Cascade signal amplification for ultrasensitive electrochemical DNA detection
Jin
Xu
,
Qiong
Wang
,
Yun
Xiang
*,
Ruo
Yuan
and
Yaqin
Chai
Key Laboratory on Luminescence and Real-Time Analysis, Ministry of Education, School of Chemistry and Chemical Engineering, Southwest University, Chongqing 400715, P.R. China. E-mail: yunatswu@swu.edu.cn; Fax: +86-23-68252277; Tel: +86-23-68254000
First published on 9th October 2013
Abstract
In this work, by integrating multiple signal enhancement approaches, a new cascade signal amplification strategy is described to achieve highly sensitive electrochemical DNA detection. The presence of the target DNA leads to the unfolding of the biotin-modified hairpin probes on the sensor surface. With the addition of the primer sequences and polymerase, the target DNA is recycled and reused through isothermal strand-displacement polymerase reactions (ISDPR) to unfold a large number of the probes, which offer numerous binding sites to capture alkaline phosphatase (ALP)-loaded nanoparticle labels. These surface captured ALP enzymes catalyze the conversion of p-aminophenylphosphate to p-aminophenol, which generates amplified catalytic current responses due to the redox-recycling process during the potential sweep in the presence of the co-reactant NADH. With the synergistic signal amplifications by ISDPR-assisted target recycling, multi-ALP enzyme labels and redox-recycling, the proposed method offers highly sensitive detection of DNA down to 0.1 fM with single-base discrimination capability. Due to the significantly high sensitivity, the developed cascade signal amplification strategy can be potentially extended to detect various DNA targets at ultralow levels for early diagnoses of different diseases.
Introduction
Sensitive and selective detection of sequence specific DNA is essential in pathogen analysis,1–4 molecular diagnostics,5,6 genetics therapy,7 early screening of cancers,8 antiterrorism and forensic analysis.9 Signal amplification has proven to be one of the most effective ways to enhance the signal output and achieve high sensitivity for DNA detection. Over the past decades, numerous signal amplification methods have been reported. Although polymerase chain reaction (PCR)10 and rolling circle amplification (RCA)11,12 represent the most widely used signal amplification methods with powerful amplification capabilities, these two approaches encounter the drawbacks of requirement of sophisticated instrumentation and skilled technicians, potential contamination and high cost.13,14 In recent years, a number of new hybridization-based signal amplification schemes, such as biobarcode,15–17 multi-label,18,19 redox-recycling20,21 and hybridization chain reaction,5 have been suggested as alternatives to compete with PCR/RCA in DNA detection. These approaches are primarily based on the involvement of numerous amplification labels in each single DNA hybridization event to amplify the signal output to achieve impressive sensitivities. Another class of hybridization-based signal amplification means developed recently, however, innovatively involves the reuse of the target DNA or target DNA recycling to realize signal enhancement. In these approaches, the hybridizations between the target and probe sequences lead to catalytic digestion of the hybridized-probe sequences by nucleases22–24 or displacement of the target DNA in the presence of a primer and polymerase.25 The target sequence will then be released to initiate the target DNA recycling amplification process. Despite that these newly developed signal amplification methods can offer high sensitivity, the detection of ultralow levels of sequence-specific DNA with PCR-like sensitivity remains a major challenge in current DNA biosensing technologies.To explore the development of ultrasensitive DNA sensing methods, by integrating multiple signal amplification routes into one single assay protocol, we report on a new cascade signal enhancement strategy for electrochemical DNA detection. Our new approach combines isothermal strand displacement polymerase reaction (ISDPR), multi-enzyme labels and redox-recycling to achieve significant hybrid signal amplification. The ISDPR leads to direct recycling of the target DNA and subsequent attachment of multi-enzyme loaded nanoparticles on the sensing electrode. In the presence of nicotinamide adenine dinucleotide (NADH), the enzymatic products are recycled on the electrode surface to generate amplified current responses for DNA detection down to the sub-femtomolar level.
Experimental
Chemicals and materials
Tris–HCl, 6-mercapto-1-hexanol (MCH), HAuCl4, NADH, alkaline phosphatase (ALP), streptavidin (STV) and streptavidin-conjugated alkaline phosphatase (STV–ALP) were purchased from Sigma (St Louis, MO, USA). Disposable screen-printed carbon electrodes (SPCEs), comprised of a carbon working electrode, carbon counter electrode, and silver reference electrode, were from Zensor R&D (Taichung, Taiwan). Gold-coated Fe3O4 nanoparticles (Fe3O4@Au) were provided by Xi'an GoldMag Nanobiotech Co., Ltd (Xi'an, China). p-Aminophenylphosphate (p-APP) was obtained from LKT Laboratories Inc. (St Paul, MN, USA). Deoxynucleoside triphosphates (dNTP), polymerase Klenow Fragment and all oligonucleotides were ordered from Shanghai Sangon Biological Engineering Technology and Services Co., Ltd (Shanghai, China). The oligonucleotide sequences were as follows:Hairpin probe (HP): 5′-SH-(CH2)6-TCTTGGACACAGTAAA GAGAGGTGCGCCCATTGTGTCCAAGA-biotin-3′;
Target DNA (tDNA):
5′-ATGGGCGCACCTCTCTTTACTGTGTC-3′;
Single-base mismatched DNA (sDNA):
5′-ATGGGCGCACC TCTCTTT![[G with combining low line]](https://www.rsc.org/images/entities/char_0047_0332.gif) CTGTGTC-3′;
CTGTGTC-3′;
Non-complementary DNA (nDNA):
5′-GACCCTCTGTTATATAAAGTACACAT-3′;
Primer: 5′-TC TTGGAC-3′.
All reagents were analytical grade and solutions were prepared using ultrapure water (specific resistance of 18 MΩ cm).
Preparation of ALP-Fe3O4@Au-STV bioconjugates
The immobilization of ALP and STV onto Fe3O4@Au was completed in a single-step process.26,27 In brief, an aliquot (20 μL of 5 mg mL−1) of Fe3O4@Au was transferred into a centrifuge vial and washed with 40 μL of the coupling buffer (2 mM Tris–HCl, 5 mM NaCl, pH 8.2). After magnetic separation, the Fe3O4@Au were suspended in 30 μL of the coupling buffer containing ALP and STV at a 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (ALP:STV) molar ratio. The mixture was incubated at 37 °C for 30 min under 180 rpm rotation to achieve the attachment of ALP and STV onto the Fe3O4@Au surfaces via physical adsorptions. The resulting ALP-Fe3O4@Au-STV conjugates were magnetically separated, washed three times with washing buffer (10 mM PBS, 0.025% Tween-20, pH 7.4), separated again and resuspended in 30 μL of the hybridization buffer (10 mM PBS, 0.2 M NaCl, pH 7.4) and stored at 4 °C for further use.
:1 (ALP:STV) molar ratio. The mixture was incubated at 37 °C for 30 min under 180 rpm rotation to achieve the attachment of ALP and STV onto the Fe3O4@Au surfaces via physical adsorptions. The resulting ALP-Fe3O4@Au-STV conjugates were magnetically separated, washed three times with washing buffer (10 mM PBS, 0.025% Tween-20, pH 7.4), separated again and resuspended in 30 μL of the hybridization buffer (10 mM PBS, 0.2 M NaCl, pH 7.4) and stored at 4 °C for further use.
Fabrication of the sensing electrode
The working electrode of the SPCE was first preconditioned in 20 mM Tris–HCl buffer (pH 7.4) by cycling the potential between −0.6 V and 0.6 V at a scan rate of 0.5 V s−1. A AuNP layer was formed on the working electrode of the SPCE in a single step through controlled electrodeposition as described previously.28,29 In brief, the SPCE was placed in a solution of 1% HAuCl4 and controlled electrodeposition of AuNPs was performed at −0.66 V for 30 s to obtain the AuNPs-deposited SPCE (noted as AuNPs/SPCE). Subsequently, the AuNPs/SPCE was rinsed with water and dried in a nitrogen stream. Finally, a droplet (10 μL) of 1 μM HP in the immobilization buffer (10 mM Tris–HCl, 1 mM EDTA, 10 mM TCEP, 0.1 M NaCl, pH 7.4) was placed on the working electrode and incubated overnight at room temperature (25 °C) followed by incubation with 1 mM MCH for 2 h to obtain the HP/MCH/AuNPs/SPCE.Cascade signal amplification DNA sensing protocol
Ten μL of different concentrations of the target mixed with 2 μM primer, 5 U polymerase Klenow fragment, 100 μM dNTPs, 6% DMSO, 1 mM DTT and 5 mM MgCl2 in 50 mM Tris–HCl (pH 8.0) was dropped onto the HP modified electrodes and incubated at 37 °C for 2 h in a humid environment. After washing and drying, the sensors were further incubated with 10 μL of ALP-Fe3O4@Au-STV conjugates for 30 min at room temperature, followed by washing with washing buffer and drying with nitrogen. Subsequently, 50 μL mixture of 1 mM p-APP and 5 mM NADH in Tris buffer (50 mM Tris–HCl, 10 mM KCl, 1 g L−1 MgCl2, pH 9.0) was dropped on the sensor surface and the enzymatic reaction was allowed to proceed for 10 min in the dark. Finally, differential pulse voltammetry (DPV) measurements were performed on the sensors.Electrochemical measurements
Cyclic voltammetry (CV) and DPV measurements were performed on a CHI 852C electrochemical workstation (CH Instruments, Shanghai, China).Results and discussion
In this work, based on the combination of ISDPR, ALP-Fe3O4@Au-STV labels and the reducing agent NADH-triggered redox-recycling of the electroactive products, we proposed a new cascade signal amplification strategy to achieve ultrasensitive detection of sequence-specific DNA. As illustrated in Scheme 1, a mixed layer of 5′-thiol and 3′-biotin dual modified HPs and MCH is first self-assembled on the AuNPs-electrodeposited SPCE via S–Au bonding. In the presence of the target DNA, the HP recognizes and hybridizes with the target DNA and undergoes a conformational change, leading to the stem separation. Following this, the primer anneals with the open stem and triggers a polymerization reaction in the presence of dNTPs and polymerase Klenow Fragment, which results in the release of the target. Subsequently, the released target DNA hybridizes with another HP to trigger the ISDPR process, leading to the unfolding of a large number of the hairpin-structured probes and the attachment of massive ALP-Fe3O4@Au-STV conjugate labels on the sensor surface through strong biotin–STV interactions. Upon the addition of the ALP enzyme substrate p-APP and co-reactant NADH, ALP catalyzes the conversion of p-APP to an electroactive product, p-aminophenol (p-AP), which is electrochemically oxidized to p-quinone imine (p-QI) on the electrode surface. With the synergic effect of NADH, the electro-oxidized product p-QI is reduced back to p-AP. This leads to the occurrence of a redox-recycling cycle between p-QI and p-AP and the current response is thus amplified. Such a catalytic current response is dependent upon the amount of the surface-attached ALP-Fe3O4@Au-STV, which in turn reflects the amount of the target DNA in the testing buffer. In contrast, in the absence of the target DNA, the hairpin-structured probes remain folded and fail to anneal with the primer to induce the ISDPR process, in which the 3′-biotin termini of the HPs are protected from interacting with the ALP-Fe3O4@Au-STV conjugate labels due to the large steric hindrance effect and the generation of the catalytic current is inhibited. The expected substantial difference in catalytic current intensity in the presence/absence of the target DNA can thus offer quantitative signals for DNA determination.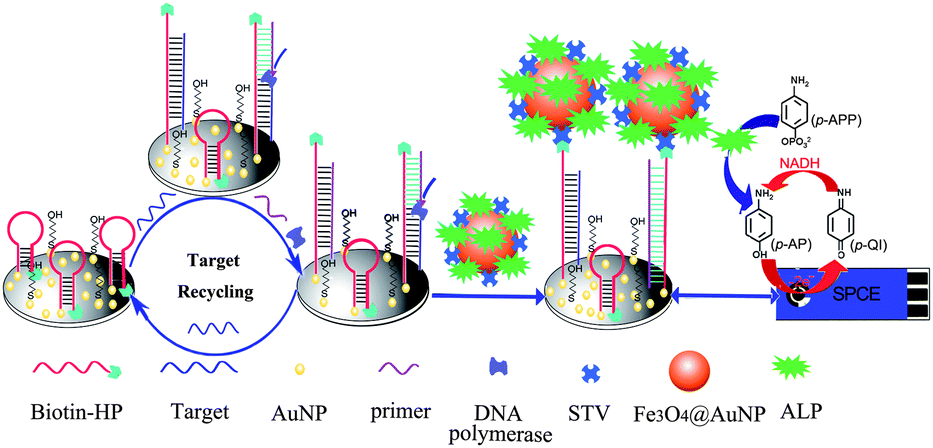 | ||
| Scheme 1 Principle of the cascade signal amplification strategy for ultrasensitive electrochemical detection of DNA. | ||
The EC redox-recycling behavior of p-AP in the presence of NADH was investigated in detail previously by several groups,30–32 and this mechanism has been shown to be useful in amplifying the current signal response for the detection of different biomolecules.21,29,30 In order to validate the feasibility of redox-cycling of p-AP by NADH on our sensing surface, the current responses of p-AP on the HP/MCH/AuNPs/SPCE were investigated by CV in the presence/absence of NADH. As can be seen from Fig. 1, when NADH (5 mM) is present in buffer (curve b), no observable peak can be obtained compared with the blank solution (curve a, buffer only). Meanwhile, in the presence of p-AP (1 mM) in buffer, a pair of reversible peaks of p-AP can be observed (curve c). However, when both NADH and p-AP are present in buffer, the shape of the voltammogram of p-AP changes to a catalytic sigmoidal curve with an enhanced anodic current and a decreased cathodic current (curve d), which is in agreement with previous reports21,29 and indicates the redox-recycling of p-AP on the sensor surface.
 | ||
| Fig. 1 Cyclic voltammograms of the HP/MCH/AuNPs/SPCE in 50 mM Tris buffer (10 mM KCl, 1 g L−1 MgCl2, pH 9.0) containing (a) buffer only, (b) 5 mM NADH, (c) 1 mM p-AP, and (d) the mixture of 5 mM NADH and 1 mM p-AP at a scan rate of 5 mV s−1. | ||
In order to verify the dramatic cascade signal amplification capability of the proposed DNA sensing method, different amplification schemes, including the conventional single enzyme, single enzyme/ISDPR and ALP-Fe3O4@Au-STV/ISDPR approaches, were systematically compared. In the conventional single enzyme-labeled signal amplification approach, the presence of the target DNA (100 pM) leads to direct unfolding of the HPs and subsequent attachment of the STV–ALP. With addition of the ALP substrate p-APP and the co-reactant NADH, a redox-recycling cycle between the enzymatic product (p-AP) and p-QI is formed and an apparent catalytic current (ΔI) (where ΔI is the current intensity difference between the target (i, b and c) and the background (i0, a)) is observed (Fig. 2, curve b) compared with that of the blank test (Fig. 2, curve a). Upon the incorporation of the ISDPR into the conventional single ALP-label DNA detection scheme, further increase (192.7%) in current intensity is evident (Fig. 2, curve c), due to the target recycling or reuse aided by ISDPR for the unfolding of more HPs and the attachment of more ALP. More importantly, when ALP-Fe3O4@Au-STV instead of single ALP are used as labels and coupled with ISDPR to achieve cascade signal amplification, a significantly amplified (376.6%) catalytic current is obtained (Fig. 2, curve c). These comparisons clearly indicate that the coupling of multiple amplification approaches can dramatically amplify the signal response for DNA detection.
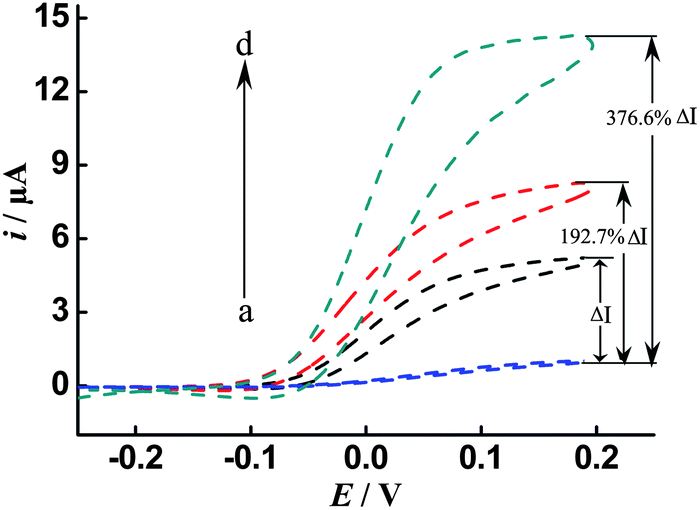 | ||
| Fig. 2 Cyclic voltammograms of different signal amplification schemes for DNA detection: single enzyme (STV–ALP) label in the presence (b) and absence (a) of 100 pM target; (c) single enzyme/ISDPR and (d) ALP-Fe3O4@Au-STV/ISDPR in the presence of 100 pM target. EC measurements were performed in Tris–HCl buffer containing 1 mM p-APP and 5 mM NADH (100 mM Tris–HCl, 0.2 g L−1 MgCl2, pH 9.0) at a scan rate of 5 mV s−1 from −0.25 V to 0.2 V. | ||
Compared with the conventional CV sweep technique, DPV can offer excellent resolution for current responses and provide much higher sensitivity. DPV was therefore employed to investigate the dependence of the catalytic current response upon various concentrations of the target DNA by using our new cascade signal amplification method. From the results displayed in Fig. 3, we can see that with increasing concentration of the target DNA from 0 fM to 100 pM, the catalytic current response increases accordingly (curve a to g). The corresponding calibration plot of log[c] vs. the current intensity difference (Δi) between the target and the background (inset in Fig. 3) exhibits a dynamic range from 1 fM to 100 pM and an estimated detection limit of 0.1 fM (3σ/slope) (where σ is the relative standard deviation of a blank solution, n = 11) is obtained. Such a low detection limit, which shows about 3.6 to 200-fold improvement over other reported strategies for electrochemical detection of DNA,17,33–36 can be basically ascribed to the integration of multiple signal amplification approaches in one single assay protocol. Six repetitive analyses of the target DNA at 100 fM yielded a relative standard deviation of 11.3%, suggesting a good reproducibility of the method.
 | ||
| Fig. 3 Typical DPV responses for the detection of different concentrations of target DNA with the proposed cascade signal amplification method. From bottom to top (a to g): 0, 1 fM, 10 fM, 100 fM, 1 pM, 10 pM and 100 pM. Inset: the resulting calibration plot for logc vs. Δi (where Δi is the current intensity difference between the target (i) and the background (i0)) in the range from 1 fM to 100 pM. DPV measurements were performed in Tris–HCl buffer (100 mM Tris–HCl, 0.2 g L−1 MgCl2, pH 9.0) containing 1 mM p-APP and 5 mM NADH with a pulse amplitude of 0.05 V and a pulse width of 0.1 s. Error bars, SD, n = 3. | ||
The selectivity of the proposed method was evaluated by comparing its catalytic current responses to different DNA sequences, including the target DNA (tDNA), single-base mismatched DNA (sDNA) and non-complementary DNA (nDNA) at an identical concentration of 100 fM. As shown in Fig. 4, it is obvious that the current response of the tDNA (curve d) is much higher than (3.9-fold) that of the sDNA (curve c), and the current response of nDNA (curve b) is close to that of the blank solution (curve a). These comparisons demonstrate that the catalytic current response is specifically triggered by the ISDPR process only in the presence of the perfectly matched DNA, which indicates that the proposed cascade signal amplification strategy can offer high specificity. The excellent selectivity of such an amplified electrochemical DNA biosensor may thus support its potential in the analysis of single nucleotide polymorphism.
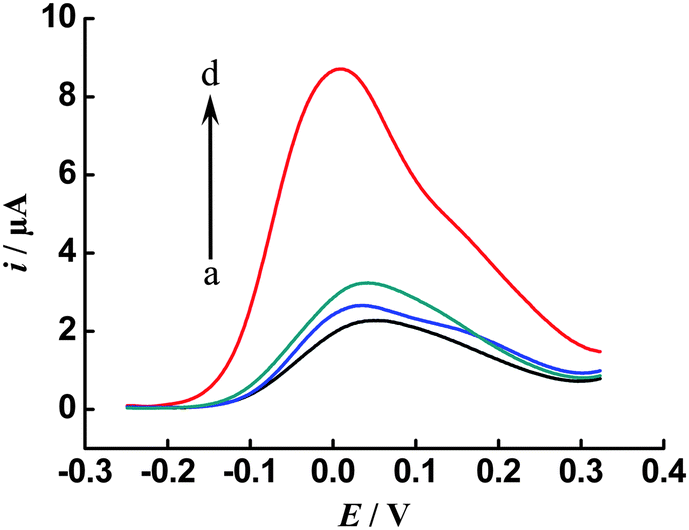 | ||
| Fig. 4 Selectivity investigation of the proposed method for target DNA against non-complementary (nDNA) and single-base mismatched DNA (sDNA) sequences: (a) blank, (b) 100 fM nDNA, (c) 100 fM sDNA and (d) 100 fM target DNA. Other conditions, as in Fig. 3. | ||
To investigate the applicability of the proposed cascade signal amplification strategy to real samples, we performed the measurements in a complex sample matrix, 10% human serum (diluted with buffer). As shown in Fig. 5, the DPV response obtained from the serum sample (in the absence of the spiked target DNA, b) decreases slightly compared to that in buffer solution (a). When the serum sample was spiked with 100 fM target DNA, an amplified current response (c) is observed, which indicates that the developed cascade signal amplification method can be applied to real samples.
 | ||
| Fig. 5 Typical DPV responses of the proposed sensor for (a) buffer solution, (b) 10% human serum and (c) 100 fM DNA in 10% human serum. Other conditions, as in Fig. 3. | ||
In conclusion, we have demonstrated a highly sensitive EC sensing platform for detecting sequence-specific DNA based on a cascade signal amplification strategy. Our new approach couples ISDPR target recycling and multi-enzyme labels with redox-recycling to achieve hybrid, dramatic signal amplification for DNA detection down to 0.1 fM. The developed method also shows single-base mismatch discrimination capability toward the target DNA. Considering the significant signal amplification feature, this method can be applied for the detection of other trace levels of DNA in general and holds great potential for designing new cascade signal amplification strategies to achieve ultrahigh sensitivity for the detection of proteins and other important biomolecules.
Acknowledgements
This work was supported by National Nature Science Foundation of China (no. 21275004, 20905062, 21075100 and 21275119), the New Century Excellent Talent Program of MOE (NCET-12-0932), Fundamental Research Funds for the Central Universities (XDJK2012A004) and research funds from Southwest University (SWUB2008078).Notes and references
- N. Rosi and C. Mirkin, Chem. Rev., 2005, 105, 1547–1562 CrossRef CAS PubMed.
- S. Nakayama, L. Yan and H. Sintim, J. Am. Chem. Soc., 2008, 130, 12560–12561 CrossRef CAS PubMed.
- S. Nakayama and H. Sintim, J. Am. Chem. Soc., 2009, 131, 10320–10333 CrossRef CAS PubMed.
- C. Riccardi, C. Kranz, J. Kowalik, H. Yamanaka, B. Mizaikoff and M. Josowicz, Anal. Chem., 2008, 80, 237–245 CrossRef PubMed.
- J. Huang, Y. Wu, Y. Chen, Z. Zhu, X. Yang, C. Yang, K. Wang and W. Tan, Angew. Chem. Int. Ed., 2011, 50, 401–404 CrossRef CAS PubMed.
- H. Dong, W. Gao, F. Yan, H. Ji and H. Ju, Anal. Chem., 2010, 82, 5511–5517 CrossRef CAS PubMed.
- Y. Huang, Y. Zhang, X. Xu, J. Jiang, G. Shen and R. Yu, J. Am. Chem. Soc., 2009, 131, 2478–2480 CrossRef CAS PubMed.
- S. Bi, J. Zhang and S. Zhang, Chem. Commun., 2010, 46, 5509–5511 RSC.
- M. Heller, Annu. Rev. Biomed. Eng., 2002, 4, 129–153 CrossRef CAS PubMed.
- R. Saiki, S. Scharf, F. Faloona, K. Mullis, G. Horn, H. Erlich and N. Arnheim, Science, 1985, 230, 1250–1354 Search PubMed.
- P. Lizardi, X. Huang, Z. Zhu, P. Bray-Ward, D. Thomas and D. Ward, Nat. Genet., 1998, 19, 225–232 CrossRef CAS PubMed.
- B. Schweitzer, S. Wiltshire, J. Lambert, S. O'Malley, K. Kukanskis, Z. Zhu, S. Kingsmore, P. Lizardi and D. Ward, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 10113–10119 CrossRef CAS PubMed.
- M. Kubista, J. Andrade, M. Bengtsson, A. Forootan, J. Jonak, K. Lind, R. Sindelka, R. Sjoback, B. Sjogreen and L. Strombom, Mol. Aspects Med., 2006, 27, 95–125 CrossRef CAS PubMed.
- S. Weissenborn, U. Wieland, M. Junk and H. Pfister, Nat. Protoc., 2010, 5, 1–13 CrossRef CAS PubMed.
- E. Goluch, J. Nam, D. Georganopoulou, T. Chiesl, K. Shaikh, K. Ryu, A. Barron, C. Mirkin and C. Liu, Lab Chip, 2006, 6, 1293–1299 RSC.
- J. Nam, S. Stoeva and C. Mirkin, J. Am. Chem. Soc., 2004, 126, 5932–5933 CrossRef CAS PubMed.
- J. Xu, B. Jiang, J. Su, Y. Xiang, R. Yuan and Y. Chai, Chem. Commun., 2012, 48, 3309–3311 RSC.
- W. Gao, H. Dong, J. Lei, H. Ji and H. Ju, Chem. Commun., 2011, 47, 5220–5222 RSC.
- Q. Wang, J. Lei, S. Deng, L. Zhang and H. Ju, Chem. Commun., 2013, 49, 916–918 RSC.
- J. Wu, K. Chumbimuni-Torres, M. Galik, C. Thammakhet, D. Haake and J. Wang, Anal. Chem., 2009, 81, 10007–10012 CrossRef CAS PubMed.
- A. Walter, J. Wu, G. Flechsig, D. Haake and J. Wang, Anal. Chim. Acta, 2011, 689, 29–33 CrossRef CAS PubMed.
- X. Zuo, F. Xia, Y. Xiao and K. Plaxco, J. Am. Chem. Soc., 2010, 132, 1816–1818 CrossRef CAS PubMed.
- J. Su, H. Zhang, B. Jiang, H. Zheng, Y. Chai, R. Yuan and Y. Xiang, Biosens. Bioelectron., 2011, 29, 184–188 CrossRef CAS PubMed.
- J. Chen, J. Zhang, Y. Guo, J. Li, F. Fu, H. Yang and G. Chen, Chem. Commun., 2011, 47, 8004–8006 RSC.
- Q. Guo, X. Yang, K. Wang, W. Tan, W. Li, H. Tang and H. Li, Nucleic Acids Res., 2009, 37, e20 CrossRef PubMed.
- Y. Cui, Y. Wang, W. Hui, Z. Zhang, X. Xin and C. Chen, Biomed. Microdevices, 2005, 7(2), 153–156 CrossRef CAS PubMed.
- A. Yu, T. Geng, Q. Fu, C. Chen and Y. Cui, J. Magn. Magn. Mater., 2007, 311, 421–424 CrossRef CAS PubMed.
- J. Ho, J. Chiu, J. Hong, C. Lin, K. Hwang and J. Hwu, J. Nanosci. Nanotechnol., 2009, 9, 23–24 Search PubMed.
- B. Jiang, M. Wang, Y. Chen, J. Xie and Y. Xiang, Biosens. Bioelectron., 2012, 32, 305–308 CrossRef CAS PubMed.
- S. Kwon, H. Yang, K. Jo and J. Kwak, Analyst, 2008, 133, 1599–1604 RSC.
- K. Mak, U. Wollenberger, F. Scheller and R. Renneberg, Biosens. Bioelectron., 2003, 18, 1095–1110 CrossRef CAS.
- B. Limoges, D. Marchal, F. Mavré and J. Savéant, J. Am. Chem. Soc., 2008, 130, 7259–7275 CrossRef CAS PubMed.
- D. Wu, B. Yin and B. Ye, Biosens. Bioelectron., 2011, 28, 232–238 CrossRef CAS PubMed.
- C. Wang, H. Zhou, W. Zhu, H. Li, J. Jiang, G. Shen and R. Yu, Biosens. Bioelectron., 2013, 47, 324–328 CrossRef CAS PubMed.
- J. Zhuang, L. Fu, M. Xu, H. Yang, G. Chen and D. Tang, Anal. Chim. Acta, 2013, 783, 17–23 CrossRef CAS PubMed.
- Y. Chen, Q. Wang, J. Xu, Y. Xiang, R. Yuan and Y. Chai, Chem. Commun., 2013, 49, 2052–2054 RSC.
| This journal is © The Royal Society of Chemistry 2014 |