DOI:
10.1039/C4AN00777H
(Communication)
Analyst, 2014,
139, 3860-3865
Bimetallic Pd–Pt supported graphene promoted enzymatic redox cycling for ultrasensitive electrochemical quantification of microRNA from cell lysates†
Received
1st May 2014
, Accepted 22nd May 2014
First published on 22nd May 2014
Abstract
The expression of microRNAs (miRNAs) is related to some cancer diseases. Recently, miRNAs have emerged as new candidate diagnostic and prognostic biomarkers for detecting a wide variety of cancers. Due to low levels, short sequences and high sequence homology among family members, the quantitative miRNA analysis is still a challenge. A novel electrochemical biosensor with triple signal amplification for the ultrasensitive detection of miRNA was developed based on phosphatase, redox-cycling amplification, a bimetallic Pd–Pt supported graphene functionalized screen-printed gold electrode, and two stem-loop structured DNAs as target capturers. The proposed biosensor is highly sensitive due to the enhanced electrochemical signal of Pd–Pt supported graphene and sufficiently selective to discriminate the target miRNA from homologous miRNAs in the presence of loop-stem structure probes with T4 DNA ligase. Therefore, this strategy provided a new and ultrasensitive platform for amplified detection and subsequent analysis of miRNA in biomedical research and clinical diagnosis.
1 Introduction
MicroRNAs (miRNAs), small endogenous noncoding RNAs with approximately 22 nucleotides, have attracted numerous researchers' attention due to their important roles in a variety of biological processes such as cell differentiation, apoptosis, proliferation, and immunological response. Recently, miRNAs have emerged as new candidate diagnostic and prognostic biomarkers for detecting a wide variety of cancers.1,2 Many methodologies, including Northern blotting,3 miRNA arrays,4,5 and real-time reverse transcription polymerase chain reaction,6 have been developed to detect miRNA expression. Unfortunately, these methods may be of low sensitivity, labor-intensive, or require a large amount of RNA samples, etc. Another great challenge for quantitative miRNA analysis is high sequence homology among family members. The let-7 family is one of the well-studied miRNA families, which consists of 9 highly homologous miRNAs (let-7a to let-7g, let-7i, and mir-98).7–9 Because their sequences are similar to each other and nucleotide variations exist at several positions, it is difficult to exclusively discriminate let-7 family members, which interferes with our deep understanding of the specific function of particular let-7 miRNAs. Therefore, it is of great importance to develop some simple, sensitive, and specific methods for miRNA detection in complex biological samples.
The electrochemical technique offers attractive advantages such as high sensitivity, inherent simplicity, and low cost, and has been widely used in bioassays.10,11 Various signal amplification strategies, such as gold nanoparticle-based assay,12,13 enzymatic assay,14,15 rolling circle amplification,16,17 and polymerization-based assay,18 have been used in developing sensitive electrochemical genosensors for the detection of low-abundance miRNAs.19,20 However, single amplification could still not satisfy the requirements of ultrasensitive detection of miRNAs, thus multiple signal amplifications were employed. An enzyme amplification technology coupling with a redox-cycling reaction to acquire ultrasensitive detection of miRNAs was reported.21,22 He et al. presented a label-free and highly sensitive electrochemical genosensor for miRNA detection with the triple signal amplification of gold nanoparticles (AuNPs), alkaline phosphatase (ALP) and p-aminophenol (p-AP) redox cycling. Capturer DNA was directly anchored on the working electrode that may disturb the electrochemical signal output.
Herein, we designed a novel electrochemical biosensor with triple signal amplification for ultrasensitive let-7b detection in two steps. Firstly, an electrochemical signal source is obtained by enzyme amplification technology. Secondly, a Pt/Pd/RGO modified screen-printed gold electrode (Pt/Pd/RGO/SPGE) was used to detect and enhance the electrochemical signal with a redox-cycling reaction. The detection limit of 3.55 fM is similar to electrochemical detection of microRNAs based on triple signal amplification of multifunctional gold nanoparticles, enzymes and redox-cycling reaction21 with a detection of 3 fM, but the linear range of our sensor from 10 fM to 0.1 nM is wider. This method significantly increases the analysis sensitivity and provides a new concept in the development of a highly selective microRNA biosensor for gene expression profiling and molecular diagnostics.
2 Experimental
2.1 Materials
Dopamine hydrochloride, tris(2-carboxyethyl) phosphine hydrochloride (TCEP) and 1-naphthyl phosphatedisodium salt (p-NPP) were purchased from Sigma-Aldrich. Tris(hydroxymethyl) aminomethane, iron trichloridehexahydrate (FeCl3·6H2O), anhydrous NaAc, ethylene glycol (EG), and 1,6-diaminohexane were purchased from Tianjin Chemical Research Institute (Tianjin, China). All chemical reagents were of analytical grade and used as received without further purification. Water was purified with a Millipore Milli-Q water system to achieve a final resistance of 18.2 MΩ. All solutions were prepared using ultrapure water treated with 0.1% DEPC. The used oligonucleotides were purchased from Shanghai Sangon Biological Engineering Technology & Services Co. (Shanghai, China). Let-7a, let-7b, let-7c, let-7d, and miR-21 were obtained from Shanghai Genepharma Co., Ltd. (Shanghai, China). The sequences are illustrated in Table S1.†
2.2 Apparatus
UV-vis spectra were recorded on a UV-3600 spectrophotometer (Shimadzu, Kyoto, Japan). Fluorescence spectra were recorded on a RF-5301PC spectroflurophotometer (Shimadzu). Transmission electron micrographs were measured on a JEOL JEM 200CX transmission electron microscope, using an accelerating voltage of 200 kV. Scanning electron micrographs were obtained with a Hitachi S4800 scanning electron microscope. X-ray-photoelectron spectroscopy (XPS) analysis was carried out with an ESCALAB 250 spectrometer (Thermo-VG Scientific, USA). Cyclic voltammetry (CV) and differential pulse voltammetry (DPV) were performed on a CHI 660C electrochemical work station.
2.3 Modification of the screen-printed gold electrode (SPGE)
The screen-printed gold electrodes were purchased from Drop Sens (Spain) including a gold electrode as the working electrode, a platinum electrode as the counter electrode, and a silver electrode as the reference electrode. Prior to experiments, the screen-printed gold electrode (SPGE) was first scanned in 0.5 M H2SO4 from 0 V to 1.5 V at 100 mV s−1 until a reproducible cyclic voltammogram (CV) was achieved. For electrodeposition of multilayered graphene films, 1.0 mg mL−1 graphene oxide (GO) in carbonate buffer solution (10 mM, pH = 9.0) was prepared. The colloidal graphene oxide was electrochemically reduced to yield multilayered graphene films on the SPGE surface by cyclic voltammetry from 0.6 V to −1.5 V with a scanning rate of 25 mV s−1 and a potential step of 1 mV for 50 rounds. Then a mixture containing 2 mM PtCl6− and 2 mM PdCl6− was dropped onto the RGO/SPGE surface to obtain the Pt/Pd/RGO/SPGE by co-electrodeposition using an amperometric i–t curve method at −0.2 V for 100 s.
2.4 Preparation of Fe3O4@PDA
Fe3O4 beads were prepared following a previously reported protocol with minor modification.23 Briefly, 1.0 g of iron trichloridehexahydrate (FeCl3·6H2O), 2.0 g of sodium acetate anhydrous, and 6.5 g of 1,6-diaminohexane were added to 30 mL of ethylene glycol with the help of ultrasound at room temperature. Then, the mixture was maintained at 473 K for 6 h in a Teflon-lined stainless steel autoclave (50 mL capacity). The black Fe3O4 were separated by a magnetic field, washed with deionized water and ethanol alternately several times, and dried at 323 K in a vacuum drying oven. 10 mg of Fe3O4 nanospheres was dispersed in 30 mL Tris–HCl (10 mM, pH 8.5) and ultrasounded for 30 min. After that, 20 mg of dopamine was added and shaken for 4 h to obtain polydopamine coated Fe3O4 (Fe3O4@PDA), which was separated by a magnetic field and washed with deionized water and ethanol alternately several times. The obtained Fe3O4@PDA was dispersed in Tris–HCl buffer (10 mM, pH = 9.0) for further use.
2.5 Preparation of Fe3O4@PDA–DNA1
The thiol–DNA1 was activated with 10 mM TCEP in pH 5.2 acetate buffer for 1 h to prevent the terminal cysteine from forming disulphide bonds. Afterwards, 420 μL of 10 μM thiol–DNA1 was mixed with 1 mL Fe3O4@PDA (1 mg mL−1) and reacted overnight at 25 °C, then at 4 °C for 12 h. Subsequently, Fe3O4@PDA–DNA1 was separated by a magnetic field and washed with Tris buffer solution several times. The obtained Fe3O4@PDA–DNA1 was dispersed in Tris–HCl (pH = 7.4, 10 mM) for further use.
2.6 RNA hybridization assay and electrochemical measurements
In a typical miRNA assay, 5 μL 1 mg mL−1 Fe3O4@PDA–DNA1 was mixed with 5 μL 5 μM DNA2, 1 μL let-7b at various assigned concentrations, 1.0 μL 40 U μL−1 RNase inhibitor and 1 μL 1000 U μL−1 T4 DNA ligase at 37 °C for 4 h to perform the hybridization reaction. Fe3O4@PDA–DNA1/let-7b/DNA2 was separated by a magnetic field and washed with Tris–HCl buffer several times. Then 10 μL 10 μg mL−1 SA-ALP was added into the obtained Fe3O4@PDA–DNA1/let-7b/DNA2 and reacted for 30 min. Afterward, the Fe3O4@PDA–DNA1/let-7b/DNA2/SA-ALP composite was washed with 1% BSA and then filled with a Tris buffer solution (pH = 9.6, 100 μL, containing 10 mM Mg2+) containing 5 mM p-NPP. The supernatant was collected and the differential pulse voltammetric (DPV) response was then recorded after 15 min from −0.2 to +0.5 V using the Pt/Pd/RGO/SPGE electrode with a step potential of 4 mV, and a scan rate of 50 mV s−1 in the absence or presence of 1 mM TCEP, respectively. The TCEP solution was freshly prepared for each experiment. All measurements were repeated for a minimum of three times with separate electrodes to obtain statistically meaningful results.
2.7 Polyacrylamide gel electrophoresis analysis
8% polyacrylamide gel was employed to verify the binding of let-7b with DNA1 and DNA2. Firstly, 2 μL DNA1 (10 μM) and 2 μL DNA2 (10 μM) were incubated with 2 μL let-7b (10 μM) at 37 °C for 4 h to form a stable DNA hybrid by a base paring hybridization in Tris buffer (10 mM, pH 7.4). Electrophoresis was carried out in TBE buffer (pH 8.0) at 100 V for 45 min at room temperature. After separation, the gel was stained with ethidium bromide and imageio-Rad imaging system (Hercules, CA).
2.8 Cell culture and cell lysis
A human breast adenocarcinoma cell line (MCF-7), human cervical cancer cells (HeLa cells) and human lung carcinoma cells (A549) were grown at 37 °C in DMEM (Dulbecco's Modified Eagle Medium) supplemented with 10% (v/v) fetal bovine serum (FBS), 100 U mL−1 penicillin, and 100 mg mL−1 streptomycin in a 5% CO2 environment. After growing to 90% confluence, the cells were washed three times with PBS and the cell number was estimated using a hemocytometer. A 1.0 mL amount of ∼5 × 105 suspended cells was added into a 6-well plate for 12 h. Total RNA from these cells was extracted using TRIzol reagent according to the manufacturer's recommended protocol. RNA concentration was determined by UV-vis absorption at 260 nm.
2.9 Let-7b detection by qRT-PCR
The total cellular RNAs were extracted from HeLa, NIH3T3 and HL-60 cells using Trizol reagent (Invitrogen) according to manufacturer's protocols. The first-strand cDNA was generated using AMV reverse transcriptase (Takara Bio, Shiga, Japan) and a stem-loop RT primer (Invitrogen). Quantitative PCR was carried with an ABI 7300 Sequence Detection System (Applied Biosystems) using SYBR Green PCR Master Mix. For analysis of the let-7b level, we used 5′-ACACTCCAGCTGGGTGAGG TAGTAGGTTGT-3′ as the sense primer and 5′-CTCAACTGG TGTCGTGGAGTCGGCAATTCAGTTGAGAACCACAC-3′ as the antisense primer, as well as 5′-CTCGCTTCGGCAGC ACA-3′ and 5′-AACGCTTCACGAATTTGCGT-3′ for U6. The relative expression of let-7b was calculated using the 2-ΔCT method, in which ΔCT = CTlet-7b − CTU6. All RT-PCR reactions were performed in triplicate.
3 Results and discussion
3.1 The mechanism and characterization of the capture probe
Self-polymerization of dopamine (DA) easily produced a surface-adherent polydopamine (PDA) nanolayer deposited on the surface of magnetic Fe3O4 nanoparticles (Fe3O4@PDA) at weak alkaline pH24,25 (Fig. 1A). It is simple to separate and wash Fe3O4 carriers under a magnetic field in the decoration process. Transmission electron microscopy was used to characterize the modification procedure of Fe3O4@PDA. A PDA shell was observed on the surface of Fe3O4 nanoparticles (Fig. 1B and C). PDA contained the oxidized quinone form of catechol that can undergo reactions with various functional groups, including thiol, amine, and quinone itself.26 Therefore, Fe3O4@PDA can be further modified by thiol-terminated DNA1 via Michael addition. UV-vis techniques were used to characterize the modification procedure of Fe3O4@PDA–DNA1. After the conjugation of DNA1, an obvious UV emission peak around 260 nm could be observed, which was attributed to the characteristic absorbance band of the nucleic acid, indicating the successful attachment of DNA1 to Fe3O4@PDA (Fig. S1†).
 |
| | Fig. 1 (A) Preparation of Fe3O4@PDA/DNA1 and TEM images of Fe3O4 (B) and Fe3O4@PDA (C). | |
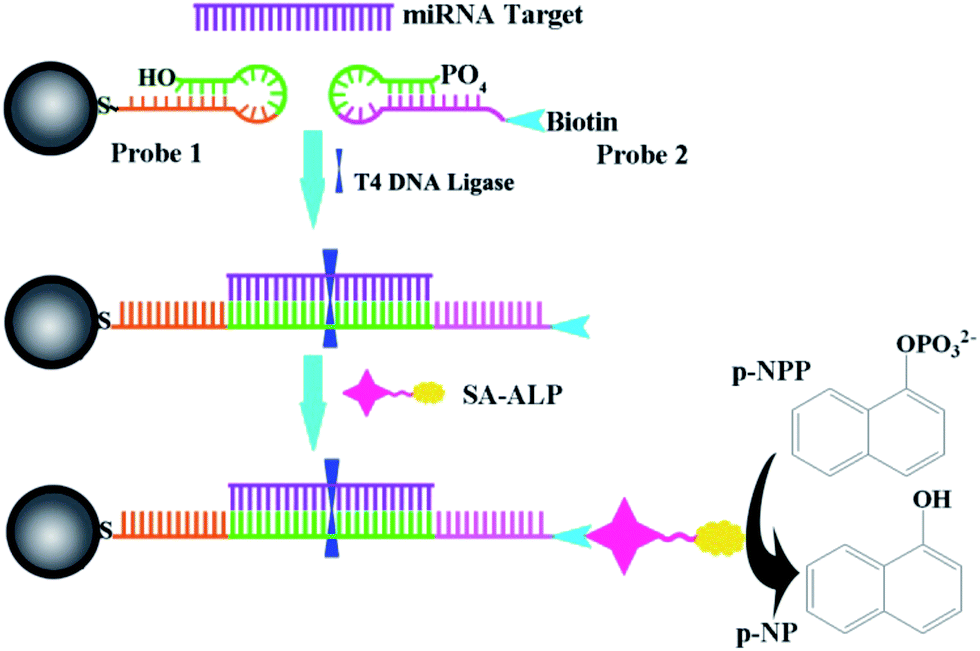
Scheme 1 illustrates the design of the genosensor for let-7b via sandwich-type hybridization assay. Two stem-loop structured DNAs were used as capturers; the stem-loop structured hairpin DNA with its excellent specificity has been widely explored for discrimination testing since stem-loop probes not only prevented their self-ligation and nonspecific hybridization with precursor miRNAs but also allowed precise distinction of a single mutant in the miRNA sequence.27,28 DNA1 has 11 bases at the 3′-end complementary to half part of let-7b as shown in Table S1.† Upon hybridization with the target let-7b, the hairpin structure of DNA1 was unfolded, and meanwhile the other half 11 bases of let-7b was further hybridized with probe 2 (DNA2) that was modified by biotin at the 3′ end. In the process, T4 DNA ligase is used to efficiently catalyze the joining between the 3′-hydroxyl group of DNA1 and the 5′-phosphomonoester group of DNA2 in the presence of let-7b like a tack.29 The hybridization reaction between the target and the capture probes was analyzed by polyacrylamide gel electrophoresis. As shown in Fig. S2,† no band appeared in Lane 2 because the sequence of single-stranded let-7b is short and difficult to be dyed by EB. Two bands in Lane 3 assigned to DNA1 and disulphide DNA1, one band in Lane 4 assigned to DNA2. In the presence of let-7b and T4 DNA ligase (Lane 8), the emission band that is higher than 300bp indicated the formation of the DNA1/let-7b/DNA2/T4 complex because T4 DNA ligase is a polypeptide with a high molecular weight so the DNA1/let7b/DNA2/T4 complex exhibited a wide emission band. Then, alkaline phosphatase-labeled streptavidin (SA-ALP) was attached to DNA2 via specific affinity between streptavidin and biotin. Alkaline phosphatase converted 1-naphthyl phosphate disodium salt (p-NPP) into electroactive 1-naphthol (p-NP).
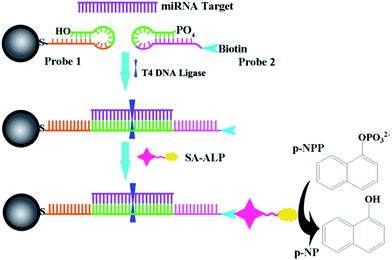 |
| | Scheme 1 Schematic illustration of the electrochemical strategy for miRNA. | |
3.2 Characterization of the electrode
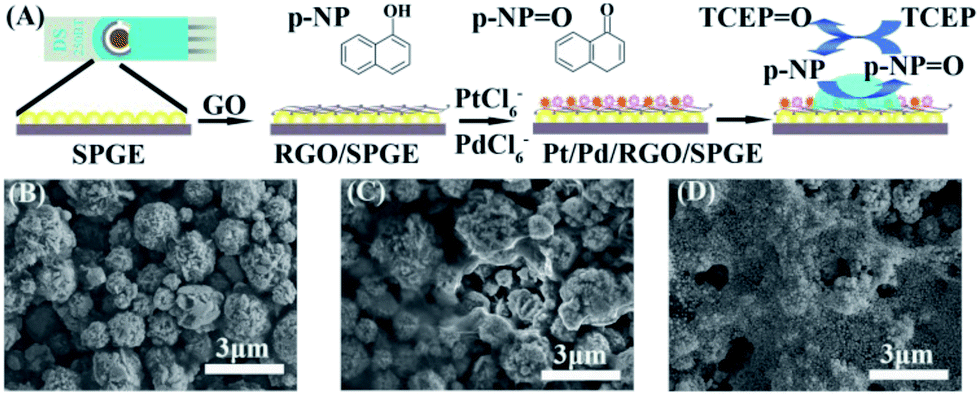
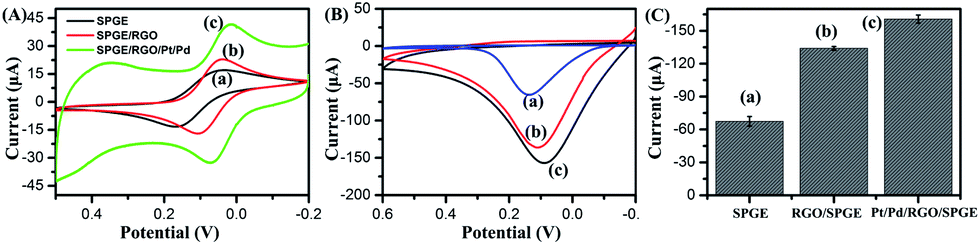
The supernatant containing p-NP was collected and dropped onto Pt/Pd/RGO/SPGE to obtain the electrochemical signal. Pt/Pd/RGO/SPGE was fabricated via the electrodeposition of RGO, Pt and Pd on SPGE (Fig. 2A). The detailed modification procedure is described in the Experimental section. The preparation process was investigated by using a scanning electron microscope (SEM). In comparison with Fig. 2B, a graphene film (RGO) could be observed on the surface of SPGE (Fig. 2C). After deposition of Pt and Pd, bimetallic Pt/Pd nanoparticles were attached on the RGO sheets as shown in Fig. 2D. The size of Pt/Pd nanoparticles is about ∼70 nm as shown in Fig. S3A†. The EDS pattern (Fig. S3B–E†) displayed the typical diffractions of C, Pt and Pd. XPS was further used to confirm the formation of bimetallic Pt/Pd/RGO modified SPGE (Fig. S4†). The three peaks at 284.2, 70.7, and 334.9 eV shown in the XPS spectrum of the electrode can be attributed to C1s, Pt4f, and Pd3d, respectively. The atomic percentages of C, Pt, and Pd were 79.11%, 12.52% and 8.36%, revealing the successful deposition of RGO, Pt, Pd. Pt/Pd/RGO modified SPGE greatly improved the conductivity of the electrochemical genosensor. As shown in Fig. 3A, the peak potential difference (ΔEp) for the Pt/Pd/RGO/SPGE (57 mV) was found to be smaller than that for both the RGO/SPGE (70 mV) and SPGE (135 mV), while the peak current is higher than the other two. Moreover, the highest signal of p-NP could be observed using Pt/Pd/RGO/SPGE (Fig. 3B and C). The peak values of SPGE, RGO/SPGE and Pt/Pd/RGO/SPGE are 0.138, 0.113, and 0.086 V, respectively. The peak potential decreased because the modification of RGO and Pt/Pd leads to the decrease of the overpotential. These results indicated that Pt/Pd/RGO/SPGE can enhance the sensing performance by promoting electron transfer on the electrode/electrolyte interface. Meanwhile tris(2-carboxyethyl) phosphine (TCEP), which can reduce electroactive organic materials such as dehydroascorbic acid and quinone at a fast rate,30,31 was added to regenerated p-NP, realizing a redox-cycling reaction (Fig. 2A). The optimal concentrations of p-NPP and TCEP were 5 mM and 1 mM, respectively (Fig. S5 and S6†). As we predicted, in the presence of TCEP, the peak current is about one time higher than that in the absence of TCEP, indicating that the constructed Pt/Pd/RGO/SPGE had strong signal amplification, which is valuable for the design of new types of sensitive electrochemical biosensors.
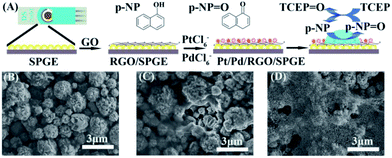 |
| | Fig. 2 (A) Preparation process of SPGE and the detection of miRNA. SEM images of SPGE (B), RGO/SPGE (C), and Pt/Pd/RGO/SPGE (D). | |
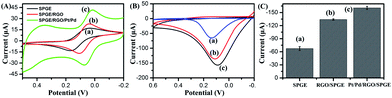 |
| | Fig. 3 (A) Cyclic voltammograms of SPGE (a), RGO/SPGE (b), Pt/Pd/RGO/SPGE (c) in 1 mM K3Fe(CN)6 at a scan rate of 100 mV s−1. (B) Cyclic voltammograms of p-NP in 5 mM p-NPP solution containing 10 μL, 0.1 mg mL−1 ALP using SPGE (a), RGO/SPGE (b) and Pt/Pd/RGO/SPGE (c) electrode respectively. (C) The peak values of the current in (B). (a) 0.138 V, (b) 0.113 V, and (c) 0.086 V. | |
3.3 Quantitative detection of let-7b
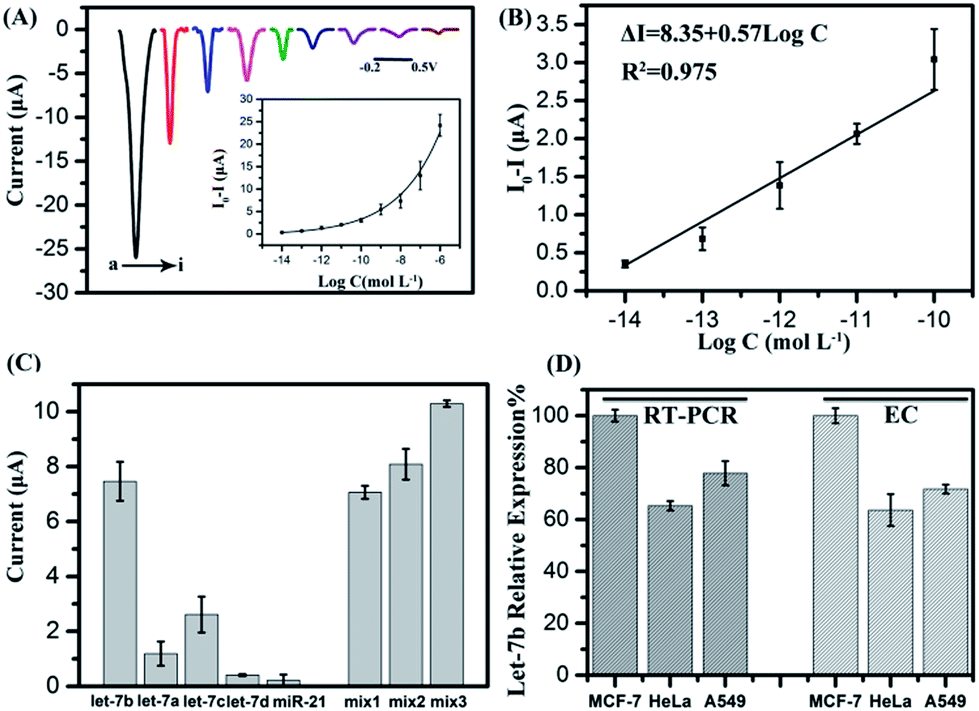
The sensitivity of the biosensor was investigated by varying the target miRNA concentrations. The current signals of p-NP after the sensor hybridized with different concentrations of let-7b were measured by differential pulse voltammetry (DPV). As shown in Fig. 4A, the absolute current value increased with the increased concentrations of let-7b in the concentration range of 10 fM–1 μM, and showed a good linear correlation with the logarithm of target miRNA concentrations in the range of 10 fM–0.1 nM (Fig. 4B) The regression equation is as follows: ΔI (I0 − I, μA) = 8.35 + 0.57 × log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) C (M), R2 = 0.975. The detection limit (S/N = 3) was calculated to be 3.55 fM.
C (M), R2 = 0.975. The detection limit (S/N = 3) was calculated to be 3.55 fM.
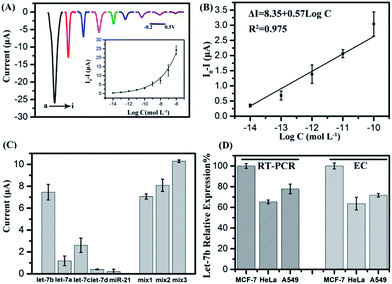 |
| | Fig. 4 (A) Differential pulse voltammetric (DPV) responses for different target let-7b concentrations: (a) 10−6 M, (b) 10−7 M, (c) 10−8 M, (d) 10−9 M, (e) 10−10 M, (f) 10−11 M, (g) 10−12 M, (h) 10−13 M, and (i) 10−14 M. The inset is the relationship between the peak current and the logarithm of the target RNA concentration. (B) The linear relationship between the peak current and the logarithm of the target DNA concentration from 10−10 M to 10−14 M. ΔI was calculated by I0 − I, where I and I0 are the peak currents observed in the presence and absence of let-7b, respectively. (C) DPV signals in the presence of let-7a, let-7b let-7c let-7d and miR-21 (all at a concentration of 10−8 M). Mix1: the mixture of let-7b (10−8 M) and let-7c (10−9 M), Mix2: the mixture of let-7b (10−8 M) and let-7c (2 × 10−9 M), Mix3: the mixture of let-7b (10−8 M) and let-7c (10−8 M). Each value is the average of three measurements. (D) The relative expression level of let-7b in various cells obtained by qRT-PCR and electrochemical biosensor (EC). | |
3.4 Specificity
To examine the selectivity of the biosensor, a comparison assay on mismatch targets and a perfect complementary target, including the perfect complementary target (let-7b) and single-base mismatch target (let-7c), two-base mismatch target (let-7a) and four-base mismatch target (let-7d), and non-complementary sequence (miR-21) at a concentration of 10 nM, was performed respectively. As shown in Fig. 4C, the complementary target let-7b leads to a current increase which is approximately two times higher than that for let-7c and five times higher than that for let-7a, while the let-7d and miR-21 caused only a small increase in the electrochemical signal, demonstrating that this biosensor has an excellent discrimination for similar miRNAs. Mixtures of let-7b and let-7c with different concentrations were detected as well. Let-7b/let-7c concentration ratios are 10:1, 5:1 and 1:1, respectively. It can be seen when the let-7b/let-7c concentration ratio decreased to 1:1, the electrochemical signal dramatically increased, demonstrating that let-7c will obstruct the signal at high concentration, while at low concentration there is no high influence on the current signal.
3.5 MicroRNA detection in cell lysates
The sensor was also directly used to analyze miRNAs in real samples, the content of let-7b extracted from HeLa cells, MCF-7 cells and A549 cells. As shown in Fig. 4D, the order of the relative expression level is MCF-7 > A549 > HeLa using the electrochemical method (EC), in compliance with the results obtained by qRT-PCR, which demonstrated the validity of the as-proposed method. These results confirmed that the biosensor could be used for the determination of let-7b in real samples, Moreover, for qRT-PCR, it is necessary to reverse-transcribe miRNA into cDNA. Then cDNA hybridized with two primers to realize amplification and finally probes were added to produce fluorescence signals. The process is complicated and skilled technicians are needed. Our proposed electrochemical biosensor can be used to detect miRNA directly, and easy to operate, providing a new, simple platform for the quantification of miRNAs and having a potential application for clinical diagnosis.
4 Conclusions
In summary, we have developed an ultrasensitive electrochemical method for the detection of let-7b by integrating phosphatase amplification with redox-cycling amplification. Moreover, the Pt/Pd/RGO fabricated SPGE of excellent conductivity was used to enhance the electrochemical signal. A proportional relationship was observed between the logarithm of target let-7b concentration and the DPV peak currents in a linear range from 10 fM to 0.1 nM with a detection limit of 3.55 fM. At the same time, this proposed biosensor is sufficiently selective to discriminate the target miRNA from homologous miRNAs and non-complementary miRNAs due to the loop-stem structure of capture probes and T4 DNA ligase. Subsequently, the proposed strategy has successfully achieved the detection of human miRNAs from cancer cell-specific total RNA extracts. Therefore, this biosensor is an attractive candidate for the development of an accurate, selective, and ultrasensitive method for miRNA expression profiling and clinical diagnostics.
Acknowledgements
The authors thank the National Basic Research Program of China (2011CB933502) and the National Natural Science Foundation of China (21335004) for financial support. FF Cheng thanks the support by the program B for outstanding PhD candidates of Nanjing University. The authors extend their appreciation to the Deanship of Scientific Research at King Saud University for funding the work through the research group project (RGP-VPP-029).
Notes and references
- J. Lu, G. Getz, E. A. Miska, E. Alvarez-Saavedra, J. Lamb, D. Peck, A. Sweet-Cordero, B. L. Ebert, R. H. Mak and A. A. Ferrando, Nature, 2005, 435, 834–838 CrossRef CAS PubMed.
- D. P. Bartel, Cell, 2004, 116, 281–297 CrossRef CAS.
- M. Lagos-Quintana, R. Rauhut, W. Lendeckel and T. Tuschl, Science, 2001, 294, 853–858 CrossRef CAS PubMed.
- I. Lee, S. S. Ajay, H. Chen, A. Maruyama, N. Wang, M. G. McInnis and B. D. Athey, Nucleic Acids Res., 2008, 36, e27 CrossRef PubMed.
- J. M. Lee and Y. Jung, Angew. Chem., Int. Ed., 2011, 50, 12487–12490 CrossRef CAS PubMed.
- C. Chen, D. A. Ridzon, A. J. Broomer, Z. Zhou, D. H. Lee, J. T. Nguyen, M. Barbisin, N. L. Xu, V. R. Mahuvakar and M. R. Andersen, Nucleic Acids Res., 2005, 33, e179 CrossRef PubMed.
- S. Bi, J. Zhang, S. Hao, C. Ding and S. Zhang, Anal. Chem., 2011, 83, 3696–3702 CrossRef CAS PubMed.
- W. Hou, Q. Tian, N. M. Steuerwald, L. W. Schrum and H. L. Bonkovsky, Biochim. Biophys. Acta, 2012, 1819, 1113–1122 CrossRef CAS PubMed.
- A. E. Pasquinelli, B. J. Reinhart, F. Slack, M. Q. Martindale, M. I. Kuroda, B. Maller, D. C. Hayward, E. E. Ball, B. Degnan and P. Müller, Nature, 2000, 408, 86–89 CrossRef CAS PubMed.
- J. J. Zhang, T. T. Zheng, F. F. Cheng and J. J. Zhu, Chem. Commun., 2011, 47, 1178–1180 RSC.
- T. T. Zheng, T. T. Tan, Q. Zhang, J. J. Fu, J. J. Wu, K. Zhang, J. J. Zhu and H. Wang, Nanoscale, 2013, 5, 10360–10368 RSC.
- M. Xu, J. Zhuang, X. Chen, G. Chen and D. Tang, Chem. Commun., 2013, 49, 7304–7306 RSC.
- T. Zheng, R. Zhang, Q. Zhang, T. Tan, K. Zhang, J.-J. Zhu and H. Wang, Chem. Commun., 2013, 49, 7881–7883 RSC.
- J. Li, Y. Jia, J. Zheng, W. Zhong, G. Shen, R. Yang and W. Tan, Chem. Commun., 2013, 49, 6137–6139 RSC.
- Y. Huang, J. Chen, S. Zhao, M. Shi, Z. F. Chen and H. Liang, Anal. Chem., 2013, 85, 4423–4430 CrossRef CAS PubMed.
- Q. Wang, H. Zheng, X. Gao, Z. Lin and G. Chen, Chem. Commun., 2013, 49, 11418–11420 RSC.
- Y. Wen, Y. Xu, X. Mao, Y. Wei, H. Song, N. Chen, Q. Huang, C. Fan and D. Li, Anal. Chem., 2012, 84, 7664–7669 CrossRef CAS PubMed.
- V. Tjong, H. Yu, A. Hucknall and A. Chilkoti, Anal. Chem., 2012, 85, 426–433 CrossRef PubMed.
- C. Pöhlmann and M. Sprinzl, Anal. Chem., 2010, 82, 4434–4440 CrossRef PubMed.
- M. Labib, N. Khan, S. M. Ghobadloo, J. Cheng, J. P. Pezacki and M. V. Berezovski, J. Am. Chem. Soc., 2013, 135, 3027–3038 CrossRef CAS PubMed.
- L. Liu, N. Xia, H. Liu, X. Kang, X. Liu, C. Xue and X. He, Biosens. Bioelectron., 2014, 53, 399–405 CrossRef CAS PubMed.
- M. R. Akanda, M. A. Aziz, K. Jo, V. Tamilavan, M. H. Hyun, S. Kim and H. Yang, Anal. Chem., 2011, 83, 3926–3933 CrossRef CAS PubMed.
- S. H. Liu, F. Lu, R. M. Xing and J. J. Zhu, Chem.–Eur. J., 2011, 17, 620–625 CrossRef CAS PubMed.
- P. H. Zhang, J. T. Cao, Q. H. Min and J. J. Zhu, ACS Appl. Mater. Interfaces, 2013, 5, 7417–7424 CAS.
- J. Si and H. Yang, Mater. Chem. Phys., 2011, 128, 519–524 CrossRef CAS PubMed.
- L. Q. Xu, W. J. Yang, K. G. Neoh, E. T. Kang and G. D. Fu, Macromolecules, 2010, 43, 8336–8339 CrossRef CAS.
- X. Meng, Y. Zhou, Q. Liang, X. Qu, Q. Yang, H. Yin and S. Ai, Analyst, 2013, 138, 3409–3415 RSC.
- S. Song, Z. Liang, J. Zhang, L. Wang, G. Li and C. Fan, Angew. Chem., Int. Ed., 2009, 48, 8670–8674 CrossRef CAS PubMed.
- J. Li, B. Yao, H. Huang, Z. Wang, C. H. Sun, Y. Fan, Q. Chang, S. L. Li, X. Wang and J. Z. Xi, Anal. Chem., 2009, 81, 5446–5451 CrossRef CAS PubMed.
- M. R. Akanda, M. A. Aziz, K. Jo, V. Tamilavan, M. H. Hyun, S. Kim and H. Yang, Anal. Chem., 2011, 83, 3926–3933 CrossRef CAS PubMed.
- J. Das, K. Jo, J. W. Lee and H. Yang, Anal. Chem., 2007, 79, 2790–2796 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Table S1 and Fig. S1–S6. See DOI: 10.1039/c4an00777h |
|
| This journal is © The Royal Society of Chemistry 2014 |
Click here to see how this site uses Cookies. View our privacy policy here.