
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Molecular phenotypic profiling of a Saccharomyces cerevisiae strain at the single-cell level†
A. Mareike
Schmidt‡
a,
Stephan R.
Fagerer
b,
Konstantins
Jefimovs
c,
Florian
Buettner
d,
Christian
Marro
b,
Erdem C.
Siringil
e,
Karl L.
Boehlen
e,
Martin
Pabst
b and
Alfredo J.
Ibáñez‡
*b
aInstitute of Molecular Systems Biology, ETH Zurich, CH-8093 Zurich, Switzerland
bDepartment of Chemistry and Applied Biosciences, ETH Zurich, CH-8093 Zurich, Switzerland. E-mail: alfredoi@ethz.ch
cLaboratory for Electronics/Metrology/Reliability, EMPA, Swiss Federal Institute for Materials Science and Technology, Überlandstrasse 129, CH-8600 Dübendorf, Switzerland
dInstitute of Computational Biology, Helmholtz-Zentrum München, 85764 Neuherberg, Germany
eLaboratory for Advanced Materials Processing, EMPA, Swiss Federal Institute for Materials Science and Technology, Feuerstrasse 39, CH-3602 Thun, Swizerland
First published on 22nd August 2014
Abstract
Studying cell-to-cell heterogeneity requires techniques which robustly deliver reproducible results with single-cell sensitivity. Through a new fabrication method for the microarrays for mass spectrometry (MAMS) platform, we now have attained robustness and reproducibility in our single-cell level mass spectrometry measurements that allowed us to combine single-cell MAMS-based measurements from different days and samples. By combining multiple measurements, we were able to identify three co-existing phenotypes in an isogenic population of Saccharomyces cerevisiae characterized by distinctively different levels of glycolytic intermediates.
Introduction
Analytical methods capable of studying individual cells play an important role in identifying and characterizing cell-to-cell heterogeneity.1–3 Cell-to-cell (phenotypic) heterogeneity is a naturally occurring characteristic that manifests itself in all organisms – including individual cells from an isogenic population – because it ensures an enhanced adaptability to fast changes and/or perturbations in the growth environment.4–7 Several causes can induce cell-to-cell heterogeneity – even stochastic reasons – which may lead to different phenotypes of otherwise genetically identical cells. Such cell-to-cell variation has medical relevance, as for example in the case of persistence. Persistence happens when non-genetic cell-to-cell heterogeneity – in an isogenic population – allows a small group of cells to endure the addition of a drug to its growth medium (i.e., because they show a better phenotypic adaptation toward the drug).6,7 Once the drug is removed, the selection pressure disappears and these surviving cells would give rise to a new (heterogeneous) – but still isogenic – population.6,7 It is for this reason that new analytical methods for identifying such cell-to-cell phenotypic variations are greatly required,1–3 in particular when developing a mathematical model of the adaptation and survival mechanisms of a cell population toward a drug.4–7A novel method for matrix-assisted laser desorption/ionization (MALDI) mass spectrometry (MS), called microarrays for mass spectrometry (MAMS), has been proven to achieve sensitivity for single-cell metabolite detection.8–11 However, the original method used for the MAMS fabrication (i.e., laser scan ablation) was inefficient in generating microarrays of consistently high quality. Thus, microarray-to-microarray comparison was difficult, since it required numerous normalization steps to compensate the technical noise originating from the microarrays.8 Furthermore; such normalization was only possible for certain metabolites. This hampered the ability to pool multiple samples to clearly identify co-existing phenotypes in an isogenic cell population.
Here, we implemented an improved MAMS fabrication process, which should be capable of reducing technical variability. To test the novel MAMS substrates, we analyzed metabolite levels in single cells of the baker's yeast Saccharomyces cerevisiae, and for the first time single-cell level measurements from multiple microarrays were combined to generate a total population of 1280 measurements (i.e., an eight-fold increase compared to previously published data).8–11 As a result of the statistical analysis of the data, three subpopulations with distinctively different levels of glycolytic intermediates were found to co-exist in the isogenic population. Thus, due to the improved MAMS fabrication process, pooling of data from multiple samples is now feasible and therefore allows using MAMS to confidently identify and characterize co-existing metabolic phenotypes.
Experimental section
MAMS are fabricated on commercial transparent indium–tin oxide coated cover glass chips (20 mm × 20 mm × 0.16 mm) with a resistivity of 8–12 Ω cm−1 (SPI Supplies, Unterfoehring, Germany). The slides were spin-coated (SuSos, Duebendorf, Switzerland) with a 1 μm thick polysilazane coating (CAG 37, marketed by Clariant, Frankfurt am Main, Germany). This polysilazane layer was structured (EMPA, Thun, Switzerland) using a projection laser ablation system equipped with an excimer laser (Exitech Ltd, Oxford, UK), with the following characteristics: 20 ns pulse, 248 nm wavelength, 50 Hz repetition rate, and an average fluence at the substrate level was 500 mJ cm−2 per pulse. The laser was collimated to illuminate an area of 16 × 16 mm2 on a mask and then focused on the sample with the de-magnification factor of 5 (Fig. 1A). By scanning the mask under the beam and the sample, a quadratic array of 13 × 13 circular recipient sites (100 μm diameter) with a site-to-site distance of 400 μm in both dimensions was created. Recipient sites of larger (1.5 mm in diameter) size were machined outside the 13 × 13 array for depositing metabolite standards to be used as mass calibrants. | ||
Fig. 1 Graphical summary of the workflow used to prepare the samples for single-cell MALDI MS analysis. Cellular metabolism is quenched by adding a cold (−20 °C) solution of 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 methanol:water with 0.85% (v/v) ammonium bicarbonate (pH 7.4). After cell handling (i.e. centrifugation and discarding the supernatant), the pellet is reconstituted in a salt free cold (−20 °C) solution of 3:2 methanol:water, and the cell suspension is spread onto a cold MAMS chip. Applying the cell suspension onto the MAMS surface will result in an automated aliquoting of the cell suspension into the hydrophilic reservoirs, without the need for a microspotter. Depending on the cell concentration employed, the number of cells on each hydrophilic reservoir can be between zero and hundreds (random Poisson distribution). The transparency of the MAMS substrate allows for microscopic analysis to determine the number of cells in each reservoir while the cells remain quenched because the entire MAMS chip is kept cold in a cryo-chamber using liquid nitrogen. After counting the cells under the microscope, 9-aminoacridine (MALDI matrix) is applied with an airbrush, and each reservoir on the plate is analyzed by MALDI MS. :2 methanol:water with 0.85% (v/v) ammonium bicarbonate (pH 7.4). After cell handling (i.e. centrifugation and discarding the supernatant), the pellet is reconstituted in a salt free cold (−20 °C) solution of 3:2 methanol:water, and the cell suspension is spread onto a cold MAMS chip. Applying the cell suspension onto the MAMS surface will result in an automated aliquoting of the cell suspension into the hydrophilic reservoirs, without the need for a microspotter. Depending on the cell concentration employed, the number of cells on each hydrophilic reservoir can be between zero and hundreds (random Poisson distribution). The transparency of the MAMS substrate allows for microscopic analysis to determine the number of cells in each reservoir while the cells remain quenched because the entire MAMS chip is kept cold in a cryo-chamber using liquid nitrogen. After counting the cells under the microscope, 9-aminoacridine (MALDI matrix) is applied with an airbrush, and each reservoir on the plate is analyzed by MALDI MS. | ||
The Saccharomyces cerevisiae mutant strain (i.e., CEN.PK.KOY.TM6*P) was used.12,13 This strain exhibits a stochastic cell growth rate, when grown in liquid culture. This observed variability between cultures is present even if the cultures originated from the same colony and were grown under exactly the same growth conditions (similar growth medium, temperature, pH, etc.).
The cell handling process is as follows, cells were taken from a liquid culture , quenched using a cold (−20 °C) methanol:water mixture (3:2 ratio) with ammonium bicarbonate (0.85% w/v, pH 7.4) to stop metabolic activity, after which the supernatant was removed and cells were washed with a methanol:water (3:2 ratio) solution (−20 °C) to remove salts. The cell suspension was then aliquoted onto the MAMS substrate (Fig. 1B). The number of cells was determined by microscopic inspection, while the entire MAMS chip was kept cold (−40 °C) in a cryo-chamber flushed with liquid nitrogen. Subsequently, 9-aminoacridine (MALDI matrix) – also cold (−20 °C) and in methanol:water (3:2) – was sprayed and the MAMS target was introduced into the MALDI-MS instrument for measurement. Data treatment and analysis were accomplished by transforming the raw data files from the AB5800 instrument to a universal data format (i.e., mzXML) using the freeware program Peak List Conversion Tool, available from http://www.proteomecommons.org. Afterwards, the spectral data (i.e., accurate mass, signal intensity, etc.) were calculated using MATLAB (MathWorks, Natick, MA, USA) peak recognition software that was kindly made available by Uwe Sauer and Nicola Zamboni (Institute of Molecular Systems Biology, ETH Zürich).14 All spectral data were normalized by a linear combination of unsaturated signals that did not correlate with signals of biological origin. A more detailed description of the cell cultivation, as well as the analysis and data processing can be found in the ESI.†
Safety considerations
9-Aminoacridine (9-AA) is a mutagenic substance and it must be handled with care. The selection of 9-AA as a matrix is due to its preferential ionization mechanism,15–17 which when coupled with the trapping of the cells in picoliter-volume reservoirs8–11 and the homogenous co-crystallization of matrix and analytes18 what allow us to reach the “single-cell” level sensitivity.Results and discussion
One key step for studying cell-to-cell heterogeneity is to identify the co-existing phenotypes in an isogenic population. Each of these co-existing phenotypes is characterized by a unique metabolic pattern. Unfortunately, when we pool phenotypes, their unique metabolic pattern is diluted due to averaging artifacts (ESI, Fig. 1†). In a previous study, we have observed naturally co-existing subpopulations in an isogenic yeast culture.8 However, due to technical limitations (i.e., mostly associated with the limited number of single-cell events), it was not possible to characterize the co-existing populations with enough statistical significance. Here, by using an improved MAMS microarray, this limitation has been overcome.Single-cell level monitoring of an S. cerevisiae isogenic population
Variations in the monitored metabolite signal intensity – at the single-cell level – do not necessarily reflect the naturally occurring differences in metabolite concentrations, since they can also result from variations in the cell size (or other trivial biological artifacts) or instrumental and sample handling variation (analytical artifacts). To illustrate the robustness of our method, as well as its ability to monitor the naturally occurring biological variability between the multiple cells within and between the set of measurements, we performed a set of control measurements (ESI, Fig. 2–5†) described in-detail in the ESI.† In summary, these control experiments gave us confidence that the metabolite signal variations were of biological origin and not artifacts influenced by trivial biological variability (cell size), or by the analysis.After having ensured that we actually monitor biological information, we used our MAMS-based technology to identify the co-existing phenotypes within an isogenic yeast strain population. The Saccharomyces cerevisiae (CEN.PK.KOY.TM6*P) strain is an excellent model, because it presents a strong stochastic behavior.12 This stochasticity is reflected in its ability to grow at different growth rates in liquid culture, even if all cultures were generated from a single colony and were grown under similar conditions. In total, four liquid cultures – each of them with a different growth rate (0.14, 0.16, 0.18, and 0.21 1/h) – were used for this study. Furthermore, the mass spectrometry single-cell data of these 4 liquid cultures were pooled together during a post-data processing step to generate an in silico “master” mixed growth-rate cell population (a total of 1280 wells) to obtain a better understanding of the CEN.PK.KOY.TM6*P strain metabolic behavior.
The total distribution of the number of cells per reservoir for the whole dataset can be seen in ESI, Table 1.† From the 159 wells containing 1 cell per reservoir, only 75 were proved suitable for further studies (n = 32, 17, 13, and 13 single-cell level measurements for the 0.21, 0.18, 0.16, and 0.14 1/h – cell growth rate samples, respectively). The selection criteria for determining if a single-cell level measurement is good or not were based on the precise mass recognition of 30 central metabolites (ESI, Table 2†) with a maximum mass deviation of 0.02 Da, and an average total mass deviation error of 25 ppm per mass spectra (see ESI† for additional details).
Interestingly, in Fig. 2A, we observed that many cells, independent of the liquid culture they originated from, had similar relative signal intensities of hexose-bisphosphate (very possibly fructose-1,6-bisphophate – F16BP).8 Therefore, to better study the distribution of abundances of hexose-bisphosphate in the CEN.PK.KOY.TM6*P yeast strain, the histogram for relative signal intensity of hexose-bisphosphate was also plotted, in Fig. 2B, for the in silico “master” cell population (i.e., the population obtained by pooling all different liquid cultures). Remarkably, the observed distribution of hexose-bisphosphate in Fig. 2B is not a symmetric uni-modal Gaussian distribution.8,19
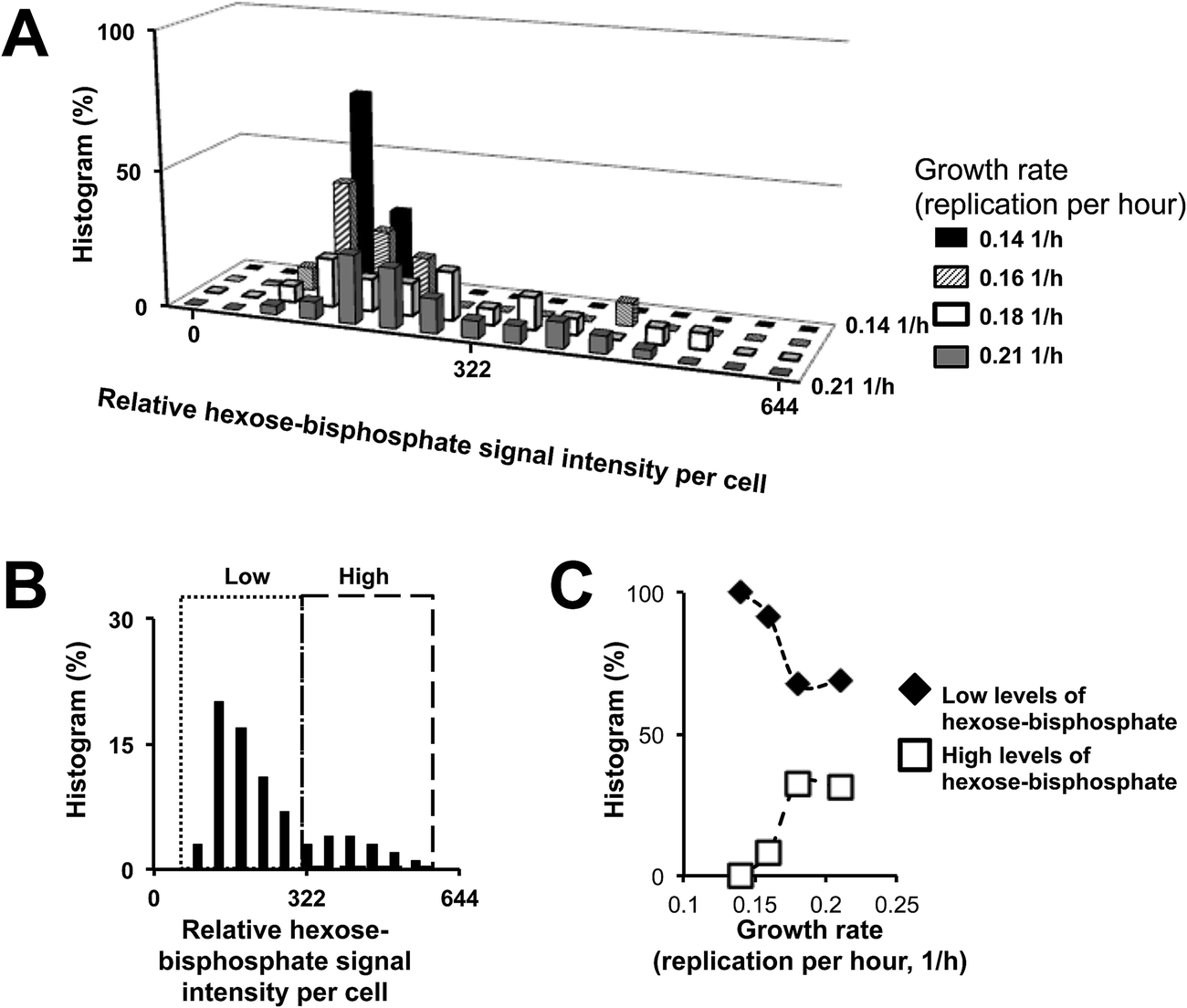 | ||
| Fig. 2 (A) The MAMS-based analytical protocol is capable of avoiding biological and technical artifacts due to averaging of a large number of cells by performing a single-cell level analysis on the liquid medium grown cells. Therefore, we observed that within a “theoretical” homogeneous clonal population of cells, there is enough cell-to-cell heterogeneity to form different phenotypes which may contribute to the observed differences in the growth rate. (B) By pooling all the cell samples together, we can observe that there is a correlation between increasing levels of F16BP and the growth rate of the cell population (C). The identification of fructose-1,6-bisphosphate (F16BP) is based on precise mass and tandem MS experiment (ESI, Fig. 4†), which was performed on a standard stainless steel target with a higher density of yeast cells (∼1000 cells). The fragmentation spectrum of hexose-bisphosphate was then compared to a commercially available sample of F16BP. Furthermore, LC-MS measurements were performed. In the LC-MS measurements, the levels of F16BP in different mutants of the CEN.PK.KOY yeast strain (each of them presenting a different growth rate) had a positive correlation; a trend that was also obtained from the different liquid cultures of the CEN.PK.KOY.TM6 strain using our MAMS platform (ESI, Fig. 5†). With this experiment, we have not only confirmed that the hexose-biphosphate signal is F16BP, but also that the level of the F16BP signal in the sample correlates with the observed cell growth. | ||
Previously, we have observed that the relative intensity of hexose-bisphosphate can correlate with the cellular levels of F16BP, which presents a bimodal distribution.8,19 The validation of the correlation between the cellular levels of F16BP and hexose-bisphosphate was done by performing a tandem MS experiment, as well as liquid chromatography coupled with mass spectrometry (LC-MS), ESI, Fig. 4 and 5† respectively. Interestingly, by classifying for example all cells in two hypothetical co-existing groups with (i) high and (ii) low levels of F16BP (Fig. 2C), a link between higher growth rates and higher levels of F16BP could be made. This was our first hint that the different growth rates could be associated with different ratios of co-existing phenotypes in each liquid culture. In the following section, we aim to describe the differences between these co-existing phenotypes based on their metabolic profiles and not only based on the relative levels of F16BP as done in a previous report.8
Identification and characterization at the metabolic level of naturally occurring molecular phenotypes in an S. cerevisiae isogenic population
Our first goal in this section is the identification of a significant source of cell-to-cell heterogeneity based on the measured mass spectrometry signals. Sources of cell-to-cell heterogeneity can be external and/or internal to the organism. An example of an external factor that contributes to cell-to-cell heterogeneity is that cells can be found in slightly different growth environments (i.e., microenvironments) even if grown under the same conditions. These microenvironments originate due to slight differences in oxygen and nutrient availability, which can impact cellular metabolism.20-22 Here, we assume that the external source of cell-to-cell heterogeneity, such as oxygen availability, is not predominant since we cannot measure it directly with our system.For this purpose, we monitor metabolite signals (or metabolite signal ratios) that are used as biomarkers for identifying the cell state. For example, a trivial source of cell-to-cell heterogeneity is cell viability (i.e., cells are either alive or dead).23 Because the mass spectra signals for adenosine monophosphate could not be identified, the ATP/ADP ratio was used instead of the energy charge for monitoring the viability of the cells.8 As it was expected, all single cells measured showed a remarkably constant ATP/ADP ratio, which indicates that all of them were equally viable at the moment of analysis (Fig. 3A). This gives additional confidence that our cell handling protocols truly quenched the metabolism of the cells without introducing analytical artifacts.
 | ||
| Fig. 3 The ratios of ATP/ADP, GTP/GDP, and F16BP/PEP were plotted to compare the different levels of biological noise associated with the cell-to-cell heterogeneity. For example, difference in viability is a source of cell-to-cell heterogeneity. The energy charge was not used because for our MS measurements the adenosine monophosphate (AMP) signal did not fulfill our signal selection criteria (explained in the ESI†). Instead the ratio between ATP/ADP was used to estimate the cell viability (see text for details). Here, we can observe that viability is not a strong source of cell-to-cell heterogeneity. Because, the ability of the cells to sense their carbon source (i.e., glucose) in the different cultures is heterogeneous, the GTP/GDP ratio is expected to have a higher CV value (the ratio of GTP/GDP is the triggering signal for cell growth and in combination with cytosolic pH is directly linked to the sensing of glucose in the cell environment). The high coefficient of variability for the GTP/GDP ratio shows that this can be one of the major sources of cell-to-cell heterogeneity. However, even in a quite noisy system (as illustrated by the GTP/GDP ratio), the distribution of the F16BP:PEP ratio falls between two conserved values. Thus, we hypothesized that the different co-existing phenotypes could be characterized in terms of their associated glucose degradation. CV is the abbreviation for coefficient of variation. | ||
Another source of cell-to-cell heterogeneity is the cell stage of development. It has recently been demonstrated that the GTP/GDP ratio in combination with cytosolic pH acts as a triggering signal for yeast growth by activating the Ras/TOR signal cascade.24 Since we did not attempt to synchronize our yeast cultures prior to collecting the samples, it is not surprising to see a greater amount of variability (biological noise) between the individual cells (Fig. 3B), in particular when compared to the previous parameter (cell viability).
Finally, another source of cell-to-cell heterogeneity is associated with how cells process the available nutrients in the growth medium (i.e., glucose). Therefore, a ratio that could be easily linked to the way fermentation is performed was plotted. One of the metabolites that we chose for this ratio was F16BP, whose levels in yeast cells have been correlated with carbon uptake via the anaerobic glycolysis pathway.25,26 The other metabolite was phosphenolpyruvate (PEP), which strongly correlates with the pentose phosphate pathway activity.27 Interestingly, when comparing the ATP/ADP and the GTP/GDP ratio data dispersion, which have a clear uni-modal distribution, the plot of the F16BP/PEP ratio (Fig. 3C) presents a bimodal distribution. It is important to state that this bimodal distribution is not associated with the previously observed bimodal distribution observed for F16BP (this will be better clarified in the following paragraphs). It is also an interesting observation that the overall dispersion (coefficient of variability, CV) for this ratio is higher than the one observed for ATP/ADP (Fig. 3A), while at the same time lower than the one observed for the GTP/GDP (Fig. 3B). This is interesting because the naturally occurring cell-to-cell heterogeneity reflected by the F16BP/PEP ratio fluctuates between two conserve values, even when the overall biological noise (reflected by the GTP/GDP ratio) is much higher. Since this finding shows that glycolysis has a role in the cell-to-cell heterogeneity observed for this yeast strain, we will attempt first to classify the cells based only on their F16BP/PEP cellular ratios. Once this classification has taken place, we will compare our results with a non-supervised classification. Based on our measurements, we tentatively assigned three different phenotypes, labeled here as: (i) Group 1, (ii) Group 2, and (iii) Group 3 (Fig. 4A). Due to the ability of mass spectrometry to simultaneously monitor multiple metabolite signals from the central metabolism, we can describe each group in terms of a metabolic pattern (Fig. 4B).
 | ||
| Fig. 4 (A) The log(F16BP/PEP) is plotted in the decreasing order. A binomial distribution can be observed, where two groups, Group 1 and Group 3, are clearly differentiated by the levels of glycolytic activity (Group 1 with low and Group 3 with high levels of anaerobic glycolytic activity, respectively). In between these two groups, few cells can be found. These cells are part of Group 2, which is an intermediate state between Groups 1 and 3. (B) Based on these three groups, a decision tree can be constructed. Using a t-test statistical approach, key metabolites that are statistically different can be retrieved. For example, for the first decision step – used to differentiate Group 1 from the rest – guanosine triphosphate and phospho-gluconolactone can be used as part of the selection criteria (p-values are 7.83 × 10−8, 8.11 × 10−6 respectively). For the second decision step, aspartate and phosphoenol pyruvate (p-values are 5.89 × 10−5 and 2.09 × 10−3, respectively) can be used. | ||
We will now compare the above result with the one obtained from a non-bias classification based on the whole metabolic profile of the cells. For this purpose, 75 single-cell mass spectra were analyzed using a non-supervise principal component analysis (MatLab, MathWorks). To have confidence that the analysis represents biological information, we identified the principal component that fulfills the following conditions: (i) it can explain most of the population variance; and (ii) its loading values for the signals of F16BP and PEP will have opposite signs (i.e., they will be anti-correlated as it is to be expected).27 In our case, both conditions were fulfilled by the principal component 4. In ESI, Fig. 6,† we plot the principal components 1 vs. 4. Each point corresponds to a single yeast cell, and is described by 332 relative ion intensity signals (normalized by a single correction factor, as described in the ESI†). On the one hand, because 97.8% of all the loading values for the PC1 (33.6% of the sample variance) were positive, we hypothesize that the PC1 represents a trivial biological trait/feature (i.e., size) or an analytical artifact, e.g., laser fluctuations that – although minimized by our analytical protocol – cannot be completely removed from the analysis. On the other hand, we hypothesize that the principal component 4 (7.9% of the sample variance) is associated with the naturally occurring cell-to-cell heterogeneity because (i) the loadings associated with the principal component 4 show different signs, in particular for metabolites from competing metabolic pathways such as PEP and F16BP and (ii) known metabolite MS signals (ESI, Table 2†) scored – either positive or negative – high loading values. Interestingly, the level of non-trivial (metabolite) cell-to-cell variability (i.e., the variance contribution of PC4) found here is realistic and in agreement with models of metabolic cell-to-cell heterogeneity that assumes variations between 5 and 10% for glycolytic intermediates.19 The groups obtained in the principal component analysis confirm to some extent our previous classification based exclusively on the F16BP/PEP signal ratio. Group 1 is clearly isolated from Group 2 and 3, due to its characteristic metabolic profile (as described in Fig. 4B), while Group 2 and 3 share a more common metabolic profile. Although, a supervised PCA approach, in which only the mass spectrometry signals of selected metabolites are detected, could have improved the clustering of the cells in Groups 2 & 3, it would not have yielded a significant improvement over the classification based on the F16BP/PEP signal ratio.
Following the tentative identification of the molecular phenotypes, we plotted the distribution of these molecular phenotypes in terms of their relative levels of F16BP – in an analogous way to the one shown in Fig. 2 (Fig. 5A), considering 100% to be the “master” population (i.e., all measured cells). Interestingly, by reducing the technical and analytical variability, we could de-convolute the overlapping populations with low F16BP levels (based on the above introduced classification, Fig. 4). Furthermore, we can clearly observe that only Group 3 (i.e., with high levels of glycolytic activity) presents the above mentioned F16BP bi-stability – the presence of both high and low level F16BP populations. In Fig. 5B and C, we demonstrate that the different growth rates observed for this yeast strain can now be clearly correlated with an increasing number of yeast cells with high glycolytic activity, and in more detail with those yeast cells that showed high levels of F16BP. This result – the correlation between glycolysis and cell growth – is in accordance with the Warburg effect, which states the increased utilization of glucose via glycolysis as a cellular resource for fast cell growth.26
 | ||
| Fig. 5 Three (3) core groups (or phenotypes) could be identified over the previous ones shown in Fig. 2B, based on the mass spectrometry signals of the central metabolism (as shown in Fig. 4). (A) The histogram of fructose-1,6-bisphosphate for all measured yeast samples (i.e., pooled data – shown in Fig. 2B) is further de-convoluted using these three groups that are characterized by different levels of glycolytic activity. By de-convoluting the co-existing populations, it is possible to avoid overlapping of three different phenotypes that present similar levels of F16BP, but show different glycolysis activity (100% = 75 cells, which is the total population of the measured yeast cells). (B) The co-existence of these three groups in different ratios is also associated with the growth rates observed in liquid growth medium (100% = 32, 17, 13, and 13, for the 0.21, 0.18, 0.16, and 0.14 1/h – cell growth rate samples, respectively). (C) The Group 3 (i.e., the group characterized by a high glycolytic activity) can be further subdivided in two sub-populations, one with high and another with low levels of fructose-1,6-bisphosphate (100% as in (B)). (B and C) show that it is the relative abundance of these four subgroups in the overall population (and not only 2 based on a high and low F16BP phenotypes) which can explain the growth rates observed for the CEN.PK.KOY.TM6*P populations. | ||
Finally, in ESI, Fig. 7,† we can take advantage of the analytical power associated with the pooling of single-cell level measurements and compare the mass spectra of one particular cell vs. another cell (ESI, Fig. 7A†), or for example between the pooled mass spectra of two different phenotypes (ESI, Fig. 7B†) and perform an in-depth statistical analysis to find the key differences between the two populations. For example, when compared with our first attempt to characterize the high and low F16BP populations using our single-cell platform,8 the possibility of accumulating larger numbers of single-cell data, with a lower technical/analytical variability, allowed us to perform a 2-sample t-test statistical study on both populations. Furthermore before performing this comparison, we can remove from the high and low F16BP populations (characterized by a high glycolysis activity) other cells that might present similar levels of F16BP but present different levels of glycolytic activity. Thus, in addition to the trivial difference in F16BP, the high and low F16BP populations can be differentiated for example in terms of adenosine triphosphate, uracil triphosphate, and 3-deoxy-D-arabino-heptulosonic-acid-7-phosphate (p values equal 6 × 10−5, 2.69 × 10−3, and 1.89 × 10−3, respectively).
Furthermore, we can now also observe that metabolite–metabolite correlations present unique information about the metabolic networks' underlying system architecture for each phenotype. In Fig. 6A, the “average” partial rank correlation for those metabolites that have been described in ESI, Table 2.† While, Fig. 6B–D, the partial rank correlations for a selected cluster of metabolites belonging to Group 1, Group 3b, and Group 3a are shown. Interestingly, for cells associated with Group 1 (low glycolytic activity) the partial correlation between oxaloacetate and coenzyme A is quite strong. However, the correlation between these two metabolites becomes fainter for the cells from Group 3 (high glycolytic activity). In addition, cells from Group 3 show additional correlation between these two metabolites and phosphogluconolactone. The latter correlation even differs in the case if the cell levels F16BP are high or low (Group 3a and b, respectively). It would be tempting at this stage to provide a metabolic model that explains the differences between the observed phenotypes (in particular for Group 1, Group 3a and Group 3b). However, a more in-depth analysis is required prior to developing such a biological model. Fortunately, the MAMS substrates are compatible with fluorescence microscopy measurements, thus it would be possible to label transcription factors and/or proteins with fluorescent protein tags to monitor the up- or down-regulation of a particular metabolic pathway. For this reason, we believe that a MAMS-based mass spectrometric analysis at the single-cell level is a great first step in trying to understand the causes for cell-to-cell heterogeneity.
 | ||
| Fig. 6 (A) Partial rank correlation for the whole population of measured cells based on the post-measurement pooling of the mass spectra from Groups 1, 2, 3a & b characterized by low glycolytic, intermediate, and high glycolytic activity (with high and low levels of fructose-1,6-bisphosphate), respectively. (B) Partial rank correlation for a small subset of metabolites using the data obtained exclusively from Group 1 (i.e. low glycolytic activity yeast cells). (C) Partial rank correlation for a small subset of metabolites using the data obtained exclusively from Group 3b (high glycolytic activity with low cellular levels of F16BP yeast cells). (D) Partial rank correlation for a small subset of metabolites using the data obtained exclusively from Group 3a (high glycolytic activity with high cellular levels of F16BP yeast cells). | ||
In summary, this new generation of microarrays for mass spectrometry is able to show the non-genetic heterogeneity present in a clonal population, and it might prove useful as a first line of study to better understand cell-to-cell heterogeneity. The possibility of coupling these transparent substrates with a fluorescent read-out could be the next step to better identify the biological processes that allow the formation of different non-genetic phenotypes. This could be of great interest in a clinical research application, where a clonal population of cells might present non-genetic heterogeneity (e.g., cancer cells). A MAMS-based approach could identify different phenotypes, and possibly identify the effect of a drug on these populations in such a way that after measuring the cell population at different time points we could identify the surviving phenotype. This would then really lead to a “tailor-made” therapy for diseases, such as cancer.
Conclusions
In this study we exploit a new microfabrication method (i.e., projection laser ablation) for the fabrication of microarrays for mass spectrometry. By using this new generation of MAMS substrates, we reduced the technical (or analytical) variability to directly visualize four different co-existing phenotypes (characterized by the cellular levels of glycolytic intermediates) in an isogenic cell population of Saccharomyces cerevisiae strain, CEN.PK.KOY.TM6*P. The existence of these subpopulations in different ratios can be correlated with the different growth rates observed in the liquid cultures of this particular yeast strain.There is in theory no limitation to the number of single-cell level measurements that can be performed and combined to compare/characterize – with statistical significance – individual cells, a group of cells that are phenotypically similar, or the whole cell population (the latter two by pooling selected or all the individual results together, respectively). Because, we exploit the inherent variability between individual cells (e.g. due to the cell cycle, stochastic effects, cell age, cell size, etc.) as a potent system perturbation, we could observe differences in the metabolic networks' underlying system architecture. This information in combination with the ability of this platform to calculate the biological variance, and the number of co-existing phenotypes, can be later used for data driven modeling or for designing more complex biological experiments to validate the existence of the co-existing phenotypes.
Conflict of interest
S.R.F., K.J., and M.P. are developing a commercial product based on the microarrays for mass spectrometry technology.Acknowledgements
A.J.I. gratefully acknowledges the financial support of the Ambizione Program of the Swiss National Science Foundation (SNF) (PZ00P3_142615) and the support of the following ETH Zürich professors: Prof. Dr Renato Zenobi (who hosts the research of A.J.I.), Prof. Dr Petra Dittrich, Prof. Uwe Sauer, and Prof. Dr Matthias Peter, who granted access to their facilities to perform the experiments. S.R.F., K.J., and M.P. were supported by the Swiss KTI (Kommission für Technologie und Innovation) Grant no. 13123.1 PFNMNM. The authors also would like to thank: (i) Karin Elbing for providing the CEN.PK.KOY.TM6*P yeast strain, (ii) the support of AB Sciex, in particular Tobias Schibli, who granted free access to the AB5800 MALDI TOF/TOF instrument, (iii) Fabian Wahl, Gerd Hayenga, Jens Boertz and Rudolf Köhling from Sigma-Aldrich, Switzerland for their support during the microarray chip development and (iv) Prof. Dr Matthias Heinemann, Dr Reinhard Dechant, Dr Madina Mansurova, Dr. Nicola Zamboni, and Robert Steinhoff for their advice, support and thoughtful insights during the drafting of this publication.References
- R. Zenobi, Science, 2013, 342, 6163 CrossRef PubMed.
- A. Svatos, Anal. Chem., 2011, 83, 5037–5044 CrossRef CAS PubMed.
- M. Heinemann and R. Zenobi, Curr. Opin. Biotechnol., 2011, 22, 26–31 CrossRef CAS PubMed.
- S. J. Altschuler and L. F. Wu, Cell, 2010, 141, 559–563 CrossRef CAS PubMed.
- A. Raj and A. van Oudenaarden, Cell, 2008, 35, 216–226 CrossRef PubMed.
- S. V. Avery, Nat. Rev. Microbiol., 2006, 4(8), 577–587 CrossRef CAS PubMed.
- N. Q. Balaban, J. Merrin, R. Chait, L. Kowalik and S. Leibler, Science, 2004, 305, 1622–1625 CrossRef CAS PubMed.
- A. J. Ibáñez, S. R. Fagerer, A. M. Schmidt, P. L. Urban, K. Jefimovs, P. Geiger, R. Dechant, M. Heinemann and R. Zenobi, Proc. Natl. Acad. Sci. U. S. A., 2013, 110(22), 8790–8794 CrossRef PubMed.
- S. R. Fagerer, T. Schmid, A. J. Ibáñez, M. Pabst, R. Steinhoff, K. Jefimovs, P. L. Urban and R. Zenobi, Analyst, 2013, 138, 6732–6736 RSC.
- P. L. Urban, A. M. Schmidt, S. R. Fagerer, A. Amantonico, A. Ibañez, K. Jefimovs, M. Heinemann and R. Zenobi, Mol. BioSyst., 2011, 7(10), 2837–2840 RSC.
- P. L. Urban, K. Jefimovs, A. Amantonico, S. R. Fagerer, T. Schmid, S. Mädler, J. Puigmarti-Luis, N. Goedecke and R. Zenobi, Lab Chip, 2010, 10(23), 3206–3209 RSC.
- A. M. Schmidt, PhD thesis, ETH Zürich, 2013.
- K. Elbing, C. Larsson, R. M. Bill, E. Albers, J. L. Snoep, E. Boles, S. Hohmann and L. Gustafsson, Appl. Environ. Microbiol., 2004, 70, 5323–5330 CrossRef CAS PubMed.
- T. Fuhrer, D. Heer, B. Begemann and N. Zamboni, Anal. Chem., 2011, 83(18), 7074–7080 CrossRef CAS PubMed.
- D. Yukihira, D. Miura, Y. Fujimura, Y. Umemura, S. Yamaguchi, S. Funatsu, M. Yamazaki, T. Ohta, H. Inoue, M. Shindo and H. Wariishi, J. Am. Soc. Mass Spectrom., 2014, 25(1), 1–5 CrossRef CAS PubMed.
- S. R. Fagerer, S. Nielsen, A. Ibáñez and R. Zenobi, Eur. J. Mass Spectrom., 2013, 19, 39–47 CrossRef CAS.
- R. L. Vermillion-Salsbury and D. M. Hercules, Rapid Commun. Mass Spectrom., 2002, 16, 1575–1581 CrossRef CAS PubMed.
- M. Pabst, S. R. Fagerer, R. Köhling, S. K. Küster, R. Steinhoff, M. Badertscher, F. Wahl, P. S. Dittrich, K. Jefimovs and R. Zenobi, Anal. Chem., 2013, 85(20), 9771–9776 CrossRef CAS PubMed.
- J. H. van Heerden, M. T. Wortel, F. J. Bruggeman, J. J. Heijnen, Y. J. Bollen, R. Planqué, J. Hulshof, T. G. O'Toole, S. A. Wahl and B. Teusink, Science, 2014, 343(6174), 987 CrossRef CAS PubMed.
- D. A. Elias, S. L. Tollaksen, D. W. Kennedy, H. M. Mottaz, C. S. Giometti, J. S. McLean, E. A. Hill, G. E. Pinchuk, M. S. Lipton, J. K. Fredrickson and Y. A. Gorby, Arch. Microbiol., 2008, 189(4), 313–324 CrossRef CAS PubMed.
- M. R. Mashego, K. Rumbold, M. De Mey, E. Vandamme, W. Soetaert and J. J. Heijnen, Biotechnol. Lett., 2007, 29(1), 1–16 CrossRef CAS PubMed.
- N. C. Duarte, B. Ø. Palsson and P. Fu, BMC Genomics, 2004, 5(63) DOI:10.1186/1471-2164-5-6.
- A. Amantonico, P. L. Urban, S. R. Fagerer, R. M. Balabin and R. Zenobi, Anal. Chem., 2010, 82, 7394–7400 CrossRef CAS PubMed.
- R. Dechant, S. Saad, A. Ibáñez and M. Peter, Mol. Cell, 2014, 55, 1–13 CrossRef PubMed.
- D. H. Huberts, B. Niebel and M. Heinemann, FEMS Yeast Res., 2012, 12(2), 118–128 CrossRef CAS PubMed.
- H. Pelicano, D. S. Martin, R. H. Xu and P. Huang, Oncogene, 2006, 25, 4633–4646 CrossRef CAS PubMed.
- N. M. Grüning, M. Rinnerthaler, K. Bluemlein, M. Mülleder, M. M. Wamelink, H. Lehrach, C. Jakobs, M. Breitenbach and M. Ralser, Cell Metab., 2011, 14(3), 415–427 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4an01119h |
| ‡ Contributed equally to this publication. |
| This journal is © The Royal Society of Chemistry 2014 |