Species-specific isotope dilution-GC-ICP-MS for accurate and precise measurement of methylmercury in water, sediments and biological tissues
Xu
Ma
,
Yong-guang
Yin
,
Jian-bo
Shi
,
Jing-fu
Liu
* and
Gui-bin
Jiang
State Key Laboratory of Environmental Chemistry and Ecotoxicology, Research Center for Eco-Environmental Sciences, Chinese Academy of Sciences, Beijing, 100085, China. E-mail: jfliu@rcees.ac.cn; Fax: +86-10-62849192; Tel: +86-10-62849192
First published on 29th October 2013
Abstract
Species-specific isotope dilution (SSID) in combination with gas chromatography-inductively coupled plasma-mass spectrometry (GC-ICP-MS) has been developed for the separation and determination of methylmercury (MeHg) in water, sediments and biological tissues. Optimum conditions for the measurement of isotope ratios on the transient chromatographic peaks have been established. SSID analysis was performed using a laboratory synthesized enriched spike of Me198Hg. Isotope ratio precision based on the peak area measurements was 1.4% RSD for 50 pg (as Hg). The absolute detection limits obtained with GC-ICP-MS were 4.3 pg for 198Hg and 8.1 pg for 202Hg, respectively. Analytical precision was typically less than 3.6% RSD over eight repeated measurements. The accuracy of the method has been compared with the method with Tl as internal standard and further validated with certified reference materials (ERM-cc580 for sediments and Tort-2 for the biological tissues). The results obtained by SSID-GC-ICP-MS were in good agreement with the certified reference values.
Introduction
Mercury, as a global pollutant, is ubiquitous in various environmental compartments including atmosphere, soil, water, sediment, and biota.1 The global distribution and cycle of mercury via atmospheric transport have made mercury a great concern for many countries. Methylmercury (MeHg), a neurotoxin and the most toxic organomercuric compound, can be formed in anaerobic sediments through biotic methylation by sulfur-reducing bacteria2 and iron-reducing bacteria.3 Because MeHg can be bioaccumulated through the food chain, it is of great threat to top predators and humans. The negative environmental impacts and the establishment of regulations to reduce mercury pollution call for improvement in current analytical requirements to obtain an accurate and sensitive analytical speciation method, especially for MeHg.In the past decades, chromatographic separation based hyphenated methods have been developed for speciation analysis of MeHg, including gas chromatography (GC)4 and high-performance liquid chromatography5 coupled with element-selective detection, e.g., atomic absorption spectrometry,6 atomic fluorescence spectrometry,7 and inductively coupled plasma mass spectrometry (ICP-MS).8–10 Among these mentioned methods, GC-ICP-MS appears to be the most promising hyphenated technique because of its compatibility with purge-and-trap preconcentration, high sensitivity and multi-isotopic detection capabilities.
Currently, aqueous ethylation–purge and trap is becoming a routine pretreatment protocol for ultra-trace MeHg analysis.11 However, aqueous ethylation using sodium tetraethylborate (NaBEt4) suffers from serious matrix effects. Ubiquitous chloride ions and dissolved organic matter (DOM) in a sample matrix could interfere with the derivatization procedure and therefore decrease the sensitivity and reproducibility of the method.9,12,13For example, chemical transformation of MeHg was observed in different natural water samples during derivatization with NaBEt4.9 Consequently, before ethylation derivatization, water samples with high salinity or DOM should be pretreated via extraction/back-extraction or distillation. Whereas, the tedious pretreatment procedure could decrease the total recovery and also induce artifact formation of MeHg.
To tackle these above mentioned problems of losses and chemical transformation of MeHg in sample cleanup and derivatization, the isotope dilution (ID) technique is a good alternative method14,15,22 since quantitative recoveries are not necessary, once the enriched isotope is fully equilibrated with the natural isotope. Isotope dilution mass spectrometry has been considered to be an absolute method, which can play an important role as the reference method for MeHg analysis in different environmental matrices.
In this work, GC-ICP-MS in combination with the isotope dilution technique was applied to the measurement of MeHg in different environmental matrices. The synthesized Me198Hg has been used for the species-specific isotope dilution of MeHg in DOM-containing water, reference sediment ERM-cc580 and biological tissue Tort-2.
Experimental
Instrumentation
The purge and trap apparatus, including a Hg gold trap for scrubbing the N2-purging gas, a gas flow meter, glass reaction vessels and spargers, Tenax traps, and the trap desorption module, were purchased from Brooks Rand (Seattle, WA). 100 × 4 mm i.d. quartz tubes were filled with 150 mg of Tenax TA as the absorbent material. Adsorbed ethylated Hg species were desorbed at 200 °C for 30 s from the Tenax columns by electrothermal heating using nichrome wire wrapped around the tube and connected to a variable transformer, which was remotely controlled by the mercury analyzer software. The GC column was silanized and packed with 15% OV3 on Chromosorb W, AW, DMCS (Brooks Rand, Seattle, WA, USA). The GC oven was set at 36 °C. High-purity Ar was used as the carrier gas for GC at a flow rate of 25 mL min−1. This setup is essentially the same as that described initially by Hintelmann et al.16 The outflow from the GC was mixed with an Ar stream (using the dilution gas supply of the ICP-MS) and interfaced to the customized torch adapter of the Agilent 7700 ICP-MS by means of a homemade T-connector without additional heating of transfer line because of the high vapor pressure of the organomercuric compounds investigated. All gas flows were controlled by internal mass flow controllers. The operating conditions for the GC-ICP-MS coupling systems are listed in Table 1. The raw data of transient isotope signals for the different mercury species were further processed using Chromafile MS software (Agilent, Japan) to obtain the corresponding isotope ratios.| GC | |
|---|---|
| Argon flow/mL min−1 | 25 |
| Column temperature/°C | 36 |
| Adsorber material | 150 mg Tenax TA, mesh 20/35 |
| Desorption temperature | Ramp heating to 200 °C within 30 s |
| ICP-MS | |
|---|---|
| rf power | 1250 W |
| Carrier gas | 1.0 L min−1 |
| Auxiliary gas | 1.0 L min−1 |
| Plasma gas | 15 L min−1 |
| Dwell time | 80 ms (for all measured isotopes) |
Reagents and chemicals
Water was purified using a Millipore system (Milli-Q, 18 MΩ cm). HNO3 and HCl were purchased from Merck (Darmstadt, Germany). Sodium tetraethylborate (98%) was purchased from Strem Chemicals (Newburyport, MA) as individual 1 g solid samples sealed under argon. Methylcobalamin (Sigma) used for synthesis was a prepared dissolution in an acetic acid–acetate buffer solution (0.1 mol L−1, pH 5). HgCl2 as the enriched stable Hg isotope of 198Hg was obtained from Trace Sciences International (Ontario, Canada). An enriched Me198Hg solution was prepared by the reaction of an inorganic 198Hg solution with methylcobalamin as previously described.17 The relative isotopic abundance and integrity of the MeHg species spike were confirmed frequently by purge and trap GC-ICP-MS analysis.The reference materials Tort-2 (Lobster hepatopancreas) and ERM-cc580 (sediment) were obtained from the Institute for the National Measurement Standard of Research Council of Canada (Ottawa, Ontario, Canada) and the Institute for Reference Materials and Measurements (Geel, Belgium), respectively.
Extraction of MeHg from different environmental matrices
MeHg was isolated from the sediments according to the method described by Qian et al.18 In brief, to about 0.5 g sediment in 50 mL centrifuge tubes, 0.2 mL Me198Hg solution was added as a species-specific internal standard. 10 mL of 1.4 mol L−1 KBr in 5% (v/v) H2SO4 and 2 mL 1 mol L−1 CuSO4 were then added. The centrifuge tubes were shaken for 6 h, then 10 mL of CH2Cl2 was added, and shaking proceeded for additional 4 h. Samples were centrifuged for 15 min at 6000 r min−1 to facilitate separation of the aqueous phase and organic layers. The organic phase was transferred into 50 mL polypropylene tubes and 30 mL of Milli-Q water were added. The samples were purged with N2 to remove the organic solvent. The sample in the water matrix was transferred to a 100 mL glass flask along with 300 μL acetic acid–acetate buffer (pH 4.9) followed by addition of 100 μL 1% NaBEt4 in 2% KOH. The volatile organomercuric compounds were purged and trapped on a Tenax adsorber, thermodesorbed onto the GC column, isothermally separated at 36 °C, and mercury was quantified by ICP-MS. Replicates (n = 4) were prepared for the certified reference material (ERM-cc580).Extraction of MeHg from biological samples was based on the procedure reported by Cai and Bayona.4 Briefly, ∼0.5 g Tort-2 was accurately weighed into 50 mL centrifuge tubes, followed by addition of Me198Hg solution. A 5 mL volume of 25% (w/v) KOH in methanol was added. The tubes were then placed in an ultrasonic bath for 8 h at 70 °C. After the tubes were cooled to ambient temperature, the solution was centrifuged for 15 min at 4000g. From the supernatant, 0.2 mL was transferred to a 100 mL glass flask and MeHg analysis was carried out.
Isotope dilution technique
Isotope dilution (ID) is based on the addition of a known amount of the enriched isotope to a sample. Equilibration of the spiked isotope with the natural elements/species in the sample alters the isotope ratio that is measured and used for calculation. As a result, the achievement of equilibrium of the enriched spike and the sample is critical for the accuracy.The isotopically enriched MeHg was synthesized from inorganic 198Hg through methylation by methylcobalamin based on Martin-Doimeadios's work.17,23 After synthesis, Me198Hg was diluted to a proper concentration to spike the samples. The optimized spike for IDMS is on the same order of magnitude as the amount of species to be analyzed in the sample. The determination of such a Me198Hg solution using GC-ICP-MS resulted in an isotopic composition for the spike and reference isotope of 198Hg = 93.06% and 202Hg = 0.31%. The corresponding concentration of 345 ± 13 μg L−1 solution was determined by using a Brooks Rand cold vapor atomic fluorescence spectrometer model-3 mercury analyzer. The long-term stability of this enriched solution was investigated and no significant change in its concentration and isotopic composition was found within one year of storage.
The MeHg content of the sample was calculated by applying the isotope dilution equation:19
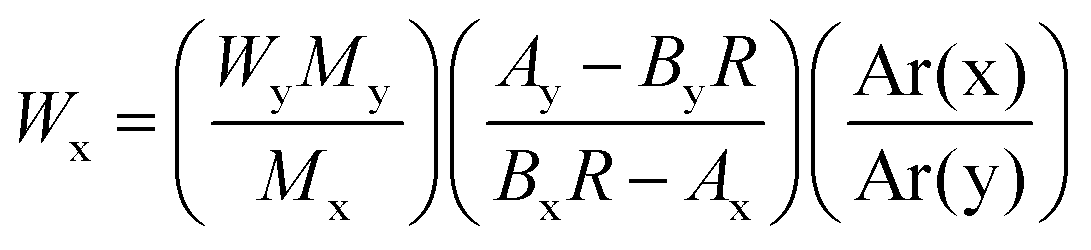
Enriched isotope equilibration experiment
To assess the kinetics of equilibration between the enriched isotope spikes and the ambient Hg species in the presence of natural organic matter (NOM), a kinetic study was conducted with Suwannee River NOM. Solutions of NOM were prepared at a concentration of 20 mg L−1 (corresponding to about 10 mg L−1 dissolved organic carbon). The NOM water samples were spiked at approximately 0.75 ng L−1 MeHg and placed in the dark for 5 days to equilibrate. After equilibration, 100 mL of each spiked NOM solution was pipetted into the reaction vessel. An approximate amount of Me198Hg for isotope dilution quantification was added. The spiked MeHg was then directly ethylated, and purged as mentioned above, without any extraction or distillation. The samples were analyzed by purge and trap-GC-ICP-MS at selected time intervals with multiple time points for the first 24 h and the analysis continued to 48 h. Duplicate experiments were conducted for each NOM sample.Results and discussion
Synthesis of 198Hg-enriched MeHg
The isotopically labeled spike can easily be synthesized in an aqueous solution by methylation of 198Hg-enriched HgCl2 with methylcobalamin as described in Materials and methods. By applying 13.6 mg of HgCl2 as the starting material, Me198Hg was obtained within a total synthesis time of about 2–3 h. However, the synthesis yield of the spike does not play an important role in trace mercury species analysis, because the added amount of spike for isotope dilution-MS should be in the same range as the amount of the species to be determined in the sample. This means that normally, ≪1 μg of the spike Me198Hg is applied per analysis, whereas 13.6 mg of enriched 198Hg produces isotopically labeled Me198Hg sufficient for several thousand isotope dilution-MS analyses, even at low synthesis yields.Optimization of the GC-ICP-MS conditions
For MeHg determination, the analyte being introduced into the plasma is of a gaseous nature, and the optimum conditions obtained with nebulisation differed greatly from the optimum conditions obtained with dry plasma (as obtained by GC-ICP-MS coupling). Thus, the optimization should be carried out on a gaseous species as well rather than aspirating a test solution directly into the plasma. On the other hand, it is impractical to perform an optimization on a transient signal like a MeHg peak. In our study, the ICP-MS operating parameters such as carrier gas flow rate and radio frequency (rf) power were optimized by continuously monitoring the Ar2+ signal daily at m/z = 80 (ref. 20) while the GC is connected. The carrier gas was always held constant at values ranging from 0.85 to 1.2 mL min−1 and the radio frequency power was scanned from 900 to 1550 W over a period of 20 min and the change in sensitivity was recorded in real time. Generally, the sensitivity increased with increasing rf power, reached a plateau at around 1200 W and decreased again. Therefore, a final setting for a carrier flow rate of 1.0 L min−1 and the rf power of 1250 W were chosen for a subsequent study.Also, the sequential detection of ICP-MS with quadrupole mass analyzers poses a potential problem when dealing with transient signals as obtained by GC separation. In order to avoid so-called spectral skew, which describes the peak shape deterioration and apparent shift on different isotopes, the integration time per isotope that may affect precision and accuracy of the isotope ratio measurements had been carefully evaluated. Long integration times provide the advantage of improved signal-to-noise ratios, but the chromatographic profile will be ill-defined, while very short integration time per isotope provides good peak definition, but suffers from higher signal noise. Thus, 80 ms for mercury isotopes (198, 202) was chosen, resulting in a total integration time of <200 ms and a total chromatographic elution time of 6 min.
Analytical performance
Reproducibility of 198Hg/202Hg isotope ratio measurements on peak area was 1.4% (50 pg, n = 4). Calibration curves in the range of 10–200 pg for Me202Hg exhibited linear regression slopes and coefficients of 1510 counts per pg and r2 = 0.994, respectively. The average absolute detection limits (n = 8), calculated using three times the standard deviation of background noise next to the chromatographic peak, were 4.3 pg for 198Hg and 8.1 pg for 202Hg or, assuming a sampling volume of 100 mL, 43 pg L−1 for 198Hg and 81 pg L−1 for 202Hg, respectively. However, it is important to note that this is the instrument detection limit for the GC-ICP-MS, representing the lowest-possible attainable detection limits based solely on the signal-to-noise ratio of ICP-MS. The real limit of detection for isotope dilution analysis depends on the precision with which isotope ratios can be measured. It is necessary to measure the isotope ratio with optimum precision. In our experimental conditions, above 10 pg of MeHg, sufficient precision with RSD < 3.6% (n = 8) in the isotope ratio measurements was obtained.Enriched isotope equilibration kinetics in DOM-containing water
In surface water, MeHg often exists as a complex with DOM through coordination of MeHg at reduced sulfur sites within the organic matter.21 Consequently, the ubiquitous DOM in water samples could decrease the derivitization efficiency of MeHg. We conducted a kinetic experiment to establish the time for the isotope spike to equilibrate with the natural MeHg complexed to the natural DOM sample. In terms of using isotope dilution for quantification of the Hg species, if equilibrium of the enriched isotope spike with the ambient Hg species is not instantaneous, the added spike will be more reactive to ethylation than the natural Hg species, leading to a higher isotope ratio and a lower calculated concentration for the natural Hg species than that of the initial added amount. As the spike equilibrates with the natural Hg species, the isotope ratio should decrease, and the predicted concentration should approach the expected concentration. The result of this kinetic experiment is shown in Fig. 1. Initially, the calculated MeHg concentration of the NOM solution was 81% of the added concentration. The enriched spike continued to equilibrate over time, and after 12 h of equilibration, the MeHg concentration plateaued at 0.76 ng L−1, which is 101% recovery of the added concentration. As a result of our kinetic study, we established a 24 h equilibration time for the enriched isotope spikes prior to speciation analysis. The enriched isotope equilibration kinetics also gives a good alternative for studying the interaction kinetics between MeHg and DOM.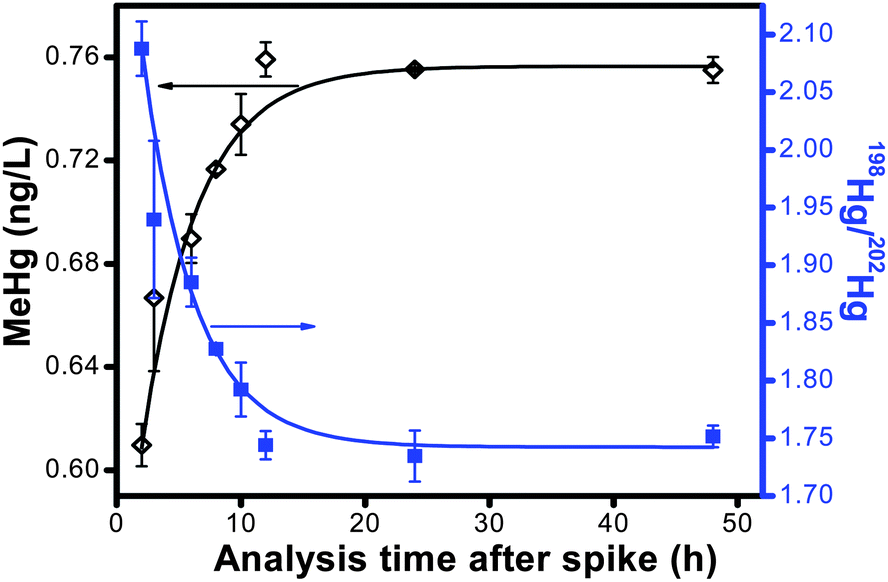 | ||
| Fig. 1 MeHg concentration and isotope ratio determined by ID-GC-ICP-MS as a function of equilibration time after the isotope spike. | ||
In addition, MeHg concentration in DOM-rich water was determined using internal standard quantification (approximately 5 μg L−1 Tl as internal standard solution to calibrate the instrumental shift during GC-ICP-MS analysis) to further validate the superiority of the isotope dilution technique over the traditional quantitative method. The results showed that the total recovery of MeHg using direct ethylation was 70.7 ± 2.3% (n = 4) and 57.2 ± 1.0% (n = 4), respectively after spiking with 0.7 ng L−1 and 1.0 ng L−1 MeHg standard solution, indicating that the incomplete recovery occurred resulting from the DOM interference during the ethylation procedure.
As a result, we can safely conclude that the isotope dilution technique is expected to be one of the most promising tools to determine ultra-trace MeHg concentration in DOM-rich freshwater, irrespective of total recovery.
Validation of the isotope dilution analysis of MeHg in certified sediment and biological samples
Speciated isotope dilution analysis was further validated by measuring MeHg in a sediment certified reference material (ERM-cc580) and a lobster tissue reference material (Tort-2). As full equilibration of isotope spike is critical for the accurate quantification, long equilibration times of the spike and sample (days) are used in combination with short extraction time (in hours) by wet extraction for sediment and ultrasonic-assisted extraction for biological samples, respectively. Optimum of the analyte-to-spike ratio is also important for minimizing errors in the final determination. Most of the analytical uncertainties can be avoided when the spiked isotope concentration matches the analyte concentration in the sample. In our case, four levels of MeHg from 0.0345 to 34.5 ng were spiked into the reference materials. Four independent spiking experiments were carried out to analyze each reference material.The chromatograms obtained for ERM-cc580 and Tort-2 spiked with 198Hg-enriched MeHg solution are shown in Fig. 2. As can be seen, the isotopic composition of MeHg has been altered drastically after Me198Hg spiking, from the natural value 198/202 of 0.333 to the measured isotope ratio of 1.598 for Tort-2 and 3.992 for ERM-cc580, respectively.
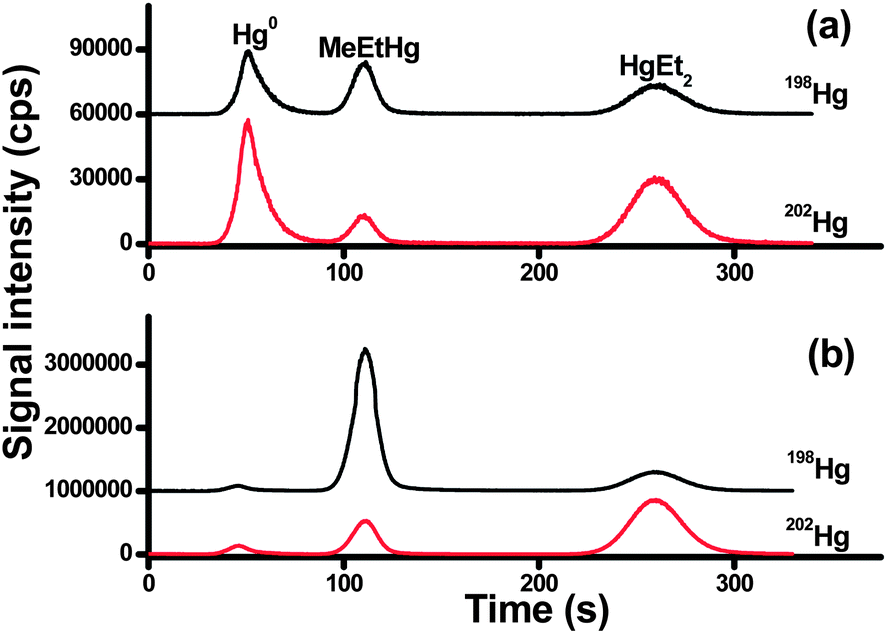 | ||
| Fig. 2 Chromatograms obtained for (a) Tort-2 and (b) ERM-cc580 extracts spiked with 198Hg-enriched MeHg solution. Note: the chromatograms at mass 198 were shifted to facilitate the comparison. | ||
Final results for the speciation of MeHg by isotope dilution analysis are presented in Table 2. As can be seen, the isotope dilution protocol provides excellent results independent of the total recovery, if an appropriate amount of spike is added to the sample and the spike fully equilibrates prior to sample pretreatment. Results within the analytical uncertainty between the certified and determined concentrations were obtained for both ERM-cc580 and Tort-2, and values close to the recommended value were found for both the reference materials used in this study.
| Material | Me198Hg spiked (ng g−1) | Determined (ng g−1) | Certified (ng g−1) |
|---|---|---|---|
| ERM-cc580 (sediment) | 73.4 | 74.9 ± 0.4 (n = 4) | 75.0 ± 4.0 |
| 7.34 | 59.2 ± 0.3 (n = 4) | ||
| 0.734 | 51.4 ± 2.2 (n = 4) | ||
| 0.0734 | 25.0 ± 3.8 (n = 4) | ||
| Tort-2 (Lobster tissue) | 156.8 | 155.3 ± 1.1 (n = 4) | 152.0 ± 13.0 |
Conclusions
An accurate and precise isotope dilution-GC-ICP-MS method has been developed for MeHg determination in water, sediments and biological tissues. As isotope dilution is considered to be a definitive method of analysis, the proposed procedure could be used as a reference method in control laboratories. In addition to the inherent benefits, the isotope dilution technique facilitates a considerably simpler and more flexible analytical protocol in terms of sample processing, compared to most other methods for trace element speciation analysis, which is a very important feature for routine applications.Acknowledgements
The authors acknowledge the joint support from the National Program on Key Basic Research Project (973 Program) (2013CB430002) and the National Natural Science Foundation of China (21120102040, 20937002, and 21025729).References
- C. T. Driscoll, R. P. Mason, H. M. Chan, D. J. Jacob and N. Pirrone, Mercury as a Global Pollutant: Sources, Pathways, and Effects, Environ. Sci. Technol., 2013, 47, 4967 CrossRef CAS PubMed.
- J. K. King, J. E. Kostka, M. E. Frischer and F. M. Saunders, Sulfate-reducing bacteria methylate mercury at variable rates in pure culture and in marine sediments, Appl. Environ. Microbiol., 2000, 66, 2430 CrossRef CAS.
- E. J. Fleming, E. E. Mack, P. G. Green and D. C. Nelson, Mercury methylation from unexpected sources: Molybdate-inhibited freshwater sediments and an iron-reducing bacterium, Appl. Environ. Microbiol., 2006, 72, 457 CrossRef CAS PubMed.
- Y. Cai and J. M. Bayona, Determination of Methylmercury in Fish and River Water Samples Using in-Situ Sodium Tetraethylborate Derivatization Following by Solid-Phase Microextraction and Gas-Chromatography Mass-Spectrometry, J. Chromatogr., A, 1995, 696, 113 CrossRef CAS.
- L. N. Liang, G. B. Jiang, J. F. Liu and J. T. Hu, Speciation analysis of mercury in seafood by using high-performance liquid chromatography on-line coupled with cold-vapor atomic fluorescence spectrometry via a post column microwave digestion, Anal. Chim. Acta, 2003, 477, 131 CrossRef CAS.
- S. Rapsomanikis and P. J. Craig, Speciation of Mercury and Methylmercury Compounds in Aqueous Samples by Chromatography Atomic-Absorption Spectrometry after Ethylation with Sodium Tetraethylborate, Anal. Chim. Acta, 1991, 248, 563 CrossRef CAS.
- Y. G. Yin, J. F. Liu, B. He, J. B. Shi and G. B. Jiang, Simple interface of high-performance liquid chromatography-atomic fluorescence spectrometry hyphenated system for speciation of mercury based on photo-induced chemical vapour generation with formic acid in mobile phase as reaction reagent, J. Chromatogr., A, 2008, 1181, 77 CrossRef CAS PubMed.
- H. Hintelmann, R. D. Evans and J. Y. Villeneuve, Measurement of Mercury Methylation in Sediments by Using Enriched Stable Mercury Isotopes Combined with Methylmercury Determination by Gas-Chromatography Inductively-Coupled Plasma-Mass Spectrometry, J. Anal. At. Spectrom., 1995, 10, 619 RSC.
- N. Demuth and K. G. Heumann, Validation of methylmercury determinations in aquatic systems by alkyl derivatization methods for GC analysis using ICP-IDMS, Anal. Chem., 2001, 73, 4020 CrossRef CAS.
- Y. X. Mao, G. L. Liu, G. Meichel, Y. Cai and G. B. Jiang, Simultaneous speciation of monomethylmercury and monoethylmercury by aqueous phenylation and purge-and-trap preconcentration followed by atomic spectrometry detection, Anal. Chem., 2008, 80, 163 CrossRef PubMed.
- M. S. Jimenez and R. E. Sturgeon, Speciation of methyl- and inorganic mercury in biological tissues using ethylation and gas chromatography with furnace atomization plasma emission spectrometric detection, J. Anal. At. Spectrom., 1997, 12, 597 RSC.
- A. De Diego, C. M. Tseng, T. Stoichev, D. Amouroux and O. F. X. Donard, Interferences during mercury speciation determination by volatilization, cryofocusing, gas chromatography and atomic absorption spectroscopy: comparative study between hydride generation and ethylation techniques, J. Anal. At. Spectrom., 1998, 13, 623 RSC.
- R. Fischer, S. Rapsomanikis and M. O. Andreae, Determination of Methylmercury in Fish Samples Using Gc/Aa and Sodium Tetraethylborate Derivatization, Anal. Chem., 1993, 65, 763 CrossRef CAS.
- R. C. R. Martin-Doimeadios, E. Krupp, D. Amouroux and O. F. X. Donard, Application of isotopically labeled methylmercury for isotope dilution analysis of biological samples using gas chromatography/ICPMS, Anal. Chem., 2002, 74, 2505 CrossRef.
- J. P. Snell, I. I. Stewart, R. E. Sturgeon and W. Frech, Species specific isotope dilution calibration for determination of mercury species by gas chromatography coupled to inductively coupled plasma- or furnace atomisation plasma ionisation-mass spectrometry, J. Anal. At. Spectrom., 2000, 15, 1540 RSC.
- H. Hintelmann and R. D. Evans, Application of stable isotopes in environmental tracer studies – Measurement of monomethylmercury (CH3Hg+) by isotope dilution ICP-MS and detection of species transformation, Fresenius' J. Anal. Chem., 1997, 358, 378 CrossRef CAS.
- R. C. R. Martin-Doimeadios, T. Stoichev, E. Krupp, D. Amouroux, M. Holeman and O. F. X. Donard, Micro-scale preparation and characterization of isotopically enriched monomethylmercury, Appl. Organomet. Chem., 2002, 16, 610 CrossRef CAS.
- J. Qian, U. Skyllberg, Q. Tu, W. F. Bleam and W. Frech, Efficiency of solvent extraction methods for the determination of methyl mercury in forest soils, Fresenius' J. Anal. Chem., 2000, 367, 467 CrossRef CAS.
- E. Prichard, G. M. MacKay and J. Points, Trace analysis: A structured approach to obtaining reliable results, The Royal Society of Chemistry, London, 1998 Search PubMed.
- J. R. Encinar, J. I. G. A. Alonso and A. Sanz-Medel, Synthesis and application of isotopically labelled dibutyltin for isotope dilution analysis using gas chromatography-ICP-MS, J. Anal. At. Spectrom., 2000, 15, 1233 RSC.
- M. Ravichandran, Interactions between mercury and dissolved organic matter – a review, Chemosphere, 2004, 55, 319 CrossRef CAS PubMed.
- L. Yang, Z. Mester and R. E. Sturgeon, Determination of methylmercury in fish tissues by isotope dilution SPME-GC-ICP-MS, J. Anal. At. Spectrom., 2003, 18, 431 RSC.
- C. Bancon-Montigny, L. Yang, R. E. Sturgeon, V. Colombini and Z. Mester, High-yield synthesis of milligram amounts of isotopically enriched methylmercury (CH3198HgCl), Appl. Organomet. Chem., 2004, 18, 57 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2014 |