
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePeptomer substrates for quantitative pattern-recognition sensing of proteases†
Mariah J.
Austin
 a,
Hattie C.
Schunk
ab,
Natalie
Ling
a and
Adrianne M.
Rosales
*a
a,
Hattie C.
Schunk
ab,
Natalie
Ling
a and
Adrianne M.
Rosales
*a
aMcKetta Department of Chemical Engineering, University of Texas at Austin, Austin, TX 78712, USA. E-mail: arosales@che.utexas.edu
bBiomedical Engineering Department, University of Texas at Austin, Austin, TX 78712, USA
First published on 17th January 2023
Abstract
The utility of active proteases as biomarkers is often limited by overlapping substrate specificity. Here, this feature is leveraged to develop a quantitative pattern-recognition sensing system driven by the degradation patterns of peptide–peptoid hybrid substrates to classify proteases and estimate their concentration by multivariate data analysis.
Pattern-recognition sensing, an approach that relies on the cross-reactivity of multiple probes to sense diverse analytes, has gained popularity for sensing biomarkers over the past two decades as researchers shifted from designing for specificity to engineering differential capacity.1 Sensing targets have included small species like volatile organic compounds,2 metal ions,3–6 and individual amino acids7–9 all the way up to proteins10,11 and cells themselves,12–15 as well as combinations of multiple components.16,17 Enzymes are often regarded as analytes that fit into the “lock-and-key” category of specific sensing. Many proteases, however, do not exhibit such defined specificity and have significant overlap in their specificity profiles.18 Thus, pattern-recognition sensing is well suited to advance the toolbox of proteomic detection, especially by utilizing their natural behavior (i.e., protein/peptide degradation).
Of the few examples, existing protease sensors developed with this approach have employed functionalized polymers19 and single-stranded DNA.20 In both investigations, sensing was achieved by fluorescent response to binding interactions between the proteases and probes, as is the case for the signaling component in many examples. While this route has proven to be robust and adaptable for sensing many different proteins, leveraging the unique capability of proteases through degradation affords opportunity to maximize chemometric capability (i.e., a large dynamic range of signal that scales with protease concentration) and readily integrate with biomaterials that already rely on degradative components. In addition, the goal of accurately identifying biomarkers has been achieved, even in mixtures,21,22 but doing so quantitatively is less common.23 Furthermore, modifying a pattern-recognition sensing approach to be sensitive to degradation enables all of the diverse tools already established to detect proteolysis (e.g., fluorescence/bioluminescence energy transfer, colorimetric detection, spectroscopic techniques)24,25 to be incorporated into similar arrays.
We have previously demonstrated the utility of peptomers (peptide–peptoid hybrids) for differentiating between matrix metalloproteinases;26 however, classification was only achieved at a single concentration condition. Here, our objectives were to (1) expand the breadth of proteases from different sources and families, (2) accurately classify protease identity over a range of concentrations, and (3) predict concentrations of proteases based on their degradative behavior. We employed four probes to do so, a peptide, and three peptomers with a single peptoid substitution each (Fig. 1a and Fig. S1, ESI†).
 | ||
| Fig. 1 (a) Peptide sequence (PAALVA) and peptoid residues employed. Peptomers are named by the position (active site residues A–F, from N to C-terminus) of substitution and the amino acid they are replacing. (b) Array-based sensing approach employed with six proteases screened at eight concentrations. Substrates included a fluorophore–quencher pair on each terminus, enabling quantitative cleavage tracking by disruption of fluorescence resonance energy transfer (FRET) as proteolysis occurred. | ||
Peptoids are peptidomimetics with sidechains attached to the amide backbone nitrogen, rather than the α-carbon, a modification which influences proteolytic recognition. To elicit markedly different cleavage rates for array-based sensing, we employed peptomers with peptoid sidechains analogous to the amino acids they were replacing in three positions within a proteolytically degradable peptide sequence (PANLVA, referred to as the Peptide). The target analytes were six proteases (Fig. 1b and Table S1, ESI†), all of which were able to cleave the Peptide, but representing unique catalytic mechanisms and distinct families. Each protease was screened under the same conditions using a Tris-HCl-based buffer at pH 7.8. Each of the proteases are active in this buffer (despite not being optimized for each individual protease), thereby producing measurable signal for multivariate data analysis. The four peptomers were individually exposed to proteases at eight different concentrations, serially diluted from 80 μg mL−1 to 0.125 μg mL−1 and conducted in triplicate, and their rates of cleavage were tracked fluorescently (Fig. 2a and Fig. S2–S6, ESI†). The substrates included a fluorophore–quencher pair which afforded fluorescence tracking of proteolysis as the fluorophore was liberated from the quencher upon the hydrolysis of a peptide bond. Each cleavage trace was then fit to an exponential plateau function to determine an exponential constant representing the rate of proteolysis. The log10 values of those rates were compiled (Fig. 2b and Table S2, ESI†) and subjected to multivariate data analysis.
 | ||
| Fig. 2 (a) Representative fluorescent cleavage tracking of the four probes exposed to eight concentrations of collagenase. Experiments were conducted in triplicate; error bars represent the standard deviation of each measurement. Degradation data for the other proteases are Fig. S2–S6 (ESI†). (b) Array of degradation rates (scaled by log10)determined for each protease-substrate combination. Features are listed in order of descending concentration for each protease. | ||
The dataset was first evaluated with principal component analysis (PCA). PCA is an unsupervised technique, meaning class labels are not considered, that uses eigenvalue decomposition to project data in low-dimensional space by quantifying the covariance between samples. PCA was effective, as over 99% of the dataset's variance was captured in three PCs. When projected into two dimensions, the samples were mapped in a logical manner, highly representative of the parameters for each feature (Fig. 3a). Specifically, PC1 projected samples in order of concentration and PC2 captured the different protease identities. All proteases were mapped in ascending order of concentration along the PC1 axis, and those with higher activity (i.e., thermolysin and papain, graphically represented as darker regions on the heatmap in Fig. 2b) span further to the right on that axis. However, as concentration increases, the datapoints frequently slant downward, moving on the PC2 axis. This disrupts the separation of different proteases along PC2 and hinders the formation of well-separated clusters. Indeed, even when projected in three dimensions (Fig. 3b) the proteases have overlapping regions. The loadings, which depict the contribution of each feature (in this case the Peptide and peptomers) to separation, confirm that they are eliciting differential behavior between the proteases, but again the high activity enzymes are spanning wide ranges, preventing tight clustering and good separation of the proteases. Therefore, we next turned to linear discriminant analysis (LDA) in attempt to induce better separation among the proteases.
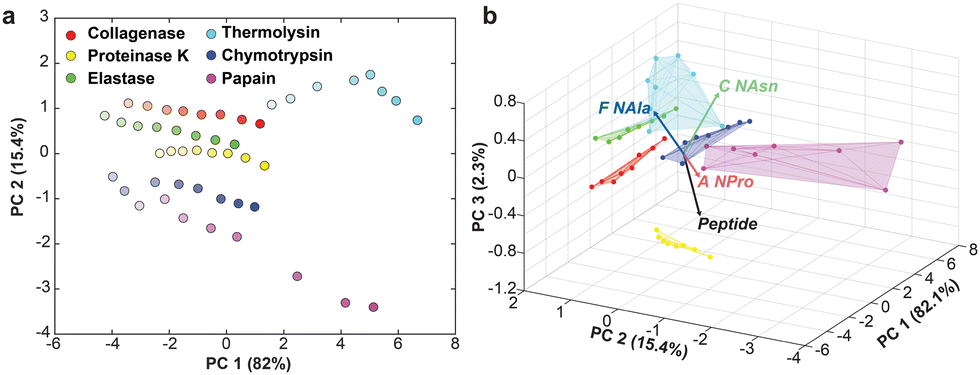 | ||
| Fig. 3 (a) Two-dimensional PCA plot depicting concentration variance captured in PC1 (represented by opacity of the data points) and protease identity mapped along PC2. (b) Three-dimensional biplot showing contribution of each probe to separation. | ||
LDA is similar to PCA in that it serves as a dimensionality reduction technique but is instead a supervised learning approach. While PCA seeks to capture the maximum variance in the minimum number of components, LDA works to minimize variance within a class (i.e., cluster samples of the same identity together) and maximize separation between different classes. LDA generates scores that are weighted combinations of the input features (i.e., cleavage rates by the different substrates) which can be visualized in lower dimensional space. Given that the samples analyzed here are not replicates, but rather distinct concentrations, we hypothesized that LDA would be more effective in separating the samples by protease. Indeed, when all eight datapoints for each sample were subjected to LDA, they were effectively clustered by protease identity (input as the class label), with only papain and chymotrypsin sharing a similar area (Fig. 4a). However, like PCA, the high activity proteases did not cluster as effectively as those that maintained a smaller range of rates over the various concentrations.
 | ||
| Fig. 4 (a) All eight protease concentration samples projections in two-dimensions using LDA. Shaded areas represent the 95% confidence interval. (b) Representative confusion matrix with 94% classification accuracy when employing stratified holdout cross-validation. (c) Representative multiple regression results depicting concentrations estimated for the testing samples classified in b. | ||
To test the classification ability of the LDA model, stratified holdout cross-validation was conducted. For each iteration, 5/8 samples from each protease were used for training and 3/8 samples were withheld for testing. Importantly, the three testing samples are chosen at random and differ for each of the proteases. Furthermore, the samples selected can change each time the cross-validation sequence is run; therefore, a representative example is presented here. An LDA model was constructed using 30 (five per protease) training samples and the coefficients computed were then applied to the testing dataset and used to predict the identity of the sample proteases. Here, one thermolysin sample was incorrectly classified as collagenase, giving a classification accuracy of 94% (Fig. 4b). The sample misclassified was the lowest concentration of thermolysin. Collagenase and thermolysin behave similarly in that they show high activity for the Peptide, A NPro, and F NAla substrates, but decreased degradation rate of C NAsn. It makes sense that this low concentration thermolysin datapoint was misclassified as it more closely resembles the cleavage signature for many of the collagenase samples, versus the higher activity thermolysin.
The dataset remained partitioned as multiple regression was employed to predict the concentration of the test samples. Multiple regression assigns weights to the value of all four features for each sample, essentially making a standard curve with four independent variables. When executed, collagenase, proteinase K, elastase, and chymotrypsin all resulted in accurate concentration estimations (Fig. 4c). For thermolysin, one of the samples was incorrectly classified as collagenase, and was therefore fit according to the regression of collagenase's training samples, resulting in an erroneously high concentration. Beyond the misclassification, concentration estimations for thermolysin and papain were less accurate. Thermolysin cleaves three of the four substrates so quickly that fitting the exponential function accurately was difficult as it heavily relied on the first few time points before saturation was reached, explaining why the regression for this sample was not as robust. This impediment could likely be improved by incorporating a peptomer probe with multiple peptoid substitutions to impede the rate of cleavage and therefore better refine the relationship between rate and concentration. For papain, all three sample concentrations were underestimated, which likely had more to do with the selection of samples here, which were all low concentrations. This means regression fitting did not have the low concentration datapoints and therefore the slope is dominated by larger concentrations.
As described, the data here is representative of the results achieved with holdout cross-validation. To evaluate the overall performance, the model was run ten times with different holdout selections each time (Table 1, Table S3 and Fig. S7, ESI†). The average classification accuracy was 96% with a standard deviation of 6%. The proteases most frequently misclassified were thermolysin (as collagenase) and papain (as chymotrypsin). Again, these are the two highest activity enzymes that do not cluster as effectively by both PCA and LDA. Importantly, LDA uses the distance from the centroid of each training cluster for classification.27,28 Thus, outer datapoints for thermolysin and papain get incorrectly classified, as they are closer to the centroid of collagenase and chymotrypsin, respectively, which are tightly clustered. Moving forward, a different form of discriminant analysis, like quadratic discriminant analysis, may better handle the fringe datapoints.
| Iteration | CA (%) | In bounds (%) | Percent error (%) |
|---|---|---|---|
| 1 | 94 | 89 | 27 |
| 2 | 100 | 89 | 25 |
| 3 | 100 | 83 | 33 |
| 4 | 100 | 72 | 87 |
| 5 | 94 | 89 | 27 |
| 6 | 94 | 78 | 27 |
| 7 | 100 | 89 | 236 |
| 8 | 83 | 78 | 22 |
| 9 | 100 | 72 | 75 |
| 10 | 89 | 83 | 64 |
| Average | 96 | 82 | 62 |
| St. Dev. | 6 | 7 | 65 |
Results from ten iterations of the model also confirmed that concentrations on the high and low ends were more likely to be erroneous, as the regression curve fitting did not include the whole range of concentrations. If this approach were to be used to identify proteases in an unknown sample, it would be crucial that the training dataset includes the full range of concentrations possible. While our goal here was to cover a large concentration range, using concentrations that are linearly spaced would also likely result in more accurate fitting. When correctly classified, the average error in concentration was 62%. However, given the three orders of magnitude spanned by the concentrations tested, this metric is not entirely representative of the model's success. 82% (148/180) of all predicted concentrations were within the bounds of the concentrations above and below the actual concentration (i.e., between half and double the actual concentration). Of the 32 samples that did not meet this qualification, eight were misclassified proteases, confirming that improvements to classification would also improve concentration estimates.
Here we provide a proof-of-concept for classifying proteases over a large range of concentrations. While PCA exhibited a logical pattern mapped by concentration and protease identity in two dimensions, LDA was more effective at clustering datapoints from different proteases. The holdout cross-validation scheme provided meaningful insight to the performance of the LDA algorithm, as it tests on datapoints that are distinctly different from the concentrations the model is trained with. Thus, an average of 96% classification accuracy and 82% of concentrations estimated in bounds substantiates the achieved objectives. Potential improvements include (1) adding a peptomer probe that is more resistant to thermolysin and papain, (2) linearly spacing concentrations for training, and (3) experimenting with different discriminant analysis techniques that may improve clustering. Moving forward, the utmost goal would be to sense proteases in complex mixtures with even more sensitive probes. Ultimately, the accuracy of a model depends on the strength of the training dataset, meaning high-throughput screening would be especially useful to generate datasets with rich enough information to differentiate individual species in a mixture. Altogether, pattern-recognition sensing has proven to be an exceptionally useful tool in biosensing and this demonstration propels it along the trajectory toward hypothesis-free universal sensors.29
M. J. A.: conceptualization, methodology, validation, formal analysis, visualization, writing – original draft. H. C. S.: conceptualization, methodology, software. N. L.: investigation, data curation. A. M. R.: conceptualization, resources, supervision, funding acquisition, writing – review & editing.
This research was supported by the NSF (Grant #2046746) and Burroughs Wellcome Fund (#1015895). The authors acknowledge the use of the Proteomics Facility at the University of Texas at Austin. Fig. 1b contains enzyme structures created with BioRender.com using the PDB 1L6J-crystal structure of human matrix metalloproteinase MMP9 (gelatinase B) published at doi.org/10.1107/s0909444902007849.
Conflicts of interest
There are no conflicts to declare.Notes and references
- J. J. Lavigne and E. V. Anslyn, Angew. Chem., Int. Ed., 2001, 40, 3118–3130 CrossRef CAS PubMed.
- D. S. Lee, J. K. Jung, J. W. Lim, J. S. Huh and D. D. Lee, Sens. Actuators, B, 2001, 77, 228–236 CrossRef CAS.
- H. Qiu, F. Pu, X. Ran, J. Ren and X. Qu, Chem. – Eur. J., 2017, 23, 9258–9261 CrossRef CAS PubMed.
- D. G. Smith, L. Mitchell and E. J. New, Analyst, 2018, 144, 230–236 RSC.
- L. Guan, Z. Jiang, Y. Cui, Y. Yang, D. Yang, G. Qian, L. L. Guan, Z. W. Jiang, Y. J. Cui, Y. Yang, D. R. Yang and G. D. Qian, Adv. Opt. Mater., 2021, 9, 2002180 CrossRef CAS.
- C. Zhai, L. Miao, Y. Zhang, L. Zhang, H. Li and S. Zhang, Chem. Eng. J., 2022, 431, 134107 CrossRef CAS.
- Y. He, Y. Liang and H. Yu, ACS Comb. Sci., 2015, 17, 409–412 CrossRef CAS PubMed.
- W. Zhang, N. Gao, J. Cui, C. Wang, S. Wang, G. Zhang, X. Dong, D. Zhang and G. Li, Chem. Sci., 2017, 8, 6281–6289 RSC.
- B. Wang, J. Han, N. M. Bojanowski, M. Bender, C. Ma, K. Seehafer, A. Herrmann and U. H. F. Bunz, ACS Sens., 2018, 3, 1562–1568 CrossRef CAS PubMed.
- H. Zhou, L. Baldini, J. Hong, A. J. Wilson and A. D. Hamilton, J. Am. Chem. Soc., 2006, 128, 2421–2425 CrossRef CAS PubMed.
- M. Okada, H. Sugai, S. Tomita and R. Kurita, Sensors, 2020, 20, 5110 CrossRef CAS PubMed.
- H. Kong, D. Liu, S. Zhang and X. Zhang, Anal. Chem., 2011, 83, 1867–1870 CrossRef CAS PubMed.
- S. Rana, A. K. Singla, A. Bajaj, S. G. Elci, O. R. Miranda, R. Mout, B. Yan, F. R. Jirik and V. M. Rotello, ACS Nano, 2012, 6, 8233–8240 CrossRef CAS PubMed.
- S. Tomita, S. Ishihara and R. Kurita, ACS Appl. Mater. Interfaces, 2019, 11, 6751–6758 CrossRef CAS PubMed.
- H. Sugai, S. Tomita, S. Ishihara, K. Shiraki and R. Kurita, Chem. Commun., 2022, 58, 11083–11086 RSC.
- M. Lin, P. Song, G. Zhou, X. Zuo, A. Aldalbahi, X. Lou, J. Shi and C. Fan, Nat. Protoc., 2016, 11, 1244–1263 CrossRef CAS PubMed.
- X. Wang, L. Qin, M. Zhou, Z. Lou and H. Wei, Anal. Chem., 2018, 90, 11696–11702 CrossRef CAS PubMed.
- P. Kasperkiewicz, M. Poreba, K. Groborz and M. Drag, FEBS J., 2017, 284, 1518–1539 CrossRef CAS PubMed.
- O. R. Miranda, C. C. You, R. Phillips, I. B. Kim, P. S. Ghosh, U. H. F. Bunz and V. M. Rotello, J. Am. Chem. Soc., 2007, 129, 9856–9857 CrossRef CAS PubMed.
- S. Tomita, H. Sugai, M. Mimura, S. Ishihara, K. Shiraki and R. Kurita, ACS Appl. Mater. Interfaces, 2019, 11, 47428–47436 CrossRef CAS PubMed.
- M. De, S. Rana, H. Akpinar, O. R. Miranda, R. R. Arvizo, U. H. F. Bunz and V. M. Rotello, Nat. Chem., 2009, 1, 461–465 CrossRef CAS PubMed.
- O. R. Miranda, H. T. Chen, C. C. You, D. E. Mortenson, X. C. Yang, U. H. F. Bunz and V. M. Rotello, J. Am. Chem. Soc., 2010, 132, 5285–5289 CrossRef CAS PubMed.
- W. Cuypers and P. A. Lieberzeit, Front. Chem., 2018, 6, 268 CrossRef PubMed.
- I. L. H. Ong and K. L. Yang, Analyst, 2017, 142, 1867–1881 RSC.
- H. C. Schunk, D. S. Hernandez, M. J. Austin, K. S. Dhada, A. M. Rosales and L. J. Suggs, J. Mater. Chem. B, 2020, 8, 3460–3487 RSC.
- M. J. Austin, H. Schunk, C. Watkins, N. Ling, J. Chauvin, L. Morton and A. M. Rosales, Biomacromolecules, 2022, 23, 4909–4923 CrossRef CAS PubMed.
- S. Stewart, M. A. Ivy and E. V. Anslyn, Chem. Soc. Rev., 2014, 43, 70–84 RSC.
- J. H. Friedman, J. Am. Stat. Assoc., 1989, 84, 165–175 CrossRef.
- W. J. Peveler, M. Yazdani and V. M. Rotello, ACS Sens., 2016, 1, 1282–1285 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Material descriptions, experimental procedures, fluorescent degradation traces, holdout cross-validation of classification and multiple regression. See DOI: https://doi.org/10.1039/d2cc06587h |
| This journal is © The Royal Society of Chemistry 2023 |