 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Novel DFO-SAM on mesoporous silica for iron sensing. Part I. Synthesis optimization and characterization of the material†
Raffaela
Biesuz
*a,
Giovanni
Emma
a,
Chiara
Milanese
a,
Giacomo
Dacarro
a,
Angelo
Taglietti
a,
Valeria Marina
Nurchi
b and
Giancarla
Alberti
a
aDipartimento di Chimica, Università di Pavia, via Taramelli 12, 27100 Pavia, Italy. E-mail: rbiesuz@unipv.it; Tel: +39 0382 987348
bDipartimento di Scienze Chimiche e Geologiche, University of Cagliari, Cittadella Universitaria, 09042 Monserrato-Cagliari, Italy
First published on 23rd May 2014
Abstract
The synthesis and the physico-chemical characterisation of a novel solid phase, designed for iron(III) sorption, are presented. The solid (indicated in the following as DFO-SAMMS) is made with a hydroxamate siderophore, the deferoxamine (DFO), covalently bound on a self-assembled monolayer on mesoporous silica (SAMMS). The data demonstrate that the DFO molecules are bound to the solid material, grafted on the surface and do not enter the silica pores. A new one-pot synthesis is presented in which DFO is dissolved in DMSO, and left to react with GPTMS with stirring overnight. In the same mixture, SAMMS is added to get the final product. The optimisation of experimental conditions of this novel one-pot synthesis is presented, with results indicating that a temperature of 90 °C, for the reaction between DFO and GPTMS, and the use of MCM-41 silica are the most convenient conditions. The kinetics of sorption reveals that the iron uptake is relatively fast, around 100 min at pH = 2.5, and from the sorption profile of iron(III), the estimated capacity of the product obtained under optimized conditions was higher than 0.3 mmol g−1. The results found in the present research are very promising for application in real biological samples.
Introduction
Iron is an essential element that plays a crucial role in cell life such as uptake and transportation of oxygen to tissues and electron transfer processes in synthesis of nucleic acids. The presence of Fe(III) in biological systems has to be efficiently moderated as both its deficiency and overloading can induce various biological disorders.1,2 Excess of iron is related, in the presence of molecular oxygen, to the ability of the Fe(II)/Fe(III) redox cycle to generate dangerous hydroxyl free radicals, via Fenton reaction. The presence of reactive radicals, able to interact with a variety of molecules, nucleic acids, proteins and lipids, results in severe peroxidative damage.Our body is not able to protect cells against iron overload, as a consequence of hereditary disorders or caused by multiple frequent blood transfusions of patients affected by different kinds of anemia, like β-thalassemia major, or sickle cell anemia.
Irreversible tissue damage and fibrosis appear in various organs, in the absence of a chelation therapy. Moreover, in the past few decades, the role of iron in neuroinflammation and progression of neurodegenerative diseases, such as Alzheimer's disease, has been clarified. Conversely iron deficiency can be equally harmful for organisms.
For these reasons, in analytical clinical chemistry, the ability to detect traces of iron(III) is extremely important. A device capable of selectively quantifying iron(III) could be also employed in studies on chelation therapy, to evaluate the ability of a sequestering agent, to estimate the half life of a drug and to measure the NTB (non-transferrin-bound) iron.
We intend to develop a sensor for signalling and quantifying iron(III), based on an active centre, a metal acceptor moiety with strong affinity for iron(III), linked to a solid phase with suitable properties.
DFO is a hydroxamate siderophore, employed in chelation therapy, which forms a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 Fe:DFO octahedral complex with the six oxygen atoms of the hydroxamate groups.3 The terminal amino group is not involved in iron complexation and is suitable to be functionalized, for example in order to be grafted into a solid phase, or on a surface. Furthermore, the complex of DFO with Fe(III) in water is coloured: it has a maximum absorption at about 425 nm (ref. 4–6) and an extinction coefficient of about 2500–2800 M−1 cm−1 a feature which seems promising for sensing properties.
:1 Fe:DFO octahedral complex with the six oxygen atoms of the hydroxamate groups.3 The terminal amino group is not involved in iron complexation and is suitable to be functionalized, for example in order to be grafted into a solid phase, or on a surface. Furthermore, the complex of DFO with Fe(III) in water is coloured: it has a maximum absorption at about 425 nm (ref. 4–6) and an extinction coefficient of about 2500–2800 M−1 cm−1 a feature which seems promising for sensing properties.
Thanks to the terminal amino groups, several strategies to immobilize DFO have been proposed on different insoluble substrates such as cellulose,7,8 nylon,9 acrylate polymers,10 Sepharose11 and silica materials.11–14
In this paper we characterized the properties of a novel solid phase for iron(III) detection, based on mesoporous silica (MS) on which DFO is covalently bound. The immobilization of the siderophore molecule has been firstly performed following a reaction reported for a glass slide.12 The synthesis of DFO self-assembled monolayers on MS (SAMMS) was then performed following a novel one-pot strategy. Two types of MS, with different pore sizes, have been considered: MCM-41 and MSU-H.
The existence of SAM on mesoporous silica in such hybrid materials is reported in several papers.15–19 An optimization of the synthesis, based on experimental design techniques, was employed to obtain the material with the best performance, i.e. the highest Langmuir sorption capacity.
The new materials were characterized with the common physico-chemical methods. The kinetics of sorption of iron(III) and the sorption isotherm profiles on the new materials are presented and discussed.
Materials and methods
Chemicals
All chemicals were of analytical grade.Mesoporous silica MCM-41 type, mesoporous silica MSU-H type, (3-glycidyloxypropyl)trimethoxysilane (GPTMS), dimethyl sulfoxide (DMSO, anhydrous), acetonitrile, KNO3, HNO3 for trace analysis and NaOH were purchased from Sigma Aldrich. Deferoxamine mesylate salt (DFO) was obtained from Novartis. All these reagents were used as received. Toluene (Carlo Erba) was distilled over Na under N2 before use. An iron standard solution for ICP of 1000 mg L−1 (Fluka) was used to obtain the proper Fe(III) concentration in the solution phase. Solutions were prepared with ultrapure water (Milli-Q).
Instrumentation
IR analyses were carried out with a FTIR Perkin Elmer 1600 apparatus with diffuse reflectance equipment. Thermal analysis was performed using a coupled differential scanning calorimeter-thermogravimetric apparatus, DSC-TGA TA Instruments SDT2960. The SEM and EDS analyses were performed using a Zeiss Evo-MA10-HR system equipped with an Oxford INCA Energy 350× Max EDS detector for microanalysis. A cobalt standard was used for the calibration of the quantitative elementary analysis. TEM characterisation was carried out in the Department of Physics, Autonomous University of Barcelona, UAB (Bellaterra, Spain), on a JEOL JEM-2011 microscope operated at 200 kV. An Orion420 pH-meter, with a combined glass electrode, was used to determine the pH of all the solutions, standardized daily in [H+] concentration. The aqueous solutions were analysed for the iron content by an ICP-AES PerkinElmer Optima 3300 DV. The calibration curves were obtained according to the constructor indications. The LOD and LOQ were respectively 0.15 μM and 0.50 μM for Fe at 238.204 nm.Gas adsorption analysis measurements using nitrogen at 77 K were conducted on a Thermo Electron\Sorptomatic 1990.
Synthesis
DFO-mesoporous silica (MS) MCM-41 type and MSU-H type were obtained according to a synthesis employed to prepare DFO monolayers on silica slides, in a two step synthesis pathway.12 Afterwards, a novel one-pot synthesis was proposed. For simplicity, the two synthesis pathways are reported in Scheme 1.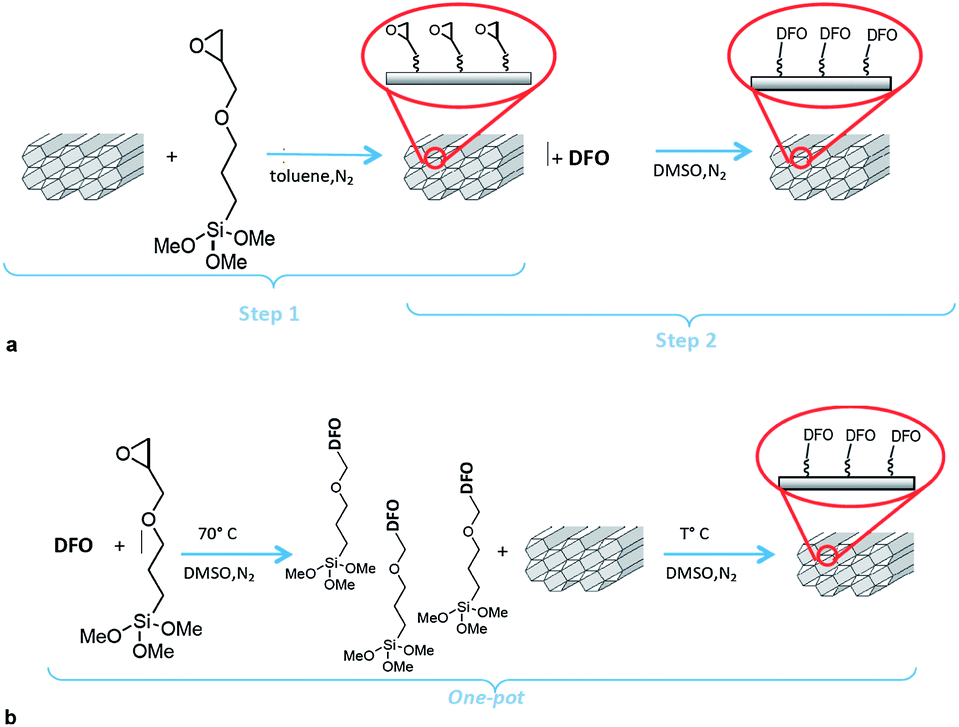 | ||
| Scheme 1 (a) Synthesis of the DFO-SAMMS following the two step synthesis. (b) Synthesis of the DFO SAMMS following the novel one-pot synthesis. | ||
Elemental analysis (MCM-41 silica):
GPTMS SAMMS: 7.61% C, 1.4% H, 0% N;
DFO-SAMMS (two steps): 15.03% C 2.49% H, 2.03% N;
The DFO was used in the one pot reaction in two forms: mesylate salt, as received, or free amine. In the second case, the mesylate salt was dissolved in methanol and an equivalent amount of NaOH was added to the solution; after 20 minutes, the solvent was removed and the residue was washed three times with acetonitrile and dried before use.
Elemental analysis (MCM-41 silica):
DFO-SAMMS (one pot under optimised conditions) l: 12.51% C, 2.52% H, 2.55% N.
Kinetic and isotherm experiments
Kinetic profiles and sorption isotherms were acquired following a continuous procedure. About 50 mg of the solid phase were put in contact with a known volume of 0.1 M KNO3 solution at different iron concentrations (from 0 to 1 mM) to obtain the sorption isotherms, or with the same iron concentration (e.g. 10 μM), to evaluate kinetic profiles. The temperature was controlled at 25.0(2) °C, the pH was adjusted to 2.50(5) and the solutions were gently stirred. At a given time, in the case of kinetic experiments, or after equilibration, in the case of isotherm experiments, a small amount of solution was collected, diluted with 0.5% v/v HNO3 in a new disposable testing tube and analysed by ICP-OES to determine the iron content. The amount of sorbed iron was determined by the difference from the total metal ion content.Results
Physico-chemical characterization
 | ||
| Fig. 1 FT-IR spectrum of DFO-SAMMS (black line) and GPTMS-MS (grey line). The MS MCM-41 type was employed. | ||
The FT-IR spectrum of DFO SAMMS presents an amide I band of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibrations, at 1630 cm−1, at a wavenumber a little lower than that expected (1695–1650 cm−1), possibly due to intra-molecular hydrogen bonding that reduces the frequency of the carbonyl stretching, according to what is observed in a deferoxamine sample or DFO grafted on a MS-coated wafer.13,16 At a higher wavenumber region, between 3200 and 3370 cm−1, the stretching vibrations of N–H and O–H overlap in a unique band. This band is also observed on DFO samples.16
O stretching vibrations, at 1630 cm−1, at a wavenumber a little lower than that expected (1695–1650 cm−1), possibly due to intra-molecular hydrogen bonding that reduces the frequency of the carbonyl stretching, according to what is observed in a deferoxamine sample or DFO grafted on a MS-coated wafer.13,16 At a higher wavenumber region, between 3200 and 3370 cm−1, the stretching vibrations of N–H and O–H overlap in a unique band. This band is also observed on DFO samples.16
The amide I band is absent in the GPTMS-MS spectrum, while the O–H stretching vibrations, present in the high wavenumber region, even if at small intensity, could be caused by a partial hydrolization of the epoxy group.
FT-IR spectra of the final products, similar to the black line reported here, were obtained also for the MS MSU-H, and in the case of products obtained by one pot synthesis.
 | ||
| Fig. 2 TEM analysis of MS MCM-41 type (above) and MS MSU-H type (below) both loaded with iron(III). On the right the results of the microelementary analysis are shown. | ||
Similar results were found on the same samples by SEM-EDS analysis: the Si/O ratio, equal to 3.1(2)/6.8(2), was as expected, and the iron percentage was estimated to be around 1.1 to 1.4 wt%.
The iron percentage corresponds to an amount of 0.2 mmol g−1, not so far from the value found for maximum sorption capacity from sorption isotherms (see below), suggesting that in the case of one-pot synthesis, the DFO-GPTMS fragment is likely larger than the pore size of both the types of SAMMS (the declared pore diameters are 2.5 nm for the MCM-41 and 7.1 nm for the MSU-H) and DFO should be present mainly on the silica surface.
In Fig. 3, the TGA profiles of GPTMS-MS and DFO-SAMMS referred to as the MCM-41 MS type, obtained with the two step synthesis, are reported with black and grey lines, respectively.
 | ||
| Fig. 3 TGA of the GPTMS-SAMMS (black line) and DFO-SAMMS (grey line). Temperature program: 1 °C min−1; N2 atmosphere. | ||
The TGA of GPTMS-MS shows a large weight loss of about 13.2%, due to GPTMS molecules bound on the surface and, in this case, also inside the pore structure. Indeed, the GPTMS can enter the hexagonal channels of the matrix. The weight loss of DFO-SAMMS increases only by 1.2%, with respect to that of GPTMS-MS, meaning that not all the GPTMS has reacted with the DFO, but only a part of it, most likely that present on the surface. This is confirmed by elemental analysis findings.
From nitrogen elemental analysis, the amount of DFO in the hybrid materials is determined. As expected, in GPTMS-MS, no nitrogen is present, while in DFO-SAMMS prepared by the two step synthesis, it corresponds to about 0.24 mmol g−1. The carbon percentage in the DFO-SAMMS exceeds that expected from the nitrogen content, meaning that there is unreacted GPTMS.
In the one-pot reaction, the DFO-GPTMS moieties, prepared before being grafted on the MS, are too large to enter the pores of the silica structure, so they react with the free –OH groups present on the surface. The TGA of one-pot DFO-SAMMS, not reported, confirms the above hypothesis: the weight loss is 8.3% w/w, lower than that of DFO-SAMMS obtained by the two step synthesis, where there was the contribution coming from the GPTMS inside the pores.
This hypothesis seems to be confirmed by elemental analysis, performed on the optimized product. From N%, the concentration of DFO on silica was found to be around 0.3 mmol g−1, consistent with other findings (see below) and in pretty good agreement with the C%, roughly assuming that the number of carbon atoms comes from the DFO-GPTMS adduct.
The results are reported in Fig. 4 and they clearly demonstrate that the pore volume of the native material compared to that of the hybrid one is not significantly different, demonstrating that the DFO-GPTMS units do not enter the silica pores.
 | ||
| Fig. 4 N2 adsorption/desorption isotherms of (a) MCM-41 and (b) DFO-SAMMS (one-pot synthesis). SBET = surface areas calculated by the BET model; DBJH average pore diameters calculated by the BJH model; V = total pore volume. | ||
Kinetic studies
The kinetics of the metal uptake has been studied according to the procedure described in the Experimental section.In Fig. 5, as an example, two kinetic profiles where the fraction of the sorbed metal, f, is reported as a function of time, are shown. They referred to SAMMS obtained with the one-pot synthesis, the experimental data are reported with symbols, while the continuous lines are the curves calculated with the fitting parameters of the model that better describe the profile. The results of the fitting of the kinetics data for the DFO-SAMMS are reported in the caption of the figure. They are referred to as the pseudo first order kinetic equation.22
 | ||
| Fig. 5 Kinetic profiles of DFO-SAMMS on MCM-41 type (black dots) and MSU-H type (white dots). All the experiments were performed at 25.0(1) °C in 0.1 M KNO3 at pH 2.50(5); V = 20 mL, w = 30 mg, cFe = 1.8 × 10−5 M. The lines represent the calculated profiles obtained by fitting of the pseudo first order equation, k = 0.048(3) min−1 and 0.043(1) min−1 for MCM-41 and MSU-H respectively. | ||
The equilibrium was reached after 200 minutes; in the following isotherm experiments, this duration was considered as the minimum to reach equilibrium conditions.
Under similar experimental conditions (pH, temperature, stirring and ionic strength), a lower equilibrium rate was observed for the DFO-SAMMS prepared by the two step synthesis for both the types of silica. The possible explanation could be that the one-pot strategy assures a better organization of the active sites that makes the solid phase more available for the metal. As is evident in Fig. 5, none of the solid phases gave a quantitative sorption, since the fraction of metal ion sorbed was around 0.7, but this pH value was chosen to prevent iron precipitation.
Sorption isotherms
From the analytical point of view, our concern in the development of the solid sorbents was to improve the maximum sorption capacity, i.e. in the present study, the number of DFO molecules on the silica surface. The Langmuir and the Freundlich models are often used to describe the relationships between the sorbed quantity, q (mmol g−1), and ceq (M) that is the remaining solute concentration, when the equilibrium has been reached. Their different performances have been reviewed many times22 and they will not be further commented here. Sorption isotherms were obtained at 25 °C in 0.1 M KNO3 at pH 2.50(5) for all solid phases.The DFO-SAMMS with a more compact pore structure (MCM-41) reveals a maximum uptake capacity, higher than that found for the DFO-SAMMS MSU-H type. The consequence is probably due to the higher surface area of the MCM-41 type (1 × 107 cm2 g−1 instead of 7.6 × 106 cm2 g−1 for the MSU-H type) that assures a more uniform coverage of the silane and finally of the DFO molecule.
The sorption uptake process is better described by the Langmuir model. The qmax is 0.14 mmol g−1 for the MCM-41 type and 0.06 mmol g−1 for the MSU-H type.
The results, in terms of maximum sorption capacity (qmax), obtained in the case of the one-pot synthesis, from the beginning, revealed to be more promising. This is the reason why we decided to optimize this synthesis, according to the experimental design technique.23,24 The results are reported in Table 1. (See also the ESI†.) The rationale of the choice of the experiments and the discussion on the results will be commented in the next paragraph.
| Langmuir | Freundlich | ||||||
|---|---|---|---|---|---|---|---|
| q max, mmol g−1 | K L, L mol−1 | Err. | n | Err. | K F, L mol−1 | Err. | |
| 1 | 0.33(6) | 2829 | 995 | 1.78 | 0.25 | 14 | 9 |
| 2 | 0.16(1) | 6560 | 1189 | 2.74 | 0.23 | 2 | 0.5 |
| 3 | 0.33(4) | 2672 | 763 | 1.68 | 0.23 | 16 | 10 |
| 4 | 0.25(3) | 930 | 211 | 1.59 | 0.15 | 9 | 3 |
| 5 | 0.22(2) | 2010 | 407 | 1.99 | 0.16 | 5 | 1 |
| 6 | 0.14(1) | 8929 | 2087 | 2.32 | 0.34 | 3 | 2 |
| 7 | 0.12(2) | 3428 | 2153 | 2.65 | 0.57 | 1 | 1 |
| 8 | 0.13(3) | 2051 | 1175 | 1.70 | 0.43 | 5 | 5 |
Experimental design for optimisation of Langmuir capacity
Preliminary results have shown that the one-pot synthesis (method b) was more convenient, in terms of type and amount of solvent, simplification of the procedure and slightly faster kinetics of the hybrid material.We selected the maximum sorption capacity, as found from the sorption isotherm experiments, described above, as the response to maximize the yield of the synthesis, being the valuable parameter for a solid performance. Before discussing the experimental design, few preliminary considerations about the solvent and the form of the reagents have to be made.
DMSO, used in the synthesis, is not a common solvent, but sometimes it is used as a mild oxidation reagent in order to convert alcohol to aldehyde or ketone25,26 or to open epoxide rings in order to form aldehyde or hydroxy ketone.27–29
The deferoxamine has a low solubility in many solvents and the DMSO seems to be the correct choice in terms of solvation of all the reagents involved in the synthesis. With this choice, the variable temperature in the second step of the one-pot synthesis assumes an important role: if it is too high, it could definitely promote the conversion of the epoxy-group of the GPTMS with formation of by-products, while for low values, the reaction does not start.
The most appropriate chemical form of DFO was also tested, in some preliminary experiments. As told before, the DFO is sold as a mesylate salt, in a slightly acid form. In principle the acid could catalyse the reactions reported in Scheme 2. For this reason, the effects of deferoxamine in the basic form and in the mesylate form were tested. In the Experimental section, the procedure to obtain what will be named, in the following tables and figures, neutral DFO is described. The DFO in the mesylate form was used as received.
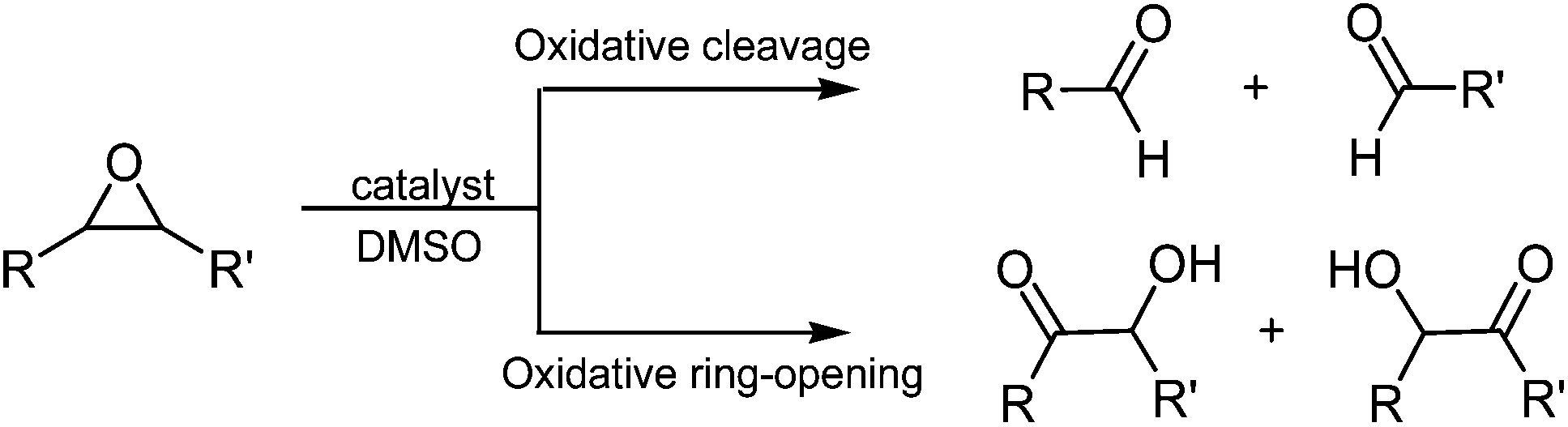 | ||
| Scheme 2 Epoxide side reaction promoted by DMSO. | ||
The effect of the pore dimensions of the silica materials on the new one-pot synthesis was also tested. In principle, the larger pores of MSU-H can allocate the DFO-GPTMS moiety inside them, at least to some extent, better than MCM-41 and this could affect the sorption capacity.
Finally, we selected three variables that reasonably have influence on the number of active sites of the solid phase: the temperature of the second step of the reaction, the form of DFO (mesylate or free ligand) and the type of silica, MCM-41 or MSU-H. The levels of the variables, which define the experimental domain, are reported in Table 2.
| Variables | −1 | +1 |
|---|---|---|
| Temperature (°C) | 90 | 120 |
| Type of DFO | Mesylate | Neutral |
| Type of silica | MCM-41 | MSU-H |
As reported above, to describe the response, we selected the maximum uptake capacity, as evaluated from the Langmuir sorption isotherm.
On these bases, a 23 full factorial design was considered.
The design of experiments and the experimental plan are presented in Table 3. The experimental response values are also reported in the last column. Any central point was planned.
| No. | 23 experimental design | Experimental plan | Response | ||||
|---|---|---|---|---|---|---|---|
| T | DFO | Silica | T | DFO | Silica | q, mmol g−1 | |
| 1 | −1 | −1 | −1 | 90 | Mesylate | MCM-41 | 0.33 |
| 2 | 1 | −1 | −1 | 120 | Mesylate | MCM-41 | 0.16 |
| 3 | −1 | 1 | −1 | 90 | Neutral | MCM-41 | 0.33 |
| 4 | 1 | 1 | −1 | 120 | Neutral | MCM-41 | 0.25 |
| 5 | −1 | −1 | 1 | 90 | Mesylate | MSU-H | 0.22 |
| 6 | 1 | −1 | 1 | 120 | Mesylate | MSU-H | 0.14 |
| 7 | −1 | 1 | 1 | 90 | Neutral | MSU-H | 0.12 |
| 8 | 1 | 1 | 1 | 120 | Neutral | MSU-H | 0.13 |
The equation of the model is:
| R = b0 + b1x1 + b2x2 + b3x3 + b12x1x2 + b13x1x3 + b23x2x3 | (1) |
In order to determine the coefficient of the 23 factorial design, m-files in MATLAB® environment were used.30 The values of the coefficients are shown in Fig. 6, where asterisks indicate the significance of the same coefficients according to the usual convention: * p < 0.05, ** p < 0.01, and *** p < 0.001.
 | ||
| Fig. 6 Values of the coefficients obtained from the multiregression of eqn (1) for the 23 full factorial design performed for the DFO-SAMMS synthesis. | ||
If we introduce the coefficients, the eqn (1) becomes:
| R = 0.208(1) − 0.038(1)x1 − 0.005(1)x2 − 0.057(1)x3 + 0.021(1)x1x2 + 0.022(1)x1x3 − 0.025(1)x2x3 | (2) |
The sign of the coefficients indicates in which way the variable has to be set in order to increase the response. In this case, T and type of silica have a strong effect on the yield and also their interaction is important. They should be set at their low level to maximise the yield (R).
Apparently x2, the form under which DFO is employed, is not significant, while the interactions of this variable with T and type of silica are. Consequently, all the variables are important and must be carefully set to have the highest sorption capacity.
The isoresponse plane shows how the response, calculated on the basis of eqn (2), varies depending on a couple of variables, keeping the third one at a fixed level.
In Fig. 7 the isoresponse plot, for the variables temperature and type of silica, when DFO is in its mesylate form, is shown. It can be seen that the highest value of the maximum sorption capacity is obtained at low temperature (90 °C), with the MCM-41 MS type. The effect of the temperature is important for both the types of silica: the maximum sorption capacity increases, decreasing T. There is a strong interaction between the two variables, this effect being definitely more important for MCM-41 than for MSU-H.
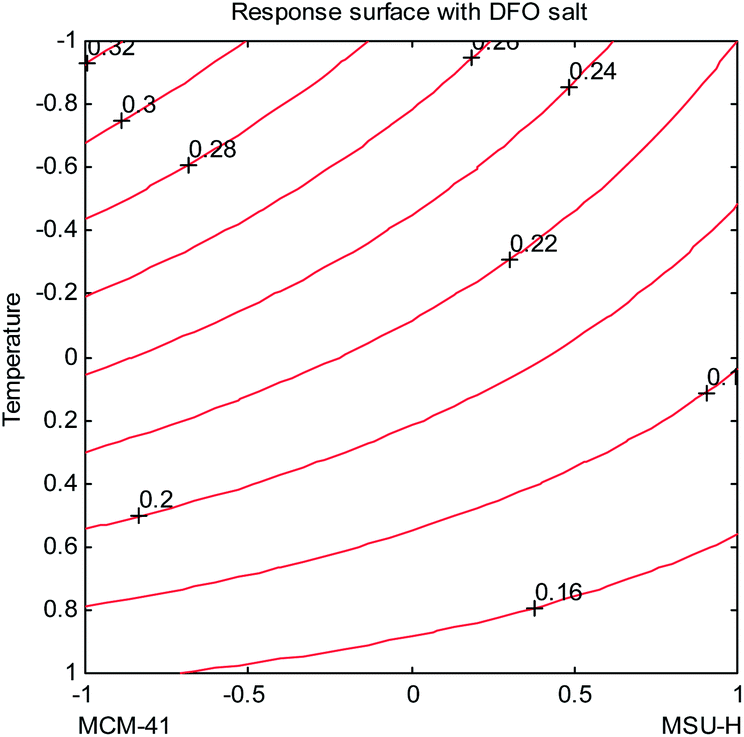 | ||
| Fig. 7 Isoresponse curves for the variables temperature and type of silica, using mesylate DFO. | ||
For the first one, the synthesis at 120 °C (T at level +1) gives a maximum uptake capacity of 0.16 mmol g−1, but it becomes 0.32 mmol g−1 for T = 90 °C (Tat level: −1). In the case of MSU-H, from 0.15 mmol g−1, obtained at a high T level, an increase to 0.22 mmol g−1 was observed for the same decrease of temperature.
Using neutral DFO, as shown in Fig. 8, the effect of the temperature is again more important for MCM-41 than for MSU-H. Actually, using MUS-H, the T is not important and the maximum sorption capacity remains around 0.15 mmol g−1, while in the case of MCM-41, the temperature at a low level gives a product with the best performance, since the response is 0.33 mmol g−1. We did not explore the reason why MSUH-based materials exhibit lower performance than those based on MCM 41, being outside the purpose of the present research.
 | ||
| Fig. 8 Isoresponse curve for the variables temperature and type of silica, using neutral DFO. | ||
In conclusion, the maximum response is obtained using the MCM-41 MS type, and 90 °C is the correct temperature for the coupling between the GPTMS and the DFO. The form of the deferoxamine is not important for the reaction with the MCM-41 silica, while it assumes significance when the MSU-H type is used. The explanation is probably related to the different pore sizes.
The validation of the model cannot be performed in the central points of the design, since two of the three variables are qualitative. We decided not to perform experiments at intermediate values of T, due to the limited availability of the ligand. The maximum sorption capacity of 0.33 mmol g−1 was considered satisfactory and totally unexpected before starting the optimization. Under these conditions, several batches have been produced, always with a reproducible value of the maximum sorption capacity, with a coefficient of variability around 6%, on data obtained from 6 different batches, by different operators.
The material is stable for at least three cycles of Fe(III) sorption/desorption. From our evidence, the recycling of the material is only limited by mechanical chipping, while it resulted being stable for months, dry or wet, in its free or bound to iron forms, when not stirred all the time. It was decided to use the DFO as a free amine instead of the mesylate salt, because all the silica synthesized with the basic form were colourless, while the other ones had a very pale yellow colour and this could be an advantage when the solid is employed as a colorimetric sensor.
Conclusions
A novel solid phase based on DFO-SAMMS with MCM-41 silica type design for iron(III) sorption was obtained. The data of physico-chemical characterisations demonstrated that the DFO molecules are bound to the solid material, which are grafted to the surface and do not enter the silica pores. The one-pot synthesis was preferred to the previously reported two step strategy. We optimized the experimental conditions of the new one-pot synthesis, resulting in the temperature of 90 °C and the use of MCM-41 silica type as the most convenient ones, while the use of neutral or the mesylate form of DFO was not an important variable. The final product showed a satisfactory coverage of active sites. We found that optimisation of preparative steps, based on DOE, such as the one presented here, is convenient and satisfactory, as observed in other cases.31 Indeed, the maximum sorption capacity (achieved from the sorption isotherm of iron(III) for the product obtained under optimized conditions) was higher than 0.3 mmol g−1. The iron uptake was relatively fast, around 100 min, at pH = 2.5.In perspective, our intent is to further characterize the iron uptake to get a quantitative descriptor of the sorption reaction, such as a distribution coefficient and the exchange constant. We do believe that it is possible, in principle, not only to quantify the total amount of iron present in a sample, but also to get information about speciation of iron in that medium. We intend to apply the same strategy, employed several times by our group with synthetic resins and a large variety of metal ions in beverage and natural waters.22,32,33 Encouraging results have been obtained and will be published soon. Since the solid phase becomes intensively reddish-brown coloured, our intent is also to directly estimate the degree of complexation from spectrophotometric measurement of the solid. Also in this case the first results are promising.
Acknowledgements
We want to thank NOVARTIS for DFO supply, FAR (Fondi Ateneo per la Ricerca) of the University of Pavia for funds, and the Servei of Microscopia of the Universitad Autonoma of Barcelona for the TEM analysis.Notes and references
- G. Cairo and A. Pietrangelo, Biochem. J., 2000, 352, 241 CrossRef CAS.
- E. Beutler, V. Felitti, T. Gelbart and N. Ho, Drug Metab. Dispos., 2001, 29, 495 CAS.
- S. D. Domagal-Goldman, K. W. Paul, D. L. Sparks and J. D. Kubicki, Geochim. Cosmochim. Acta, 2009, 73, 1 CrossRef CAS PubMed.
- B. Monzyk and A. L. Crumbliss, J. Am. Chem. Soc., 1982, 104, 4921 CrossRef CAS.
- M. P. Murphy, K. S. Echtay, F. H. Blaikie, J. sin-Cayuela, H. M. Cocheme, K. Green, J. A. Buckingham, E. R. Taylor, F. Hurrell, G. Hughes, S. Miwa, C. E. Cooper, D. A. Svistunenko, R. A. J. Smith and M. D. Brand, J. Biol. Chem., 2003, 278(49), 48534 CrossRef CAS PubMed.
- O. W. Duckworth and G. Sposito, Environ. Sci. Technol., 2005, 39, 6045 CrossRef CAS.
- Y. Takagai, H. Yamaguchi, T. Kubota and S. Igarashi, Chem. Lett., 2007, 36, 136 CrossRef CAS.
- J. Wenk, A. Foitzik, V. Achterberg, A. Sabiwalsky, J. Dissemond, C. Meewes, A. Reitz, P. Brenneisen, M. Wlaschek, W. Meyer-Ingold and K. Scharffetter-Kochanek, J. Invest. Dermatol., 2001, 116, 833 CrossRef CAS PubMed.
- Y. Takagai, A. Takahashi, H. Yamaguchi, T. Kubota and S. Igarashi, J. Colloid Interface Sci., 2007, 313, 359 CrossRef CAS PubMed.
- N. A. A. Rossi, Y. Zou, M. D. Scott and J. N. Kizhakkedathu, Macromolecules, 2008, 41, 5272 CrossRef CAS.
- Z. Yehuda, Y. Hadar and Y. Chen, J. Agric. Food Chem., 2003, 51, 5996 CrossRef CAS PubMed.
- M. Wanunu, S. Livne, A. Vaskevich and I. Rubinstein, Langmuir, 2006, 22, 2130 CrossRef CAS PubMed.
- E. G. Roy, C. H. Jiang, M. L. Wells and C. Tripp, Anal. Chem., 2008, 80, 4689 CrossRef CAS PubMed.
- B. L. Su, N. Moniotte, N. Nivarlet, L. H. Chen, Z. Y. Fu, J. Desmet and J. Li, J. Colloid Interface Sci., 2011, 358, 136 CrossRef CAS PubMed.
- Y. Lin, G. E. Fryxell, H. Wu and M. Engelhard, Environ. Sci. Technol., 2001, 35, 3962–3966 CrossRef CAS.
- C. C. P. Chan, N. R. Choudhurya and P. Majewskib, Colloids Surf., A, 2011, 377, 20 CrossRef CAS PubMed.
- L. Wei, D. Shi, Z. Zhou, P. Ye, J. Wang1, J. Zhao, L. Liu, C. Chen and Y. Zhang, Nanoscale Res. Lett., 2012, 7, 334 CrossRef PubMed.
- M. Etienne, S. Goubert-Renaudin, Y. Rousselin, C. Marichal, F. Denat, B. Lebeau and A. Walcarius, Langmuir, 2009, 25, 3137 CrossRef CAS PubMed.
- O. Cozar, N. Leopold, C. Jelic, V. Chis, L. David, A. Mocanu and M. Tomoaia-Cotisel, J. Mol. Struct., 2006, 788, 1 CrossRef CAS PubMed.
- M. H. Lee, S. J. Lee, J. H. Jung, H. Lim and J. S. Kim, Tetrahedron, 2007, 63, 12087 CrossRef CAS PubMed.
- Z. Hu, X. Zhang, D. Zhang and J. X. Wang, Water, Air, Soil Pollut., 2012, 223(5), 2743 CrossRef CAS.
- G. Alberti, V. Amendola, M. Pesavento and R. Biesuz, Coord. Chem. Rev., 2012, 256 Search PubMed.
- R. Leardi, Anal. Chim. Acta, 2009, 652, 161 CrossRef CAS PubMed.
- R. G. Brereton, in Chemometrics, ed. W. John, Sons, 2003 Search PubMed.
- W. W. Epstein and F. W. Sweat, Chem. Rev., 1967, 67, 247 CrossRef CAS.
- T. T. Tidwell, Synthesis, 1990, 857 CrossRef CAS PubMed.
- S. Antoniotti, J. Golebiowski, D. Cabrol-Bass and E. Dunach, J. Mol. Struct.: THEOCHEM, 2006, 763, 155 CrossRef CAS PubMed.
- S. Antoniotti, S. Antonczak and J. Golebiowski, Theor. Chem. Acc., 2004, 112, 290 CrossRef CAS.
- T. M. Santosusso and D. Swern, J. Org. Chem., 1975, 40, 2764 CrossRef CAS.
- R. Leardi, Anal. Chim. Acta, 2009, 652, 161 CrossRef CAS PubMed.
- G. Marrubini, P. Fattorini, C. Previderé, S. Goi, S. S. Cigliero, P. Grignani, M. Serra, R. Biesuz and G. Massolini, J. Chromatogr. A, 2012, 1249, 8 CrossRef CAS PubMed.
- G. Alberti and R. Biesuz, React. Funct. Polym., 2011, 71(5), 588 CrossRef CAS PubMed.
- G. Alberti, M. G. Guiso and R. Biesuz, Talanta, 2009, 79(3), 603 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4an00179f |
| This journal is © The Royal Society of Chemistry 2014 |