at the LOQ level and calculating the percentage relative standard deviation of the area. The LOD and LOQ concentrations were estimated to be 8 and 30 ng mL−1 for the (S)-enantiomer, respectively (Table 5). The method precision for the (S)-enantiomer at LOQ was less than 6.0% R.S.D. Therefore, this method had adequate sensitivity for the detection and estimation of the (S)-enantiomer in sitagliptin.
3.2.3. Linearity of (S)-enantiomer.
Detector response linearity was assessed by preparing six calibration sample solutions of the (S)-enantiomer covering from 30 ng mL−1 (LOQ) to 300 ng mL−1 (30, 60, 100, 150, 200 and 300 ng mL−1) in the mobile phase. The regression curve was obtained by plotting peak area versus concentration, using the least squares method. Linearity was checked for three consecutive days in the same concentration range from the same stock solution. The percentage relative standard deviation of the slope and Y-intercept of the calibration curve was calculated. Good linearity of the (S)-enantiomer was evaluated over six levels of (S)-enantiomer solutions from 30 to 300 ng mL−1, with the linear regression equation y = 0.3729x − 0.7352 (correlation coefficient r2 = 0.9996), where x is the concentration of the enantiomer in ng mL−1 and y is the corresponding peak area of the undesired enantiomer in mV s−1. The R.S.D. of the slope and Y-intercept of the calibration curve were 3.7% and 7.8% (Table 5), respectively.
3.2.4. Quantification of (S)-enantiomer in bulk sample.
The standard addition and recovery experiments were conducted for the (S)-enantiomer in bulk samples in triplicate at 0.4% (40 ng mL−1), 0.6% (60 ng mL−1) and 0.8% (80 ng mL−1) of analyte concentration. A HPLC chromatogram of the sitagliptin bulk drug sample (10 μg mL−1) spiked with the (S)-enantiomer (0.5%) is shown in Fig. 3. The recovery was calculated from the slope and Y intercept of the calibration curve obtained from the linearity study and percentage recovery ranged from 99.06% to 100.2% (Table 6). The method was proved to be accurate in estimating the amount of the undesired enantiomer of sitagliptin between 30 and 300 ng mL−1.
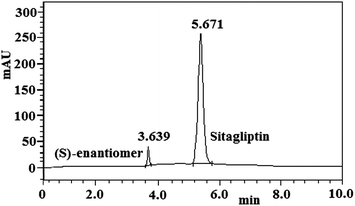 |
| | Fig. 3 Typical HPLC chromatogram of the sitagliptin bulk sample (10 μg mL−1) spiked with (S)-enantiomer (0.5%). | |
Table 6 Recovery results of the (S)-enantiomer in bulk drugsa
| Added concentration (ng mL−1) |
Concentration found (ng mL−1) ± R.S.D. (%) |
Recovery (%) |
|
n = 3 determinations.
|
| 40 |
40.08 ± 3.9 |
100.2 |
| 60 |
59.44 ± 3.1 |
99.06 |
| 80 |
80.03 ± 2.8 |
100.03 |
3.2.6. Stability.
Stability of sitagliptin in solution at analyte concentration was studied by keeping the solution in a tightly capped volumetric flask at room temperature on a laboratory bench for 2 days. The content of the (S)-enantiomer was checked at 6 h interval up to the study period. Mobile phase stability was determined by evaluating the content of the (S)-enantiomer in sitagliptin sample solutions prepared freshly at 6 h interval for 2 days. The same mobile phase was used in the experiment. No significant change in (S)-enantiomer content was observed in the sitagliptin sample during solution stability and mobile phase stability experiments. Hence, the sitagliptin sample solution and mobile phase were stable at least for 48 h.
4. Conclusion
A simple, rapid, and accurate enantioselective Reverse Phase-High Performance Liquid Chromatography (RP-HPLC) method was successfully developed, which was capable of separating the enantiomer from sitagliptin. Two different polysaccharide-based chiral columns Chiralcel OD-RH and Chiralpak AD-RH were found to be selective for the enantiomers of sitagliptin. Method validation was carried out using a Chiralcel OD-RH column due to the better chromatographic results achieved on the column. The validated method was demonstrated to be accurate, precise, selective, sensitive and robust. The developed and validated method can not only be used for the chiral purity testing of sitagliptin, but also for the quantitative determination of chiral impurities in bulk materials.
Acknowledgements
The authors thank the Directors, National Institute of Technology, Warangal and Centre for Materials for Electronics Technology, Hyderabad for providing research facilities and encouragement. Mr Thippani Ramesh thanks MHRD, Government of India for providing financial assistance.
References
- D. Kim, L. Wang, M. Beconi, G. J. Eiermann, M. H. Fisher, H. He, G. J. Hickey, J. E. Kowalchick, B. Leiting, K. Lyons, F. Marsilio, M. E. McCann, R. A. Patel, A. Petrov, G. Scapin, S. B. Patel, R. Sa Roy, J. K. Wu, M. J. Wyvratt, B. B. Zhang, L. Zhu, N. A. Thornberry and A. E. Weber, J. Med. Chem., 2005, 48(1), 141 CrossRef CAS PubMed.
- G. A. Herman, C. Stevens, K. V. Dyck, A. Bergman, B. Yi, M. Smet, K. Snyder, D. Hilliard, M. Tanen, W. Tanaka, A. Q. Wang, W. Zeng, D. Musson, G. Winchell, M. Davies, S. Ramael, K. M. Gottesdiener and J. A. Wagner, Clin. Pharm. Ther., 2005, 78(6), 675–688 CrossRef CAS PubMed.
- A. H. Barnett, Br. Med. Bull., 1994, 50(2), 397–407 CAS.
- E. Grossman and F. H. Messerli, Adv. Cardiol., 2008, 45(1), 82–106 CrossRef CAS PubMed.
- R. Nirogi, V. Kandikere, K. Mudigonda, P. Komarneni, R. Aleti and R. Boggavarapu, Biomed. Chromatogr., 2008, 22(2), 214–222 CrossRef CAS PubMed.
- R. N. Rao, P. K. Maurya and S. Khalid, Talanta, 2011, 85(2), 950–957 CrossRef CAS PubMed.
- R. I. El-Bagary, E. F. Elkady and B. M. Ayoub, Talanta, 2011, 85(1), 673–680 CrossRef CAS PubMed.
- S. Sheetal, M. Nimita, B. Priya, H. Sohail and J. Prabhat, Int. J. Pharm. Biol. Arch., 2012, 3(3), 673–678 Search PubMed.
- K. Y. Kavitha, G. Geetha, R. Hariprasad, R. Venkatnarayana and G. Subramanian, Int. Res. J. Pharm., 2012, 3(12), 123–127 CAS.
- A. B. Loni, M. R. Ghante and S. D. Sawant, Asian J. Pharm. Sci. Res., 2012, 2(8), 23–37 Search PubMed.
- M. Shyamala, S. Mohideen, T. Satyanarayana, Ch. Narasimha Raju, P. Suresh Kumar and K. Swetha, Am. J. Pharmtech. Res., 2011, 1(2), 93–101 Search PubMed.
- G. Jeyabalan and N. Narendra, J. Appl. Pharm. Sci., 2013, 3(1), 95–98 Search PubMed.
- M. M. Maste, A. N. Parate and A. R. Bhat, Asian J. Res. Chem., 2011, 4(9), 1466–1468 Search PubMed.
- V. Goyani, R. Rajeshwari, D. Ranjeet Prasad and N. Manish, Chromatographia, 2013, 76(9–10), 515–521 CAS.
- R. Sonali, P. Pallavi and Ch. Vittal, Int. J. Drug Dev. Res., 2012, 4(3), 292–297 Search PubMed.
- M. Salim, N. El-Enany, F. Belal, M. Walash and G. Patonay, Anal. Chem. Insights, 2012, 7(1), 31–46 CrossRef CAS PubMed.
- E. Ucaktürk, J. Pharm. Biomed. Anal., 2013, 74(1), 71–76 CrossRef PubMed.
- N. M. Maier, P. Franco and W. Lindner, J. Chromatogr., A, 2001, 906(1–2), 3–33 CrossRef CAS.
- R. Nageswara Rao, A. Narasa Raju and D. Nagaraju, J. Pharm. Biomed. Anal., 2006, 41(1), 280–285 CrossRef CAS PubMed.
- M. K. Srinivasu, B. Mallikarjuna Rao, B. Shyam Sundar Reddy, P. Rajeneder Kumar, K. B. Chandrasekhar and P. K. Mohakhud, J. Pharm. Biomed. Anal., 2005, 38(2), 320–325 CrossRef CAS PubMed.
- B. V. Rami Reddy, N. Raman, B. Sai Kumar and Ch. Rambabu, J. Pharm. Res., 2013, 7(1), 546–550 Search PubMed.
-
ICH Draft Guidelines on Validation of Analytical Procedures: Definitions and Terminology, Federal Register, IFPMA, Switzerland, 1995, vol. 60, pp. 11260–11268 Search PubMed.
|
| This journal is © The Royal Society of Chemistry 2014 |
Click here to see how this site uses Cookies. View our privacy policy here.