Evidence based housekeeping gene selection for microRNA-sequencing (miRNA-seq) studies†
D. P.
Tonge
* and
T. W.
Gant
Centre for Radiation, Chemical and Environmental Hazards, Public Health England, Chilton, UK. E-mail: daniel.tonge@hpa.org.uk; Tel: +44 (0)1235 822811
First published on 10th June 2013
Abstract
The accurate determination of miRNA expression by PCR requires robust data normalisation. The most commonly used approach involves the use of housekeeping or normalising factors which are assumed invariant across the range of experimental conditions investigated. This assumption is often not proven in all experimental situations, therefore we consider whether global miRNA expression data sets obtained by high throughput sequencing can be screened to identify candidate housekeeping miRNAs (HK-miRNA) for further evaluation. To establish this method, we assessed global miRNA sequencing data from a study involving 30 canine skin specimens. From an initial pool of over 200 miRNAs, we identified several candidate HK-miRNA which demonstrated stability across all study samples using both a classical statistical approach and the NormFinder algorithm. We verified these putative HK-miRNAs using real-time PCR assays to further validate their suitability in this specific experimental setup. Our analysis provides a framework to allow researchers to exploit high-throughput sequencing data (be it their own, or from the short read archive or other genomic repository) to guide HK-miRNA selection.
Introduction
MicroRNAs (miRNAs) are short non-protein-coding RNA species that have a regulatory function in modulating protein translation from specific mRNAs.1 MicroRNAs are reported to target approximately 60% of human mRNAs2 and are associated with the regulation of diverse physiological processes. Specific miRNAs appear to be dysregulated in various pathological conditions3,4 and in response to toxicant exposure,5–8 highlighting their importance in both physiological and pathological processes. Investigation into these miRNA expression signatures has the potential to provide new insights into the molecular pathways responsible for toxicity. Furthermore, these dysregulated miRNAs are detectable in various biological fluids,9 paving the way for their use as a novel class of toxicological biomarkers.10Over the past few years, various molecular techniques have been successfully applied to the detection and quantification of known miRNAs including polymerase chain reaction (PCR)11 and microarray hybridisation.12 More recently, high-throughput sequencing based techniques (miRNA-seq) have been developed and offer the advantage of detecting all miRNAs within a biological sample including novel, previously undescribed miRNA species. These techniques are particularly useful when working in less common species (exotic species, large animals etc.) which tend to have poorly characterised genomes and where analysis by PCR or microarray would be problematic and/or inefficient.
The aim of many miRNA-seq studies is to establish differences between two or more experimental groups. This is achieved by the global sequencing of all miRNAs within each biological sample, the mapping of these sequences to a reference database such as miRBase, data normalisation and P value estimation. Much like microarray based studies, candidate miRNAs identified as significantly regulated by miRNA-seq require verification by PCR which represents the gold standard for sensitivity and specificity in nucleic acid quantification.13 In order to ensure accuracy, PCR is critically dependent on thorough data normalisation14 which aims to eliminate all variation between the study samples that is not a direct consequence of the experimental treatment, pathology or other experimental variable. Common sources of technical variation include differences in the amount of starting material, the efficiency of RNA extraction and in the efficiency of enzymes used for reverse transcription.15 Furthermore, as the cellular concentration of different miRNAs may cover several orders of magnitude, different cDNA dilutions must be prepared as input into specific PCR reactions to ensure detection falls within the linear range of each assay. These cDNA dilutions may also harbour technical variance resulting from pipetting errors which also requires careful control.
Common approaches to miRNA PCR data normalisation involve the use of one or more “housekeeping” or “reference” miRNAs (HK-miRNA).11,16–18 Unlike mRNA PCR applications for which several common housekeeping genes have been described over many years, the assessment of miRNAs by PCR is still in its infancy and no such common normalisers have yet been identified. In this regard, the ideal HK-miRNA is a single nucleic acid that exhibits invariant expression across all study samples, is expressed alongside the miRNA of interest and demonstrates equivalent stability, extraction efficiency and amplification efficiency.11,19 HK-miRNA selection may have a profound effect on the final study outcome20 with no one HK-miRNA exhibiting a stable expression profile across all experimental conditions.19 It is therefore of paramount importance to select and validate appropriate normalisation genes for each experimental setup.
To this end, we investigated whether miRNA-seq data could be used to inform HK-miRNA selection for use at the PCR verification stage, and whether the commonly used PCR data analysis tool NormFinder21 could be used to effectively screen such data. We assessed global miRNA sequencing data from a study involving 30 canine skin specimens. This dataset provided several challenges in respect of HK-miRNA selection due to the presence of multiple experimental variables including drug treatment, study animals of differing ages and the presence of a subset of samples with variable skin pathology. To this end, it is similar to many other miRNA datasets that may be used for the selection of HK-miRNAs. From an initial pool of over 200 miRNAs, we identified several candidate HK-miRNA which demonstrated marked stability across all samples (in this specific experimental setup) using both a classical statistical approach and NormFinder. We verified the expression of these putative HK-miRNAs using real-time PCR assays and evaluated their suitability.
Results and discussion
Identification of putative housekeeping miRNAs (HK-miRNAs) from miRNA-seq data
The data used herein were from a global miRNA-seq study of canine skin specimens. Sequencing data was available for 24 individual animals and consisted of approximately 5 million sequencing reads per sample. Reads had been previously mapped to miRBase version 16 and differential expression between the experimental groups inferred using DESeq.22 Significantly regulated miRNAs (those with estimated P values <0.05 following false-discovery correction) were excluded prior to analysis to increase the efficiency of data processing (data not shown). In order to elucidate suitable putative HK-miRNAs, two statistical methods were utilised. The first, termed the “coefficient of variation model” involved calculation of the coefficient of variation and maximum fold change for each individual miRNA. Suitable HK-miRNAs were defined as those with the lowest coefficient of variation and a maximum fold change (MFC) between any of the experimental groups of ≤2. Such parameters have been previously utilised for housekeeping gene selection across a range of microarray and PCR platforms.23 The second model utilised a previously published algorithm known as NormFinder21 that has yet to be applied to high throughput sequencing data but has been extensively evaluated with regards to housekeeping gene selection from PCR data. In this regard, NormFinder was used to estimate intra-group variation (within a single experimental group of animals all receiving the same treatment) and inter-group variation (between the various experimental groups in the study) and combine these estimates to compute a stability value for each miRNA in question. A comprehensive overview of the NormFinder algorithm can be found in the supplementary data of Andersen et al.21 The 15 most suitable putative HK-miRNAs identified by each model are presented in Table 1 (please refer to ESI I† for a comprehensive list of all miRNAs assessed and associated descriptive statistics). The calculated coefficient of variation, MFC and stability value determined for each putative HK-miRNA is represented graphically in Fig. 1 and demonstrates the stability of the top 15 putative HK-miRNAs across the various study variables (coefficient of variation from 18.6% and maximum fold change from <1; NormFinder stability values in the range 0.23–0.28). Concordance between the top 15 most suitable HK-miRNAs identified by the two models was 60% (9 candidate miRNAs) with the coefficient of variation model identifying miR-23b as the most suitable housekeeping miRNA, and NormFinder ranking let-7f as most suitable based upon the miRNA-seq data supplied. It is not uncommon for different models to identify different housekeeping genes21,24 due to differences in how they rank stability25 however for this reason, we chose to further investigate those miRNAs identified by both independent models (see Table 1). | ||
| Fig. 1 (A) The 15 least variable miRNAs identified from miRNA-seq data. Data are coefficient of variation, expressed as a percentage, determined across all samples for which miRNA-seq data was available (n = 24). (B) Maximum fold change (MFC) determined between any of the experimental study groups for each of the 15 prospective HK-miRNAs. The red bar indicates the maximum acceptable fold change for prospective HK-miRNAs. (C) Stability values for the top 15 most stable prospective HK-miRNAs as determined by NormFinder. | ||
| Coefficient of variation model | NormFinder stability model |
|---|---|
| a Candidate housekeeping genes are listed in order of ascending coefficient of variation (column one) and descending stability (column two). MicroRNAs included in bold are present in the top 15 results generated from both statistical models. | |
| cfa-miR-23b | cfa-let-7f |
| cfa-miR-191 | cfa-miR-191 |
| cfa-miR-29b | cfa-miR-24-1* |
| cfa-miR-99a | cfa-miR-148a |
| cfa-miR-30c | cfa-miR-23a |
| cfa-miR-23a | cfa-miR-186 |
| cfa-miR-24 | cfa-miR-29b |
| cfa-let-7g | cfa-miR-15b |
| cfa-miR-628 | cfa-miR-27a |
| cfa-let-7f | cfa-miR-24-2* |
| cfa-miR-331 | cfa-miR-23b |
| cfa-miR-30e | cfa-miR-660 |
| cfa-miR-25 | cfa-miR-30c |
| cfa-miR-148a | cfa-miR-25 |
| cfa-miR-1839 | cfa-miR-99a |
Validation of putative housekeeping miRNAs (HK-miRNAs) by real-time PCR
Next, we sought to verify the expression of each of the putative HK-miRNAs by duplicate real-time PCR. Assays validated for the canine were available for all 9 of the miRNAs identified by both statistical models. The original miRNA-seq study was conducted on a restricted sample set (24 of the 30 study animals) so at this stage, samples from the remaining animals were reintroduced and PCR data generated for all 30 study animals. This arrangement is reminiscent of many research scenarios whereby sequencing is conducted on a restricted set of samples to identify a list of differentially expressed molecules of interest for validation in all study samples. Following PCR, the coefficient of variation and MFC of each putative HK-miRNA was determined (Fig. 2A and B). Stability values for each of the putative housekeeping genes were then calculated using NormFinder (Fig. 2C), which is specifically designed for the analysis of such data.21 Comprehensive PCR data for each HK-miRNA is presented in ESI II.†Fig. 2 demonstrates that the 9 putative HK-miRNAs varied considerably with regards to their suitability with variance across all study samples (%CV) ranging from approximately 20% to almost 60%, MFC ranging from 1.2 to 2.4 (exceeding the desired threshold of 2) and stability scores in the range 0.10–0.28.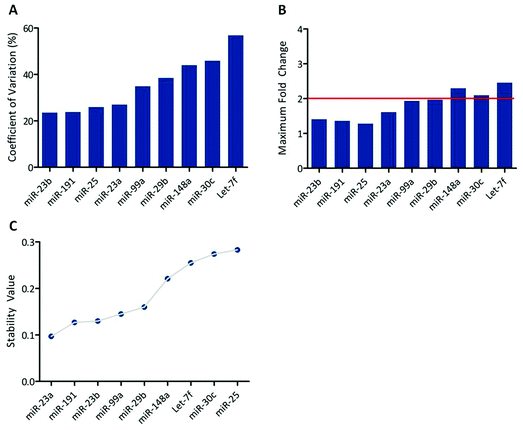 | ||
| Fig. 2 (A) Calculated coefficient of variation, expressed as a percentage, determined across all samples (n = 30) for each of the prospective HK-miRNA validated by real-time PCR. Data are based on arbitrary expression values. (B) Calculated maximum fold change (MFC) determined between any of the experimental study groups for each HK-miRNA. The red bar indicates the maximum acceptable fold change for prospective HK-miRNAs. (C) Stability values for the 9 prospective HK-miRNAs validated by PCR as determined by NormFinder. | ||
Whilst the NormFinder stability value is an excellent indicator of overall HK-miRNA suitability, it gives little information about how each individual experimental group contributes to the overall stability score. In this regard we sought to further investigate how variance between each of the experimental groups contributed to the overall stability of each HK-miRNA. Based upon the PCR data, inter-group variances were plotted for each HK-miRNA on the y-axis and the mean of the intra-group variance used to generate confidence intervals as shown in Fig. 3 (HK-miRNAs are plotted in order of stability from the most to the least stable). The most stable HK-miRNA, miR-23a, revealed small inter-group variances across all 6 experimental groups. Corresponding inter-group variances based upon the restricted set of underlying miRNA-seq data are presented in Fig. 4. Inter-group variances >0 have the capacity to reduce the apparent expression of a given miRNA for that experimental group following normalisation, whilst inter-group variances <0 have the opposite effect. It is reported that the ideal housekeeping gene has a mean inter-group variance of close to 0 and despite the broad range in the suitability of HK-miRNAs assessed here (as discussed above), all fulfilled this requirement with mean inter-group variance ranging from 0.00 to −5.9 × 10−16 for the PCR data, and 9.25 × 10−18 to −2.5 × 10−16 for the miRNA-seq data. It is therefore important to consider not only total variation, but also how this variance is distributed across the various experimental groups within a study.
 | ||
| Fig. 3 Assessment of inter and intra group variation for the 9 prospective HK-miRNA subjected to verification by PCR (A–I in order of stability). Data are estimated inter-group variation (y-axis) for each experimental group (labelled 1 to 6 in panel A) determined by real-time PCR. Confidence intervals represent estimated intra-group variation. n = 5 per experimental group, n = 30 per individual HK-miRNA. | ||
 | ||
| Fig. 4 Assessment of inter and intra group variation based upon the underlying miRNA-seq data for the 9 prospective HK-miRNA subsequently subjected to verification by PCR. Data are estimated inter-group variation (y-axis) for each experimental group (labelled 1 to 6 in panel A) determined by miRNA-seq. Confidence intervals represent estimated intra-group variation. n = 4 per experimental group, n = 24 per individual HK-miRNA. | ||
In considering the most suitable HK-miRNA (post-validation), miR-23b was identified as the least variable when taking into account %CV alone, whilst NormFinder ranked miR-23a as the most stable HK-miRNA based upon the sum of inter and intra group variation (NormFinder stability value). Both statistical models placed miR-191 as the second most suitable HK-miRNA. Interestingly, despite the stark differences in selection method complexity (the first method being based purely on %CV across all samples whilst the second comprising a complex algorithm based on intra and inter group variation estimation) 5 out of the top 6 HK-miRNAs were concordant (miRs 23a, 191, 23b, 99a and 29b) although the rankings were not consistent. Furthermore, those HK-miRNAs exceeding the MFC limit of 2 (miRs 148a, 30c and Let-7f) also tended to be the least desirable candidates identified by NormFinder with higher stability scores.
In considering which statistical method was most successful in predicting a suitable HK-miRNA from the underlying miRNA-seq data, we compared the top 6 HK-miRNA rankings from each data type. Using the classical method, the candidate HK-miRNAs were ranked miR-23b < miR-191 < miR-29b < miR-99a < miR-30c < miR-23a based on the sequencing data, and miR-23b < miR-191 < miR-25 < miR-23a < miR-99a < miR-29b following PCR validation (least to most variable) showing concordance in 5/6 of the top scoring HK-miRNAs. In the case of NormFinder, the candidate HK-miRNAs were ranked let-7f > miR-191 > miR-148a > miR-23a > miR-29b > miR-23b based on the sequencing data, and miR-23a > miR-191 > miR-23b > miR-99a > miR-29b > miR-148a following PCR validation (most to least stable; top ranking 6 HK-miRNAs shown in each case), again showing concordance in 5/6 of the top scoring HK-miRNAs. Nevertheless, both models identified putative HK-miRNAs from the miRNA-seq data that were subsequently found to be undesirable following PCR validation (miR-30c using the classical method, and Let-7f using NormFinder). It is for this reason that we recommend identifying a number of putative HK-miRNAs from miRNA-seq data for verification by real-time PCR.
Conclusions
A growing body of evidence suggests a pivotal role for miRNAs in mediating the effects of various toxicants. Moreover, miRNAs have the potential to serve as novel biomarkers of toxicity with early reports showing increased sensitivity over classical markers of toxicity.10 The study of miRNA expression requires robust data normalisation to control for technical error. We confirm that both the popular housekeeping gene selection algorithm NormFinder and a classical statistical approach are able to successfully identify a list of putative HK-miRNAs from miRNA-seq data for further analysis. Verification of these putative HK-miRNAs by real-time PCR revealed several suitable HK-miRNAs with low inter and intra group variation. We therefore recommend identifying a number of putative HK-miRNAs from miRNA-seq data for downstream analysis by real-time PCR. In summary, our analysis not only reports a list of HK-miRNAs for use in the context of this study, but also provides a framework to allow researchers to exploit high-throughput sequencing data (be it their own, or from similar studies conducted elsewhere and lodged in the short read archive) to guide HK-miRNA selection.Materials and methods
Study outline
The analyses herein were performed on archival skin specimens obtained from a time course study of female beagle dogs dosed orally once per day with either vehicle or a proprietary matrix metalloproteinase inhibitor (MMPi) known to induce fibrotic skin lesions, for between 4 and 17 days. The in vivo experiments were conducted by AstraZeneca Ltd and have been discussed in detail elsewhere.26 Briefly, the sample comprised 6 experimental groups and included control skin specimens from animals treated with vehicle only (animals 1–5), and skin specimens exhibiting various severities of fibrosis following oral MMPi administration over 4, 8, 11, 14 and 17 days (animals 6–10, 11–15, 16–20, 21–25 and 26–30 respectively) (n = 5 per group).RNA preparation
Total RNA was isolated from dorsal cervical subcutaneous skin tissue using the miRNeasy extraction kit (Qiagen), following the manufacturers standard protocol. RNA was quantitated using a NanoDrop ND-1000 spectrophotometer (Thermo Fisher) and quality was assessed using the Agilent RNA 6000 Nano Kit (Agilent). In order to enrich for small RNA molecules, total RNA samples were passed through PureLink miRNA extraction columns (Invitrogen) according to the manufacturers standard instructions. The resulting enriched RNA samples were then assessed using the Agilent Small RNA Kit (Agilent) and the percentage of small RNA (10–40 nucleotides) determined relative to the mass of total RNA.Preparation of SOLiD sequencing libraries
Global miRNA expression profiling of cervical subcutaneous tissue samples was performed by next-generation sequencing using the SOLiD 4.0 system (Applied Biosystems). Briefly, 5 ng of enriched RNA was ligated with SOLiD sequencing adapters overnight and reverse transcribed to synthesise complementary DNA (cDNA). The resulting cDNA was resolved on 10% v/v TBE-urea gels against a 10 bp ladder (Invitrogen) and the 60–80 nt region, containing miRNA-sized RNA species with adapters, excised. Gel slices were amplified by in-gel PCR using a common 5′ primer (SOLiD 5′), and a specific 3′ primer (SOLiD Barcodes 1–24) to incorporate a molecular barcode into each sample so as to permit sample multiplexing. Equimolar amounts of each library were pooled at this stage, and template preparation performed using the SOLiD EZ-Bead system and E80 emulsion PCR reagents (PN 4452722). Sequencing was performing in-house using 35 base pair (bp) chemistry (Applied Biosystems).Bioinformatic analysis
Raw sequencing reads were exported in colour-space fasta (csfasta) format into CLC Genomics Workbench (CLC). An “extract and count” routine was utilised to condense the millions of sequencing reads into a tally table of each nucleotide sequence, and the number of times that particular sequence was encountered. The resulting data were imported into miRanalyzer V0.227 and reads were mapped to miRBase version 16.0. Reads mapping to known miRNAs were counted and a relative expression value determined by dividing the number of reads mapping to each particular miRNA by the total number of reads mapped. Differential expression and P value estimation was performed using the DESeq package for R statistical language which models count-based data with negative binomial distributions and uses the method of Benjamini and Hochberg to control for type I error.22Quantitative PCR analysis
Complementary DNA was synthesised from 1 μg total RNA using the qScript miRNA cDNA synthesis kit (QuantaBio) in a total reaction volume of 20 μl. Each PCR reaction consisted of 5 μl SYBR Green master mix (QuantaBio), 0.2 μl miRNA Assay (Integrated DNA Technologies), 0.2 μl Universal PCR Primer (QuantaBio), 2.6 μl water and 2 μl template cDNA (diluted 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :100 in 1 × TE). PCR was performed using a Rotor Gene Q thermocycler (Qiagen) with an initial denaturation step at 95 °C for 2 minutes followed by 40 cycles of 95 °C 5 seconds, 60 °C 15 seconds and 70 °C 15 seconds. A melt curve was performed at the end of each run and reviewed to ensure only one specific product was formed during the PCR. The following miRNA Assays sourced from Integrated DNA Technologies were utilised: hsa-let-7f, hsa-miR-148a, hsa-miR-25, hsa-miR-29b, hsa-miR-30c, hsa-miR-99a, hsa-miR-23a, and hsa-miR-23b.
:100 in 1 × TE). PCR was performed using a Rotor Gene Q thermocycler (Qiagen) with an initial denaturation step at 95 °C for 2 minutes followed by 40 cycles of 95 °C 5 seconds, 60 °C 15 seconds and 70 °C 15 seconds. A melt curve was performed at the end of each run and reviewed to ensure only one specific product was formed during the PCR. The following miRNA Assays sourced from Integrated DNA Technologies were utilised: hsa-let-7f, hsa-miR-148a, hsa-miR-25, hsa-miR-29b, hsa-miR-30c, hsa-miR-99a, hsa-miR-23a, and hsa-miR-23b.
Author contributions
DPT conceived the study, performed the experiments and prepared the manuscript. TWG read and approved the final manuscript.Acknowledgements
We wish to acknowledge AstraZeneca Ltd for the supply of the archival canine skin specimens used in this study. We also wish to acknowledge Mrs Joan Riley for her expert assistance with configuring and running of the SOLiD 4.0 sequencer.References
- L. He and G. J. Hannon, MicroRNAs: small RNAs with a big role in gene regulation, Nat. Rev. Genet., 2004, 5(7), 522–531 CrossRef CAS.
- T. Yokoi and M. Nakajima, Toxicological implications of modulation of gene expression by microRNAs, Toxicol. Sci., 2011, 123(1), 1–14 CrossRef CAS.
- M. A. Cortez, et al., MicroRNAs in body fluids—the mix of hormones and biomarkers, Nat. Rev. Clin. Oncol., 2011, 8(8), 467–477 CrossRef CAS.
- M. V. Iorio and C. M. Croce, MicroRNA dysregulation in cancer: diagnostics, monitoring and therapeutics. A comprehensive review, EMBO Mol. Med., 2012, 4(3), 143–159 CrossRef CAS.
- P. Sathyan, H. B. Golden and R. C. Miranda, Competing interactions between micro-RNAs determine neural progenitor survival and proliferation after ethanol exposure: evidence from an ex vivo model of the fetal cerebral cortical neuroepithelium, J. Neurosci., 2007, 27(32), 8546–8557 CrossRef CAS.
- I. P. Pogribny, et al., Induction of microRNAome deregulation in rat liver by long-term tamoxifen exposure, Mutat. Res., 2007, 619(1–2), 30–37 CrossRef CAS.
- T. Fukushima, et al., Changes of micro-RNA expression in rat liver treated by acetaminophen or carbon tetrachloride—regulating role of micro-RNA for RNA expression, J. Toxicol. Sci., 2007, 32(4), 401–409 CrossRef CAS.
- O. F. Laterza, et al., Plasma microRNAs as sensitive and specific biomarkers of tissue injury, Clin. Chem., 2009, 55(11), 1977–1983 CAS.
- J. A. Weber, et al., The microRNA spectrum in 12 body fluids, Clin. Chem., 2010, 56(11), 1733–1741 CAS.
- K. Wang, et al., Circulating microRNAs, potential biomarkers for drug-induced liver injury, Proc. Natl. Acad. Sci. U. S. A., 2009, 106(11), 4402–4407 CrossRef CAS.
- H. J. Peltier and G. J. Latham, Normalization of microRNA expression levels in quantitative RT-PCR assays: identification of suitable reference RNA targets in normal and cancerous human solid tissues, RNA, 2008, 14(5), 844–852 CrossRef CAS.
- R. Ach, H. Wang and B. Curry, Measuring microRNAs: comparisons of microarray and quantitative PCR measurements, and of different total RNA prep methods, BMC Biotechnol., 2008, 8(1), 69 CrossRef.
- T. D. Schmittgen, et al., Real-time PCR quantification of precursor and mature microRNA, Methods, 2008, 44(1), 31–38 CrossRef CAS.
- C. Tricarico, et al., Quantitative real-time reverse transcription polymerase chain reaction: normalization to rRNA or single housekeeping genes is inappropriate for human tissue biopsies, Anal. Biochem., 2002, 309(2), 293–300 CrossRef CAS.
- J. Vandesompele, et al., Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes, Genome Biol., 2002, 3(7), research0034.1–research0034.11 CrossRef.
- R. Garzon, et al., MicroRNA signatures associated with cytogenetics and prognosis in acute myeloid leukemia, Blood, 2008, 111(6), 3183–3189 CrossRef CAS.
- I. Alevizos, et al., MicroRNA expression profiles as biomarkers of minor salivary gland inflammation and dysfunction in Sjögren's syndrome, Arthritis Rheum., 2011, 63(2), 535–544 CrossRef CAS.
- V. F. Viprey, M. V. Corrias and S. A. Burchill, Identification of reference microRNAs and suitability of archived hemopoietic samples for robust microRNA expression profiling, Anal. Biochem., 2012, 421(2), 566–572 CrossRef CAS.
- N. Janssens, et al., Housekeeping genes as internal standards in cancer research, Mol. Diagn., 2004, 8(2), 107–113 Search PubMed.
- J. Song, et al., Identification of suitable reference genes for qPCR analysis of serum microRNA in gastric cancer patients, Dig. Dis. Sci., 2012, 57(4), 897–904 CrossRef CAS.
- C. L. Andersen, J. L. Jensen and T. F. Orntoft, Normalization of real-time quantitative reverse transcription-PCR data: a model-based variance estimation approach to identify genes suited for normalization, applied to bladder and colon cancer data sets, Cancer Res., 2004, 64(15), 5245–5250 CrossRef CAS.
- S. Anders and W. Huber, Differential expression analysis for sequence count data, Genome Biol., 2010, 11(10), R106 CrossRef CAS.
- H. J. de Jonge, et al., Evidence based selection of housekeeping genes, PLoS One, 2007, 2(9), e898 Search PubMed.
- Z. Tong, et al., Selection of reliable reference genes for gene expression studies in peach using real-time PCR, BMC Mol. Biol., 2009, 10(1), 71 CrossRef.
- F. Pinto, et al., Selection of suitable reference genes for RT-qPCR analyses in cyanobacteria, PLoS One, 2012, 7(4), e34983 CAS.
- R. Westwood, et al., Characterization of fibrodysplasia in the dog following inhibition of metalloproteinases, Toxicol. Pathol., 2009, 37(7), 860–872 CrossRef.
- M. Hackenberg, N. Rodriguez-Ezpeleta and A. M. Aransay, miRanalyzer: an update on the detection and analysis of microRNAs in high-throughput sequencing experiments, Nucleic Acids Res., 2011, 39(Web Server issue), W132–W138 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c3tx50034a |
| This journal is © The Royal Society of Chemistry 2013 |