Ultra-trace determination of polycyclic aromatic hydrocarbons using solid-phase extraction coupled with HPLC based on graphene-functionalized silica gel composites
Ke-Jing
Huang
*,
Yu-Jie
Liu
,
Jing
Li
,
Tian
Gan
and
Yan-Ming
Liu
College of Chemistry and Chemical Engineering, Xinyang Normal University, Xinyang 464000, P.R. China. E-mail: kejinghuang@163.com; Tel: +86-376-6390611
First published on 30th October 2013
Abstract
A graphene (Gr) functionalized silica gel (Gr–SiO2) sorbent was prepared and used as a new sorbent for solid-phase extraction (SPE) of polycyclic aromatic hydrocarbons (PAHs) including naphthalene, acenaphthylene, fluorene, phenanthrene, fluoranthene, and pyrene. The analytes were separated and detected by high-performance liquid chromatography (HPLC) with a UV detector. Factors affecting the extraction efficiency including the eluent type and its volume, adsorbent amount, sample volume, sample pH and flow rate were optimized. Under the optimized conditions, the present method showed a wide linear range (0.01–600 μg L−1), acceptable repeatability of the extraction (RSDs of 1.7–8.2%, n = 3) and satisfactory detection limits (0.0029–0.052 μg L−1). The recovery studies using this new sorbent were performed by three consecutive extraction steps of water and milk samples at three spiked levels. The average recoveries of the analytes in environmental water and milk samples were 89.0–115.4%, which demonstrated the applicability of the developed method.
1 Introduction
Polycyclic aromatic hydrocarbons (PAHs) are a large class of organic compounds which contain two or more fused aromatic rings. They are ubiquitous products of the incomplete combustion of organic materials resulting from burning of fossil fuels, waste incineration, landfill, etc.1,2 The PAH residues in the environment can result in food pollution and can be concentrated in the human body through both aquatic and terrestrial food chains.3 In view of the possible harmful effects of PAHs on human health and food safety, the development of sensitive methods for PAH analysis is of great importance.Some analytical techniques have been developed for the determination of PAH compounds in different samples, such as synchronous fluorescence spectroscopy,4 gas chromatography,5 capillary electrophoresis,6 and high-performance liquid chromatography (HPLC).7–10 However, clean-up and enrichment procedures are usually required because of the complexity of the matrix and the low content of PAHs in environmental samples. Because of its advantages of high enrichment factor, high recovery, rapid phase separation, low cost, and low consumption of organic solvents, solid-phase extraction (SPE) has been the most common technique used for preconcentration of analytes in various samples.11 The choice of appropriate adsorbent is a critical factor to obtain good recovery and a high enrichment factor in the SPE procedure.12 For sample pretreatment, carbon nanoparticles represent some exciting prospects, such as the ease of material processing, low cost of synthesis, high surface area, good chemical stability, and strong mechanical strength,13,14 and they have been widely applied in sample pretreatment procedures and good analytical performance is obtained.15,16
As one of the carbon nanomaterials, graphene (Gr) attracts an enormous amount of interest from scientists.17–20 The two-dimensional structure of Gr provides numerous unexpected properties for sample pretreatment: both sides of the planar sheets are available for molecule adsorption, suggesting high sorption capacity;21 the high specific surface area (close to 2630 m2 g−1) means high sorption capacities; easy synthesis and processing imply low cost. These unusual and robust properties are useful in sample treatment procedures. Most recently, we reported a high-performance liquid chromatography (HPLC) method for sensitive determination of γ-aminobutyric acid, taurine and glycine in rat brain using Gr as a sorbent for SPE.22 The results revealed the potential of Gr as a sorbent in the analysis of biological samples. Wu et al. reported a Gr-based magnetic nanocomposite used as an adsorbent for the preconcentration of the five carbamate pesticides (metolcarb, carbofuran, pirimicarb, isoprocarb and diethofencarb) in environmental water samples prior to high performance liquid chromatography-diode array detection.23 Zhang et al. reported gas chromatography-mass spectrometry coupled with micro-solid-phase extraction for the determination of polycyclic aromatic hydrocarbons (PAHs) in water based on sulfonated Gr sheets.24 However, some problems still need to be solved when using Gr as an adsorbent in sample pretreatment. Gr usually congregates when packed into SPE cartridges, which leads to blockage in the extraction column. Therefore, many interesting and unique properties of Gr can only be realized after it is integrated into more complex assemblies. A useful technique to incorporate Gr into such assemblies is through chemical modification, which enables chemical covalent bonding between the Gr and the material of interest.25
In this work, a new sorbent of SPE was prepared based on Gr functionalized silica gel (Gr–SiO2). Six PAHs including naphthalene (Nap), acenaphthylene (Acy), fluorene (Flo), phenanthrene (Ph), fluoranthene (Fl) and pyrene (Py) were selected to test the extraction performances of Gr–SiO2 sorbents. The six PAHs were separated and detected by the HPLC-UV technique after extracted by Gr–SiO2 packed SPE cartridges. The results obtained with the Gr–SiO2 were compared with those of a commercial C18 sorbent. Finally, the Gr–SiO2 was tested in the environmental water and milk samples.
2 Experimental
2.1 Chemicals
PAHs (Nap, Acy, Flo, Ph, Fl, Py) and (3-aminopropyl) triethoxysilane (APS) were obtained from Aladdin (analytical reagent grade, Aladdin Reagent Co., Ltd., Shanghai, China). Graphite powder (spectral pure), N,N′-dicyclohexyl carbodiimide (DCC) (analytical reagent grade) and P2O5 (analytical reagent grade) were purchased from Sinopharm chemical reagent Co. Ltd. (Shanghai, China). Ultrapure water (18.2 MΩ) was obtained from a Milli-Q water purification system and used in all experiments.2.2 Apparatus
An Agilent 1200 Series of HPLC system, which consisted of a quaternary pump, a vacuum degasser and a UV detector, was used. A reversed-phase EclipseXDB-C18 column (150 mm × 4.6 mm i.d., 5 μm, Agilent, USA) was used for separation at ambient temperature and a manual sample injector with a 20 μL loop was applied for sample injection. Agilent ChemStation for the HPLC system was employed to acquire and process chromatographic data. The mobile phase consisting of 15% water and 85% methanol was delivered at a flow rate of 0.5 mL min−1. The detection wavelength was set at 254 nm. The SPE cartridge (1 mL) was obtained from Agilent (Agilent, Palo Alto, CA, USA). The pH measurements were performed with a pH meter (MP 230, Mettler-Toledo, Greiffensee, Switzerland). The images of scanning electron microscopy (SEM) were obtained from a Hitachi S-4800 (Japan). Fourier-transform infrared (FT-IR) spectra (KBr pellets) were recorded using a Bruker TENSOR27 instrument (Ettlingen, Germany). The N2 adsorption–desorption isotherms of the samples were measured using a NOVA 2000 (Quantachrome, USA) in order to determine the specific surface areas. The specific surface area was calculated from the Brunauer–Emmett–Teller (BET) plot of the nitrogen adsorption isotherm.2.3 Preparation of graphene-functionalized silica gel composites
Natural graphite powder was oxidized to graphite oxide by the modified Hummers method.26 Specifically graphite powder (5.0 g) was dispersed in a solution containing 87.5 mL concentrated H2SO4 and fuming HNO3 (45 mL) in an ice bath. 55 g KClO3 was then added gradually. The mixture was stirred for 96 h and then diluted with deionized water. The obtained graphite oxide (GO) was re-dispersed in deionized water and then exfoliated to GO sheets by ultrasonication. A brown homogeneous supernatant was obtained by repeated centrifugation and washing.The preparation of Gr functionalized-silica gel included four steps: (a) 5 g silica gel was added to HCl solution (6 mol L−1) under stirring, and then incubated at 97 °C for 4 h. After cooling naturally, the white precipitate was collected and washed with water and ethanol, respectively, and then dried in a vacuum oven at 80 °C; (b) the resultant composite was dispersed in 75 mL toluene. 5 mL APS was then added into the above mixture and stirred at 97 °C for 12 h. The preliminary activation of silica gel was performed after washing with water and ethanol; (c) 1.0 g preliminary activated silica gel was added into 150 mL GO solution. Subsequently, 2.5 g DCC was added immediately. The mixture was stirred and kept at 97 °C until white precipitates turned to gray; (d) 0.8 g sodium borohydride was added to the above mixture and incubated at 70 °C for 8 h. The precipitate was collected and washed thoroughly with water and methanol, respectively. A Gr–SiO2 composite was obtained after being dried at 60 °C under vacuum.
2.4 Solid-phase extraction procedures
A Gr–SiO2 packed mini-column was prepared by modifying a SPE column (1 mL, polypropylene), which was purchased from Agilent Corporation. The C18 packing was evacuated and a 30 mg Gr–SiO2 composite was packed in the column. The polypropylene upper frit was reset at the upper end of the column to prevent the loss of Gr–SiO2 during the operation process. In order to reduce the possible contamination, the SPE column was equilibrated with 10 mL of methanol and water, respectively. The aqueous sample (100 mL) was uploaded into the cartridge at a flow rate of 1.5 mL min−1. After washing with 5 mL water, the cartridge was washed with 0.2 mL acetonitrile to desorb the analytes from the cartridge. The extract was filtered through a 0.45 μm Millipore membrane. Finally, 20 μL of the eluate was injected into the HPLC system for analysis. After extraction, the cartridge was washed with 10 mL methanol and 10 mL water, respectively. In this way, the cartridge was available for next extraction immediately. The SPE procedure is listed in Scheme 1. | ||
| Scheme 1 The SPE procedure. | ||
2.5 Sample preparation
Two environmental water samples were chosen to evaluate the developed SPE-HPLC method. Pond water was taken from Xinyang Normal University, Xinyang, Henan Province, China (114°1′58.4′′E 32°8′18.5′′N). The river water was collected from Shihe River, Xinyang, Henan Province, China (114°1′48.0′′E 32°7′54.1′′N). Milk (whole milk, skimmed milk) used in this study was commercially available. 10.0 g milk powders were added into a 100 mL centrifuge tube, then 40 g H2O (40 °C) was added, and the mixture was shaken continuously. Finally, the mixture was cooled to room temperature. After centrifugation for 10 min (8000 rpm), a fat layer was removed and the supernatant was collected. Before the experiment, the collected water and milk samples were filtered through a Millipore membrane with pore size 0.45 μm immediately after sampling and were maintained in amber glass bottles at 4 °C.3 Results and discussion
3.1 Characterization of the Gr–SiO2 composites
Fig. 1 is the SEM images of Gr, silica gel and Gr–SiO2. A typical SEM image of the as-synthesized Gr shows 2D nanosheet morphologies (Fig. 1a), exhibiting a few thin wrinkles on the surface. Fig. 1b is the SEM image of silica gel. It exhibits a complanate surface of silica gel. Fig. 1c shows the SEM image of Gr–SiO2 composites. It exhibits the folded and winkled Gr sheets that are integrated on the surface of the silica gel. The multi-layers Gr is anchored on the silica gel, indicating the enlarged specific surface area of the Gr–SiO2 composites, which is benefit to the SPE of PAHs. | ||
| Fig. 1 SEM images of Gr (a), silica gel (b) and Gr–SiO2 (c). | ||
EDX was used to determine the composition of the Gr–SiO2 composites. The results showed that the atomic ratio percentage C/Si was about 3 in the samples, indicating the successful anchoring of Gr onto the surface of the silica gel. The specific surface areas calculated by the BET method of silica gel and Gr–SiO2 composites are 481.5 m2 g−1 and 578.2 m2 g−1, respectively. Obviously, the BET specific surface area of Gr–SiO2 composites is much higher than that of silica gel, which means that the Gr sheets anchoring onto the silica gel can effectively increase the specific surface areas of the silica gel.
3.2 Optimization of SPE procedures
To achieve accurate and sensitive chromatographic quantification of ultra-trace PAHs in complex samples, the optimum conditions for SPE were investigated, including the eluent type and its volume, adsorbent amount, sample volume, sample pH, ionic strength and sample flow rate.As far as the SPE method is concerned, the selection of organic solvent used as an eluent plays the most important role in the desorption efficiency. Five eluents including acetonitrile, methanol, acetone, ethanol and cyclohexane were tested. The experimental results demonstrated that acetonitrile, methanol and acetone give the higher elution efficiency than ethanol and cyclohexane (Fig. 2A). However, when acetone was used as an eluent, some impurity peaks appeared in the chromatogram, which interfered the separation of the PAHs (Fig. 2B). So acetonitrile was chosen as the eluent for further experiments.
 | ||
| Fig. 2 (A) Effect of eluent type on the peak area of PAHs; (B) the chromatogram of PAHs using acetone (a) and acetonitrile (b) as eluent. Eluent volume: 0.2 mL and sample volume: 40 mL. PAH concentrations: 100 μg L−1. | ||
The amount of eluent affects the desorption of PAHs. Herein, the volume of acetonitrile was optimized in the range of 0.15–0.5 mL. The results are shown in Fig. 3A. It is obvious that the extraction performance of the Gr–SiO2 packed cartridge is affected by the amount of the eluent. The peak areas of 6 objectives increase when the volume of acetonitrile varies from 0.15 to 0.2 mL, and then decrease when the eluent volume exceeds 0.2 mL. Therefore, 0.2 mL acetonitrile was selected in the further experiments.
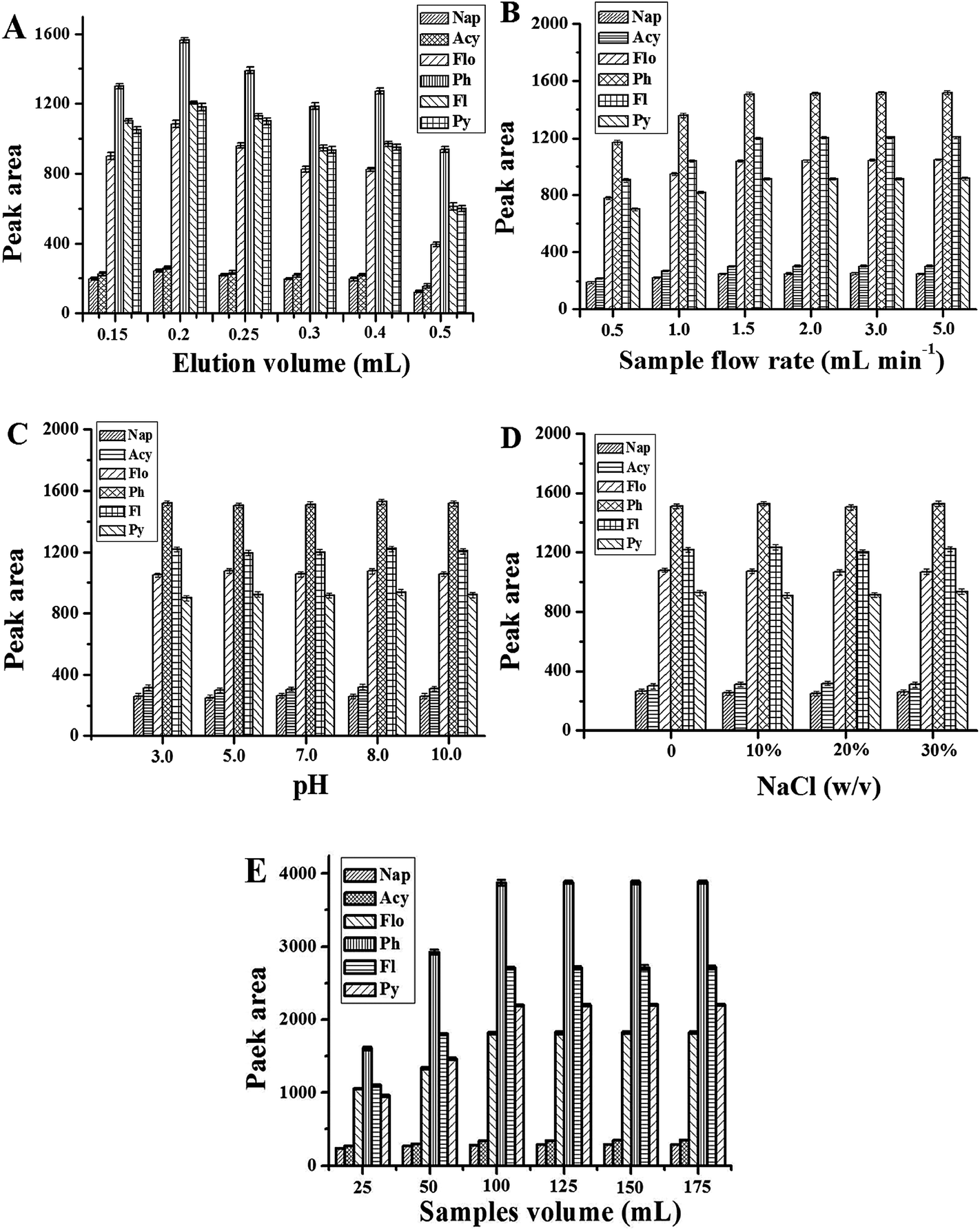 | ||
| Fig. 3 Effect of eluent volume (A), sample flow rate (B), sample pH (C), amount of NaCl (D), and sample volume (E) on the peak area of PAHs. The other conditions are same as in Fig. 4. | ||
For SPE, mass transfer of analytes from the sample to the sorbent materials is time-dependent and equilibrium based rather than an exhaustive extraction process. The sample flow rate is a key factor for extraction efficiency. In this work, the sample flow rate is estimated within the range of 1–5 mL min−1 (Fig. 3B). The experimental results show that the peak areas of PAHs increase with the sample flow rate up to 1.5 mL min−1 with no significant improvement thereafter. A sample flow rate of 1.5 mL min−1 was therefore selected for further work.
Sample pH plays an important role in the SPE procedure, because the pH value of the solution determines the present state of analytes in solution as an ionic or a molecular form, and thus determines the extraction efficiency of the target analytes. Herein, a range of sample pH values from 3 to 10 is evaluated (Fig. 3C). No significant effect is observed with sample pH change, suggesting that a non-electrostatic interaction such as pi–pi interaction plays an important role in the adsorption of PAHs to the Gr–SiO2 sorbent. Hence, all the environmental water samples can be preconcentrated directly without adjusting acidity because the pH value of environmental water hardly exceeds the range of pH 3–10.
The effect of salt addition has been used in SPE to decrease the solubility of analytes in the aqueous sample and enhance their partitioning into the organic solvent. For this investigation, various amounts of sodium chloride (ranging from 0 to 30%, w/v) are added to the sample to study the phenomenon (Fig. 3D). 100 mL of sample solution was used. No significant variation in the extraction efficiency is observed with the change of the NaCl concentration indicating that the ionic strength is negligible for the next analytical procedure.
The amount of adsorbent is another important parameter that affects the extraction efficiency. A quantitative retention is not obtained when the amount of Gr–SiO2 is less than optimum. On the other hand, an excess amount of adsorbent prevents the quantitative elution of the retained PAHs by a small volume of eluent. In order to examine the optimum amounts of solid phase, 10–50 mg of Gr–SiO2 was packed into the columns and the same volume of sample solution (100 mL) was treated by applying the general procedure. The results showed that the biggest peak areas of 6 PAHs were obtained when 30 mg Gr–SiO2 was used.
The adsorption capacity of a sorbent is an important parameter in assessing its ability to retain selected analytes. Breakthrough volume and optimal sample volume were investigated in order to obtain a higher extraction efficiency. In this study, different sample volumes (25, 50, 100, 125, 150, 175 mL) were employed and the peak areas of the target compounds were determined under the following conditions: 30 mg Gr–SiO2 sorbent, a flow rate of 1.5 mL min−1, and 0.2 mL acetonitrile as the eluent. The results are shown in Fig. 3E. For all analytes, the peak areas increase greatly in the sample volume range of 25–100 mL. However, a small change of the peak area is observed when the sample volume is over 100 mL. In practice, the sample volume is chosen according to the required sensitivity and the time acceptable for a whole analysis. Generally, further increasing the sample volume is not desirable for routine analysis since the total time needed for one analysis would be longer. So a sample volume of 100 mL was selected for subsequent analysis.
3.3 Detection of target PAHs
The external standard method for quantification was used in this analytical process. A calibration plot of the peak area as a function of the concentration of each analyte was used for quantification of the PAHs in the water samples. The analytical characteristic data for the graphene-functioned silica gel sorbent coupling with HPLC are shown in Table 1. Calibration curves were obtained by analyzing 12 concentrations of standard mixtures, with three replicates at each concentration level. A wide linear range for each analyte was obtained (0.01–600 μg L−1) with correlation coefficients higher than 0.9933. The precision of the method was assessed by determining the intraday and interday relative standard deviations (RSDs) at three concentration levels. The intraday and interday RSDs were below 7.4% and 8.2%, respectively. The sensitivity was evaluated by the limit of detection (LOD) and the limit of quantitation (LOQ). LOD and LOQ were defined as concentrations where the signal/noise ratios were equal to 3 and 10, respectively. As shown in Table 1, the LODs and LOQs were in the range of 0.0029–0.052 μg L−1 and 0.0096–0.17 μg L−1, respectively. For the repeatability study, one cartridge was used for six replicate extractions of an aqueous sample containing 5 μg L−1 PAHs under the same conditions. The RSDs were obtained below 6.0%. All these results gave evidence to demonstrate that the method was unbiased and could be applied to test the samples. Furthermore, six parallel-made Gr–SiO2 cartridges were used to detect 5 μg L−1 PAHs, respectively. The RSD was 5.8%, indicating a satisfactory reproducibility. Some reported methods for the PAH determination are compared in Table 2. The present work showed higher sensitivity and a wider linear range.27–31| Analytes | Regression equation | Linear range (μg L−1) | R | RSD | LOD (μg L−1) | LOQ (μg L−1) | |
|---|---|---|---|---|---|---|---|
| Intraday (n = 3) | Interday (n = 3) | ||||||
| Nap | y = 0.486x + 13.723 | 0.10–300 | 0.9933 | 3.6 | 7.3 | 0.052 | 0.17 |
| Acy | y = 0.545x + 29.113 | 0.10–300 | 0.9958 | 5.2 | 8.2 | 0.024 | 0.079 |
| Flo | y = 5.164x + 274.471 | 0.01–400 | 0.9976 | 1.7 | 3.3 | 0.0029 | 0.0096 |
| Ph | y = 31.206x + 988.506 | 0.01–400 | 0.9998 | 1.9 | 4.1 | 0.0082 | 0.027 |
| Fl | y = 28.007x + 118.494 | 1–600 | 0.9996 | 3.4 | 8.0 | 0.014 | 0.046 |
| Py | y = 23.291x + 103.660 | 1–600 | 0.9998 | 4.5 | 7.1 | 0.012 | 0.040 |
| Adsorbents | LOD (μg L−1) | Linear range (μg L−1) | RSD% | References |
|---|---|---|---|---|
| Multi-walled carbon nanotubes | Nap: 0.021, Acy: 0.058, Flo: 0.026, Ph: 0.009, Fl: 0.034, Py: 0.036 | Nap, Acy: 0.4–100, Flo: 0.1–25, Ph: 0.04–10, Fl, Py: 0.2–50 | Nap: 4.8, Acy: 4.0, Flo: 4.6, Ph: 3.0, Fl: 4.6, Py: 4.7 | 27 |
| Magnetic C18 microspheres | Nap: 0.8, Acy: 4.1, Flo: 3.9, Ph: 3.4, Py: 5.1 | Nap, Acy, Flo, Ph, Py: 10–800 | Nap: 4.1, Acy: 5.0, Flo: 4.9, Ph: 5.2, Py: 4.0 | 28 |
| TiO2 nanotubes | Acy: 0.031, Flo: 0.026, Ph: 0.053, Fl: 0.017, Py: 0.059 | Acy: 10–500, Fl, Flo: 50–400, Ph: 50–800, Py: 10–800 | — | 29 |
| Zeolite imidazolate framework 8 | Nap: 0.083, Acy: 0.050, Flo: 0.067, Ph: 0.050, Fl: 0.029, Py: 0.044 | Nap, Acy, Ph, Py: 0.1–50, Flo: 0.5–50, Fl: 0.05–50 | Nap: 3.1, Acy: 4.1, Flo: 8.5, Ph: 2.2, Fl: 2.7, Py: 2.1 | 30 |
| Titanate nanotube array modified by cetyltrimethylammonium bromide | Nap: 0.27, Acy: 0.19, Ph: 0.013, Fl: 0.048, Py: 0.069 | Nap, Acy, Ph, Fl, Py: 0.2–100 | Nap: 8.3, Acy: 9.2, Ph: 8.4,Fl: 1.7, Py: 2.8 | 31 |
| Graphene functionalized silica gel | Nap: 0.052, Acy: 0.024, Flo: 0.0029, Ph: 0.0082, Fl: 0.014, Py: 0.012 | Nap, Acy: 0.10–300; Flo, Ph: 0.01–400; Fl, Py: 1–600 | Nap: 3.6, Acy: 5.2, Flo: 1.7, Ph: 1.9, Fl: 3.4, Py: 4.5 | This work |
3.4 Sample analysis and comparison study
The developed method was applied to the analysis of six PAHs in real environmental water samples including pond and river water. The chromatograms of unspiked and spiked river samples pretreated with the Gr–SiO2 cartridge are shown in Fig. 4. The commercial C18 cartridge is also compared for PAHs in different water samples. No signal is detected for six PAHs in the pond and river water samples. After the water samples are spiked with PAH standard solution, both the commercial C18 cartridge and the as-prepared Gr–SiO2 cartridge show the enrichment effect for the six PAHs. However, the Gr–SiO2 cartridge shows the stronger retention ability than the C18 cartridge for the target compounds. The good performances of the Gr–SiO2-packed SPE cartridge may be due to the synergistic effect of Gr and silica gel. Gr has a large specific surface area, which suggests a high sorption capacity. The special structure makes both sides of the planar sheets of Gr available for molecule adsorption. Furthermore, the hexagonal arrays of carbon atoms in the Gr sheets may have a strong pi–pi interaction with the target molecules. The loading of Gr on the silica gel surface effectively enlarges the specific surface area of the composite and improves the penetrability of solution, which readily enhance the retention of PAHs. This characteristic is not only favorable for the adsorption of target molecules, but also helpful for the elution of analytes from the SPE cartridge.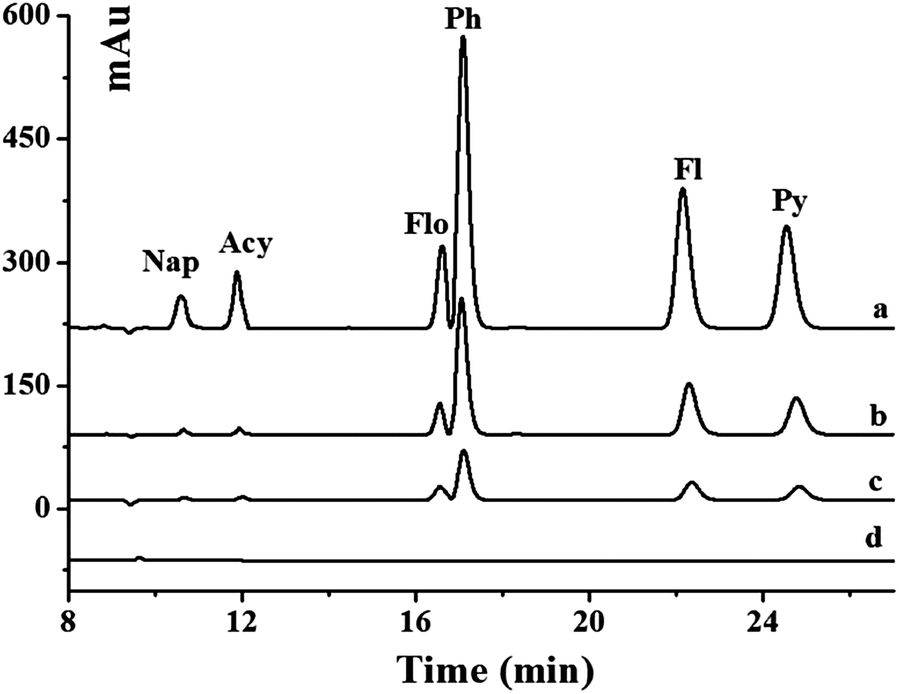 | ||
| Fig. 4 Typical chromatogram of PAHs in river water. (a) SPE of the spiked water sample with GR–SiO2 cartridges; (b) SPE of the spiked water sample with commercial C18 cartridges; (c) spiked water sample; (d) blank. Spiked PAH concentration: 50 μg L−1. | ||
The recoveries of PAHs in the spiked environmental water samples obtained from the Gr–SiO2 cartridge and C18 cartridge were 89.2–114.0% and 80.2–123.4%, respectively (Table 3), indicating that Gr–SiO2 can be an excellent SPE sorbent for PAH pretreatment and enrichment from environmental water samples. The developed method was also used to detect PAHs in milk samples. No PAHs was found in these milk samples. The recoveries of PAHs in the spiked milk samples obtained from the Gr–SiO2 cartridge and C18 cartridge were 89.0–115.4% and 82.5–125.6%, respectively (Table 4).
| SPE cartridge | Analyte | Added (μg L−1) | Pond water samples | River water samples | ||
|---|---|---|---|---|---|---|
| Recovery (%) | RSD (%) | Recovery (%) | RSD (%) | |||
| Gr–SiO2 cartridge | Nap | 5 | 94.0 | 2.1 | 114.0 | 2.6 |
| 50 | 109.2 | 2.7 | 108.2 | 3.7 | ||
| Acy | 5 | 98.0 | 3.9 | 98.0 | 3.1 | |
| 50 | 109.8 | 1.7 | 98.6 | 2.2 | ||
| Flo | 5 | 110.0 | 2.3 | 102.0 | 4.2 | |
| 50 | 102.2 | 3.6 | 101.6 | 3.6 | ||
| Ph | 5 | 110.0 | 4.4 | 106.0 | 3.0 | |
| 50 | 113.0 | 4.3 | 94.0 | 2.5 | ||
| Fl | 5 | 100.0 | 3.9 | 112.0 | 2.0 | |
| 50 | 101.6 | 3.7 | 89.2 | 3.7 | ||
| Py | 5 | 92.0 | 3.3 | 110.0 | 3.1 | |
| 50 | 103.2 | 2.9 | 101.4 | 3.9 | ||
| C18 cartridge | Nap | 5 | 92.4 | 3.5 | 110.3 | 4.0 |
| 50 | 85.6 | 3.6 | 123.4 | 3.5 | ||
| Acy | 5 | 88.3 | 2.9 | 109.4 | 3.2 | |
| 50 | 109.3 | 4.1 | 96.4 | 2.2 | ||
| Flo | 5 | 115.6 | 2.5 | 92.5 | 2.9 | |
| 50 | 87.2 | 3.3 | 88.4 | 3.5 | ||
| Ph | 5 | 80.2 | 3.9 | 107.2 | 3.7 | |
| 50 | 107.5 | 4.3 | 105.3 | 2.9 | ||
| Fl | 5 | 120.7 | 2.2 | 87.6 | 2.5 | |
| 50 | 110.2 | 3.6 | 85.3 | 3.1 | ||
| Py | 5 | 96.3 | 3.8 | 91.4 | 4.2 | |
| 50 | 90.1 | 3.0 | 88.9 | 3.2 | ||
| SPE cartridge | Analyte | Added (μg L−1) | Whole milk samples | Skimmed milk samples | ||
|---|---|---|---|---|---|---|
| Recovery (%) | RSD (%) | Recovery (%) | RSD (%) | |||
| Gr–SiO2 cartridge | Nap | 5 | 89.0 | 3.2 | 98.0 | 3.6 |
| 50 | 102.2 | 1.9 | 95.2 | 4.2 | ||
| Acy | 5 | 96.0 | 3.4 | 106.0 | 1.1 | |
| 50 | 104.8 | 1.8 | 115.4 | 2.2 | ||
| Flo | 5 | 112.0 | 2.4 | 106.0 | 3.2 | |
| 50 | 107.4 | 2.3 | 94.2 | 4.0 | ||
| Ph | 5 | 112.0 | 3.4 | 94.0 | 3.1 | |
| 50 | 102.6 | 2.4 | 97.4 | 2.3 | ||
| Fl | 5 | 91.6 | 3.4 | 102.0 | 2.6 | |
| 50 | 96.2 | 4.2 | 90.8 | 3.7 | ||
| Py | 5 | 98.0 | 2.4 | 104.0 | 3.2 | |
| 50 | 102.4 | 4.2 | 105.6 | 1.8 | ||
| C18 cartridge | Nap | 5 | 85.3 | 4.5 | 88.2 | 3.8 |
| 50 | 90.4 | 3.2 | 91.6 | 2.5 | ||
| Acy | 5 | 87.6 | 3.6 | 125.6 | 3.5 | |
| 50 | 92.5 | 3.8 | 111.2 | 2.9 | ||
| Flo | 5 | 82.5 | 2.1 | 119.3 | 4.1 | |
| 50 | 115.3 | 2.9 | 87.2 | 4.8 | ||
| Ph | 5 | 110.6 | 3.9 | 85.6 | 4.5 | |
| 50 | 84.6 | 3.4 | 86.2 | 3.7 | ||
| Fl | 5 | 109.2 | 4.3 | 94.3 | 3.1 | |
| 50 | 89.3 | 2.2 | 108.2 | 2.7 | ||
| Py | 5 | 96.5 | 3.7 | 114.5 | 3.8 | |
| 50 | 120.4 | 3.3 | 87.8 | 3.5 | ||
4 Conclusions
A simple procedure for the synthesis of a graphene-functionalized silica gel sorbent was developed by grafting graphene on silica gel. The prepared graphene-functionalized silica gel sorbent exhibited high extraction performance for PAHs, which enabled the sorbent very suitable for SPE of trace PAHs. This novel SPE sorbent has been successfully applied to the quantification of PAHs in water and milk samples. The experimental results revealed that the developed method provided high sensitivity, low solvent consumption, and good linearity over the investigated concentration range.Acknowledgements
This work was supported by the National Natural Science Foundation of China (U1304214, 21375114) and the Program for University Innovative Research Team of Henan (2012IRTSHN017).References
- C. R. Estrellan and F. Iino, Chemosphere, 2010, 80, 193–207 CrossRef CAS PubMed.
- K. Srogi, Environ. Chem. Lett., 2007, 5, 169–195 CrossRef CAS PubMed.
- C. Ding, H. G. Ni and H. Zeng, Environ. Pollut., 2012, 168, 80–86 CrossRef CAS PubMed.
- B. T. Bogolte, G. A. C. Ehlers, R. Braun and A. P. Loibner, Eur. J. Soil Biol., 2007, 43, 242–250 CrossRef CAS PubMed.
- G. Purcaro, P. Morrison, S. Moret, L. S. Conte and P. J. Marriott, J. Chromatogr., A, 2007, 1161, 284–291 CrossRef CAS PubMed.
- M. Marlow and R. J. Hurtubise, Anal. Chim. Acta, 2004, 526, 41–49 CrossRef CAS PubMed.
- S. Moret and L. S. Conte, J. Sep. Sci., 2002, 25, 96–100 CrossRef CAS.
- G. Purcaro, S. Moret and L. S. Conte, J. Sep. Sci., 2008, 31, 3936–3944 CrossRef CAS PubMed.
- B. Hu and C. H. Yu, J. Sep. Sci., 2010, 33, 2176–2183 CrossRef PubMed.
- X. N. Zhao, X. J. Liu, Z. X. Zhao, C. J. Huang, M. H. Zhang, H. L. Wang and X. D. Wang, J. Sep. Sci., 2009, 32, 2051–2057 CrossRef CAS PubMed.
- Y. Q. Cai, G. B. Jiang, J. F. Liu and Q. X. Zhou, Anal. Chem., 2003, 75, 2517–2521 CrossRef CAS.
- Z. P. Zang, Z. Hu, Z. H. Li, Q. He and X. J. Chang, J. Hazard. Mater., 2009, 172, 958–963 CrossRef CAS PubMed.
- L. M. Ravelo-Perez, A. V. Herrera-Herrera, J. Hernandez-Borges and M. A. Rodriguez-Delgado, J. Chromatogr., A, 2010, 1217, 2618–2641 CrossRef CAS PubMed.
- S. Zhu, W. Niu, H. Li, S. Han and G. Xu, Talanta, 2009, 79, 1441–1445 CrossRef CAS PubMed.
- K. J. Huang, Q. S. Jing, C. Y. Wei and Y. Y. Wu, Spectrochim. Acta, Part A, 2011, 79, 1860–1865 CrossRef CAS PubMed.
- X. L. Dong, J. S. Cheng, J. H. Li and Y. S. Wang, Anal. Chem., 2010, 82, 6208–6214 CrossRef CAS PubMed.
- L. M. Zhu, L. Q. Luo and Z. X. Wang, Biosens. Bioelectron., 2012, 35, 507–511 CrossRef CAS PubMed.
- H. F. Dong, Z. Zhu, H. X. Ju and F. Yan, Biosens. Bioelectron., 2012, 33, 228–232 CrossRef CAS PubMed.
- K. J. Huang, L. Wang, Y. J. Liu, H. B. Wang, Y. M. Liu and L. L. Wang, Electrochim. Acta, 2013, 109, 587–594 CrossRef CAS PubMed.
- S. Y. Niu, J. Sun, C. C. Nan and J. H. Lin, Sens. Actuators, B, 2013, 176, 58–63 CrossRef CAS PubMed.
- M. D. Stoller, S. J. Park, Y. W. Zhu, J. H. An and R. S. Ruoff, Nano Lett., 2008, 8, 3498–3502 CrossRef CAS PubMed.
- K. J. Huang, S. Yu, J. Li, Z. W. Wu and C. Y. Wei, Microchim. Acta, 2012, 176, 327–335 CrossRef CAS.
- Q. H. Wu, G. Y. Zhao, C. Feng, C. Wang and Z. Wang, J. Chromatogr., A, 2011, 1218, 7936–7942 CrossRef CAS PubMed.
- H. Zhang, W. P. Low and H. K. Lee, J. Chromatogr., A, 2012, 1233, 16–21 CrossRef CAS PubMed.
- S. Park and R. S. Ruoff, Nat. Nanotechnol., 2009, 4, 217–224 CAS.
- K. J. Huang, L. Wang, H. B. Wang, T. Gan, Y. Y. Wu, J. Li and Y. M. Liu, Talanta, 2013, 114, 43–48 CrossRef CAS PubMed.
- W. D. Wang, Y. M. Huang, W. Q. Shu and J. Cao, J. Chromatogr., A, 2007, 1173, 27–36 CrossRef CAS PubMed.
- Y. Liu, H. F. Li and J. M. Lin, Talanta, 2009, 77, 1037–1042 CrossRef CAS PubMed.
- B. B. Kefi, L. L. E. Atrache, H. Kochkar and A. Ghorbel, J. Environ. Sci., 2011, 23, 860–867 CrossRef CAS.
- D. Ge and H. K. Lee, J. Chromatogr., A, 2011, 1218, 8490–8495 CrossRef CAS PubMed.
- Y. R. Huang, Q. X. Zhou and G. H. Xie, J. Hazard. Mater., 2011, 193, 82–89 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2014 |