Passage determines toxicity and neuronal markers expression in PC12 cells with altered phenotype
Manuel Mejía,
Mariana Salgado-Bustamante,
Claudia G. Castillo and
María E. Jiménez-Capdeville*
Departamento de Bioquímica, Facultad de Medicina, Universidad Autónoma de San Luis Potosí, México. E-mail: mejimenez@uaslp.mx; Tel: +(52-444) 826 23 45, ext 6630
First published on 10th June 2013
Abstract
PC12 cells, a catecholaminergic cell line widely used in neurotoxicology, developed variants that have lost some of their characteristic features such as response to NGF and controlled cell division. The present study was undertaken to evaluate the responses obtained between two passages (early and late) of PC12 cells with altered phenotype in viability, percentage of cells in synthesis phase and expression of tyrosine hydroxylase. We hypothesized that being unaware of working with a cell line bearing an altered phenotype could be responsible for the high variability reported in viability assays in the scientific literature. We used sodium arsenite (5–1000 μg L−1), lead acetate (5–1000 μg dL−1), 3-nitropropionic acid (100–5000 μM) and 6-hydroxydopamine (50–300 μM) for viability assays. The results showed that PC12 cells with altered phenotype do not respond uniformly to a variety of stimuli, therefore the requirement to ascertain the true identity of cell lines before testing hypothesis is an important issue to be addressed.
Introduction
The undeniable link between environmental conditions and human health has led to the growing development of instruments and policies geared toward research into the safety of chemical substances. According to the Environmental Protection Agency Toxic Substances Control Act,1 there are more than 83![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 chemicals in commerce, most of them not even tested for general toxicity.1,2 In this context the need to analyze the toxic properties of an ever growing body of compounds entails a serious challenge that has given rise to many models for functional, morphological and biochemical screening. Among these models, in vivo testing of chemicals faces important limitations, due to ethical, environmental and economical concerns. In light of the growing need for toxicological screening there is a global trend to use in vitro models. Although recommended by several authors and agencies3–7 data obtained through in vitro experiments require further translation into in vivo models.
000 chemicals in commerce, most of them not even tested for general toxicity.1,2 In this context the need to analyze the toxic properties of an ever growing body of compounds entails a serious challenge that has given rise to many models for functional, morphological and biochemical screening. Among these models, in vivo testing of chemicals faces important limitations, due to ethical, environmental and economical concerns. In light of the growing need for toxicological screening there is a global trend to use in vitro models. Although recommended by several authors and agencies3–7 data obtained through in vitro experiments require further translation into in vivo models.
One of the models most widely used for evaluating neurotoxicity is based on PC12 cells (Table 1). These cells, derived from a rat adrenal medullar pheochromocytoma, became a useful model as they exhibit changes of their in vitro phenotype after exposure to several different substances. Classic reports demonstrate that these cells produce a branching network of neurites under nerve growth factor (NGF) stimulation – resembling thus sympathetic neurons – or differentiate into chromaffin-like cells – producing and releasing norepinephrine – under corticosteroids stimulation.8–11 Due to these changes, PC12 cells became one of the most suitable and common models for morphological, biochemical and physiological analysis of substances with putative effects on the nervous system.
| Toxic substance | Culture medium | Viability assay | Exposure period (hours) | Concentration | Viability (%) | Reference |
|---|---|---|---|---|---|---|
| Arsenic (mg L−1) | Kaighn's modification of Ham's F12 | MTS assay | 48 | 750 | 50 | Frankel, 200928 |
| RPMI | Resazurin reduction | 24 | 500 | 90 | This work | |
| 1000 | 70 | |||||
| Lead (mg dL−1) | DMEM | MTT assay | 24 | 21 | 80 | Xu, 200630 |
| 2072 | 60 | |||||
| 21–207 | 80 | Sharifi, 200529 | ||||
| 3730 | 50 | |||||
| RPMI | Resazurin reduction | 24 | 500 | 100 | This work | |
| 1000 | 100 | |||||
| 3-Nitropropionic acid (μM) | DMEM | Trypan blue | 48 | 4000 | 60 | Mandavilli, 200535 |
| 8000 | 40 | |||||
| RPMI | MTT assay | 0.25 | 20 | 90 | Lee, 200533 | |
| DMEM-serum free | LDH assay | 24 | 20 | 60 | Wei, 200932 | |
| DMEM | MTT/LDH assays | 5000 | 25 | Kawamoto, 201234 | ||
| RPMI | Resazurin reduction | 24 | 2500 | 95 | This work | |
| 5000 | 95 | |||||
| 6-OHDA (μM) | RPMI | Neutral red staining | 6 | 20 | 70 | Ha, 200337 |
| 60 | 40 | |||||
| DMEM | Trypan blue | 24 | 25 | 60 | Eminel, 200436 | |
| MTT/LDH assays | 0.25 | 150 | 50 | Hanrott, 200641 | ||
| MTT assay | 24 | 50 | 90 | Takata, 200538 | ||
| 150 | 60 | |||||
| 300 | 20 | |||||
| 150 | 40 | Yamamoto, 200740 | ||||
| 25 | 90 | Tao, 201139 | ||||
| 75 | 50 | |||||
| 150 | 20 | |||||
| F-12K | MTT/LDH assays | 12 | 1000 | 50 | Wang, 201142 | |
| RPMI | Resazurin reduction | 24 | 75 | 70 | This work | |
| 150 | 50 | |||||
| 300 | 20 | |||||
Despite the large number of data obtained from PC12 cells since their initial description during the seventies, a growing number of reports generated in parallel demonstrate the existence of PC12 cells showing altered phenotypes associated with accelerated cell division, fibroblast-like phenotype, increased resistance to toxics and lack of differentiation under NGF stimulation,12–15 and even cells that differentiate spontaneously in the absence of NGF or other neurotrophic factors.16 This diversity of circulating cell lines with a common PC12 ancestry but altered phenotypes may explain the non-replicative data found in the scientific literature where toxic doses and effects are reported based upon experimental outcomes such as changes in morphology, viability, gene expression, enzymatic activity, among others.
Since this heterogeneity of PC12 cells creates concern and an important need to assess the true identity of cells used for the evaluation of hypothesis, the purpose of this work was to demonstrate that data generated in a PC12 cell line with altered phenotype bear limitations that make it difficult to draw general conclusions about some cellular effects. We undertook a series of experiments using three compounds known by their toxic effects in all cell types, namely arsenite, lead and 3-nitropropionic acid, plus a substance widely used to induce catecholaminergic cell death, 6-hydroxydopamine. The exposure concentrations were chosen in a range that includes those reported in most viability assays, from low concentrations within which some functional or clinical reports exist, to high concentrations that have no functional or clinical relevance because they are concentrations far high to be found in human exposition or in models for studying toxicity mechanisms.17–19 This exposure panel generated information about resistance properties associated with our altered phenotype cell lines across cumulative passages in culture. In addition, we analyzed the expression of specific PC12 genes and proteins, and all the information obtained was analyzed in light of the current practices of in vitro testing of chemical compounds.
Materials and methods
Reagents
The reagents used in this protocol were obtained from the following commercial sources: sodium arsenite, sodium resazurin, 6-hydroxydopamine (6-OHDA), 3-nitropropionic acid (3-NPA), mouse monoclonal anti-tyrosine hydroxylase antibody, L-glutamine, poly-L-lysine (PLL), ethanol, sodium borohydride, bis-benzimide, ascorbic acid and other reagents for buffer solutions were obtained from Sigma-Aldrich (St. Louis, MO, USA), lead acetate from Químicos Monterrey (Monterrey, NL, México), rabbit anti-mouse FITC-labeled secondary antibody from AbD Serotec (Kidlington, OX, UK), cell culture factors RPMI 1640, horse serum, NGF and fetal bovine serum, were acquired from GIBCO (Gaithersburg, MD, USA). RT Superscript II, reaction buffer and dNTPs from Invitrogen (Carlsbad, CA, USA), SYBRGreen PCR Master Mix from Applied Biosystems (Foster City, CA, USA). Stock solutions of arsenite (100 mg L−1), lead (1000 mg L−1) and 3-NPA (500 mM) were prepared in sterile saline (0.9%). 6-OHDA stock solution (30 mM) was prepared in sterile saline and ascorbic acid (0.2 mg mL−1) to prevent oxidation.Cell culture
PC12 cells (ATCC, Manassas, VA) were expanded in complete or proliferation media (RPMI 1640 with 10% horse serum (HS), 5% fetal bovine serum (FBS), 2 mM glutamine and penicillin/streptomycin 0.1% mix) on 10 μg mL−1 PLL-coated Petri dishes, until 80% confluence. The cell passages compared in this work were 5 and 16. For the differentiation protocol 50 ng mL−1 of NGF were added, while HS and FBS were removed from the media. Under both conditions, proliferation and differentiation, media were changed every other day and cells were incubated at 37 °C in a 5% CO2 water-jacketed incubator.Cell cycle analysis
3 × 105 cells per well were seeded in 6-well PLL-coated plates. After 48 h in proliferation media two wells were fixed and media were changed for differentiation for the rest of the wells. At days +3 and +6 two further wells were fixed. The fixation process was carried out as follows: cells in wells were trypsinized and washed once with PBS 0.01 M, vortexed, fixed with cold 70% ethanol and stored at −20 °C for a minimum of 24 hours. After centrifugation, the fixed cells were incubated in propidium iodide/RNase staining buffer for 30 minutes at room temperature under light-protection. After this, the samples were analyzed in an FACSCalibur cytometer (Becton Dickinson, Franklin Lakes, NJ) and the results plotted.RNA isolation and reverse transcription
RNA extractions were performed from three separate experiments from duplicate cultures of PC12 cells in proliferation and after 3 days with 50 ng mL−1 NGF. To extract RNA, cells were scratched, washed with PBS 0.01 M and centrifuged. Then, 1 mL of trizol was added to the pellet followed by 300 μL of chloroform, incubated at room temperature 3 minutes and centrifuged. The aqueous phase containing RNA was recovered and mixed with 500 μL isopropanol to precipitate RNA overnight at 4 °C. After centrifugation, RNA was resuspended in 75% ethanol, centrifuged again and allowed to dry under sterile conditions. The dried RNA was finally resuspended in 20 μL DEPC treated water and quantified. For reverse transcription, 1 μg of RNA was used. The reaction tubes contained oligo dT, 1× buffer, dNTP's mix, DTT, RNasin and RT Superscript II in a final volume of 25 μL. Resultant cDNA was aliquoted in microtubes (50 μL) of 100 ng μL−1 cDNA each.Quantitative real-time polymerase chain reaction
The primers were designed from GenBank sequences for tyrosine hydroxylase (accession number NM_012740) and for neuron-specific enolase (accession number NM_139325). The primers used were TH forward (5′-TCC-GCC-ATG-CCT-CCT-CAC-CT-3′), TH reverse (5′-CGC-TGG-ATG-GTG-TGA-GGG-CT-3′), NSE forward (5′-CAT-CGA-GCG-GGC-AGT-GGA-GGA-3′), NSE reverse (5′-GTT-GTG-TCC-CGC-GAA-GCG-AGC-3′), 18 s rRNA forward (5′-CGG-CTA-CCA-CAT-CCA-AGG-AA-3′) and 18 s rRNA reverse (5′-GCT-GGA-ATT-ACC-GCG-GCT-3′). The fragment length for TH, NSE and 18 s rRNA amplicons was 490 bp, 301 bp and 180 bp respectively. The primers were resuspended to a concentration of 20 μM. Real-time quantitative PCR reaction samples were performed with a final volume of 10 μL, containing 5 μL SYBRGreen PCR Master Mix, 1 μL template cDNA (100 ng μL−1), 0.1 μL of each forward and reverse primer (20 μM) and DEPC treated water (3.8 μL). 40 cycles were performed in a Step One™ real-time thermocycler (Applied Biosystems, Foster City, CA) with an annealing temperature previously defined at 60 and 62 °C for TH and NSE respectively. Fluorescence data were acquired at the end of each cycle. A final melt analysis was run to determine specific amplifications.Real-time qPCR data analysis
Ct raw values obtained from each gene were normalized to constitutive 18 s expression to give a ΔCt value that was used for comparison and statistical purposes. For graphical representation of data, the fold variation was determined using the 2−(ΔΔCt) formula according to Livak and Schmittgen, where the ΔΔCt value represents the difference between the ΔCt values for each gene between a chosen reference – early passages and proliferation values – and a test sample – late passages and differentiation values,20 and the range was determined with the formula 2−(ΔΔCt ± ΔCtSEM). Logarithmic values were shown. The Ct mean values were obtained from two PCR replicates derived from RNA extractions from duplicate cultures and three independent experiments.Cell viability assay
The resazurin reduction method was used for viability determination. The method's principle is based on reduction of resazurin, a normally non-fluorescent compound, to resorufin, a fluorescent metabolite, due to the highly reducing milieu of a living cell.21 The assay was performed in 96-well plates. Previously, the number of cells to seed, resazurin concentration and incubation time were determined for PC12 cells. Two different incubation times (2.5 and 4 hours) and 3 resazurin concentrations (10, 20 and 30%) were tested at 4 different numbers of seeded cells (5, 10, 20, and 30 × 103 cells). The results of 2 independent experiments with 4 replicates each were plotted in order to find the conditions that assured the best parameters for reproducibility and linearity between fluorescence and number of cells. After establishing the best conditions, 2 × 104 cells were seeded on 96-well plates in proliferation media for 48 hours in order to allow the attachment of cells; then, a vehicle or a toxicant diluted in proliferation media was added (5, 10, 50, 100, 500 and 1000 μg L−1 for sodium arsenite; 5, 10, 50, 100, 500 and 1000 μg dL−1 for lead acetate; 100, 250, 500, 1000, 2500 and 5000 μM for 3-NPA and 50, 75, 100, 150, 200 and 300 μM for 6-OHDA) for 24 hours. After exposure, the cells were incubated for 2.5 hours in fresh media containing 30% resazurin (30 μg mL−1). The fluorescence of each condition (directly proportional to the number of cells metabolically active) was measured at 560 nm (excitation)/590 (emission) nm in a FlexStation II™ fluorometer (Molecular Devices, Sunnyvale, CA) and results of three independent experiments with triplicates were analyzed and plotted.Immunocytochemistry
For the immunocytochemistry experiments, 3 × 104 cells were plated onto 30 μg mL−1 PLL-coated sterile glass coverslips in proliferation media. After 48 hours in adherence, differentiation was induced by changing to differentiation media containing 50 ng mL−1 NGF. Coverslip duplicates were fixed at days 0, 3 and 6 of differentiation. The day of fixation the glass coverslips were carefully removed to another plate and washed in PBS 0.01 M, 10 minutes twice, followed by 15 minutes contact with 4% p-formaldehyde. After three washes with PBS 0.01 M, the cells were incubated with sodium borohydride for 5 minutes, washed 3 times again and incubated in blocking buffer (10% horse serum, 0.25% Triton X-100 in PBS 0.01 M) for 2 hours. Then, a mouse monoclonal anti-tyrosine hydroxylase antibody (1:1000) was incubated overnight at 4 °C. The next day, the cells were incubated for 2 hours at room temperature with a rabbit anti-mouse FITC-labeled secondary antibody (1:500, AbD Serotec), rinsed 3 times with PBS and finally incubated with bis-benzimide (2 μg mL−1) for 15 minutes. After a final wash, coverslips were dried and mounted on a Vectashield (Vector Labs, Burlingame, CA) and stored at 4 °C for 12 hours until image capture. Coverslip edges were sealed with enamel.
Image analysis
Random images from seven fields per coverslip with duplicates were taken at 40× using an Olympus BX41 fluorescence microscope (Tokyo, Japan) equipped with a Rolera-XR Fast 1394 camera (Q-imaging, Canada). Once documented, the numbers of cells positive to TH with respect to the total cells in the field were counted.Statistical analysis
To assess viability differences between groups, two-way ANOVA was used, where the first factor was the group (early and late) and the second factor was the toxicant concentration; Bonferroni post-test analysis was done in the case of significant difference within groups. For qPCR data analysis, ΔCt values were compared with Mann–Whitney's U test (PE vs. DE, PL vs. DL, PE vs. PL and DE vs. DL) with a significance level set at p < 0.025. For graphical purposes 2−(ΔΔCt) and 2−(ΔΔCt ± SEMΔCt) formulas were used instead (representing mean fold-change expression and ranges, respectively, for each of the groups mentioned). For cell cycle synthesis (S) phase analysis two-way ANOVA was performed where the first factor was the group (early and late) and the second factor was the days after NGF exposition; Bonferroni post-test analysis was done in the case of significant difference within groups. For immunocytochemistry statistical analysis only the presence or the absence of positive cells was reported, because quantitative analysis was not possible (see below). All analyses were performed using Graph Pad Prism software 4.0 (San Diego, CA, USA) and the results were considered significant at p < 0.05.Results
Morphological changes
Cells in proliferation showed no classical round and small somas described for non-differentiated naive cells. Instead, flat, heterogeneous and fibroblast-like cells filled the plates. When trying to reproduce morphological differentiation with NGF, we did not achieve the changes reported for these cells (cell morphology did not change with respect to non-stimulated cells), but some acquired a more pronounced bipolar morphology, extending short and thick prolongations, with the absence of ramifications or neurite–neurite contacts reported for differentiated cells. Moreover, many cells detached when NGF was added, the effect that was slightly greater in late than in the early passage group. The early group had initially more rounded somas and less fibroblastoid cells than the late group (Fig. 1). | ||
| Fig. 1 Morphologic changes after 3-day exposure to 50 ng mL−1 nerve growth factor in PC12 cells with altered phenotype. In the first column (panels a, c and e) early passage cells show an heterogeneous morphology and some fibroblast-like cells; occasionally cells with a well established network of neurites and ramifications are seen, and some appear to form contacts, these cells have a round soma and appear more homogeneous than the surrounding ones (arrows). In the second column (panels b, d and f) late passage PC12 cells are shown, also with a heterogeneous morphology as above and with response to NGF absent, there are many more cells with a fibroblast-like morphology, which appear to develop short neurites but without ramifications or nodes (arrowhead). In both groups abundant cells appear round and detaching, and most cells adhere very tight to the plate, spreading their borders to achieve a flat, ovoid morphology with fainting edges. Representative photomicrographs taken in an inverted phase contrast microscope with 20× lens. Scale 50 μm. | ||
Viability assays
Viability assays for the compounds tested are shown in Fig. 2. The viability tests were performed in proliferating cells. For sodium arsenite, 74.5% of variation in viability was explained by the compound concentration [F(1,6) = 51.68, (p < 0.0001)] and 3.2% variation was explained by the passage group [F(1,7) = 13.31, (p = 0.0005)]; Bonferroni post-test analysis revealed that decrease in viability was earlier and more pronounced in late than in early passages, because viable cells start to fall in 500 μg L−1 (p < 0.001) in the late group and until 1000 μg L−1 (p < 0.001) in the early group, with respect to the corresponding negative control; differences between early and late groups were evident only in 500 (p < 0.05) and 1000 μg dL−1 (p < 0.001, Fig. 2, upper left panel). For lead acetate no decrease in viability nor differential susceptibility between passages was found in the concentrations tested, despite a significant passage source of variation (11.2% [F(1,6) = 9.91, (p = 0.0024)], Fig. 2, upper right panel). After 24 h exposition to 3-nitropropionic acid (100–5000 μM) significant variation was found only due to passage [F(1,6) = 10.02, (0.0023)] and 3-NPA concentration [F(1,6) = 3.74, (0.0028)], explaining 9.4 and 21.1% of variation respectively; after Bonferroni post-test analysis the late group seems to be more susceptible to toxic insult, because viability decreased significantly starting from 1000 μM (p < 0.05) while in the early group no evidence of toxicity was noticed; no differences between passages (early vs. late) were found (Fig. 2, lower left panel). Viability to 6-hydroxydopamine was clearly affected in both groups, and the percentages of total variation found were 1.9 and 89.4% for passage [F(1,6) = 18.27, (p < 0.0001)] and 6-OHDA concentration [F(1,6) = 140.9, (p < 0.0001)] respectively. Viability for early and late groups starts to fall from 75 μM (p < 0.001 and p < 0.05 respectively), and differences between early and late groups were found only in 100 (p < 0.05) and 150 μM (p < 0.05), with the early group being more affected. The concentration at which we found 50% viability was around 100 and 150 μM for the early and late groups respectively. Even though there was a tendency for the early group to be more susceptible to the toxic effects of 6-OHDA at all concentrations tested over 75 μM, those differences did not reach statistical significance in every condition due to data dispersion (Fig. 2, lower right panel).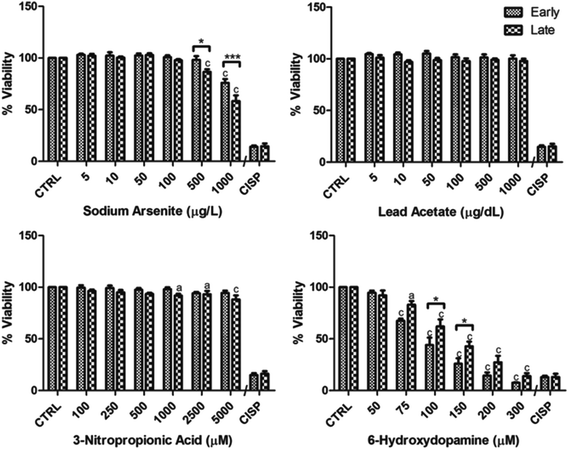 | ||
| Fig. 2 Viability assays in early and late passage groups of cells exposed to arsenic, lead, 3-NPA and 6-OHDA. (a) Upper left panel, exposure to sodium arsenite (5–1000 μg L−1) for 24 h cause a decrease in viability earlier in the late (500 μg L−1) than in the early group (1000 μg L−1) as compared to their corresponding controls. (b) Upper right panel, no alteration in viability was found between groups in the concentrations tested for lead acetate (5–1000 μg dL−1). (c) Lower left panel, viability after 24 h of 3-nitropropionic acid (100–5000 μM) exposure shows a late group slightly more susceptible, starting to decrease viability around 1000 μM 3-NPA with respect to the early passage group, where no decrease in viability was observed, and no differences between early and late groups were noticed. (d) Lower right panel, 24 h exposition to 6-hydroxydopamine (50–300 μM) causes a decrease in viability that is earlier and more pronounced in the early than in the late group, for differences between the early and the late group only in 100 and 150 μM significance are reached due to data dispersion. Data analyzed by means of 2-way ANOVA and Bonferroni post test analysis when significant values were obtained (* = p < 0.05 and *** = p < 0.001 (for differences between early and late groups); a = p < 0.05 and c = p < 0.001 (for viability decrease with respect to the control for the corresponding group)). CTRL = negative control, CISP = positive control (200 μM cisplatin). Results from 3 independent experiments with triplicates, bars represent mean ± SEM. | ||
Cell cycle analysis
One of the characteristics of PC12 cells is the decrease in the proportion of cells in S and G2/M phases of the cell cycle upon NGF exposure concomitantly with the starting of cell differentiation.22 The results obtained through our experiments were in agreement with these, since both early and late groups of mutant cells have a decrease in the proportion of cells in S and G2/M phases. For S phase analysis, time and passage were the source of variation (F(1,2) = 123.4 (p < 0.0001) and F(1,2) = 10.30 (0.0032) respectively), with significant differences between early and late groups found only at 3 days of NGF exposure (p < 0.05, two-way ANOVA with Bonferroni post-test analysis, Fig. 3). Even though at day 6 no significant difference is observed, the tendency clearly remains. For G2-M phase analysis significant variation was found only for the time factor (F(1,2) = 53.13 (<0.0001), two-way ANOVA, not shown).Tyrosine hydroxylase and neuron-specific enolase relative expression
For the relative mRNA quantification of NSE and TH expression, two conditions (0 or 3 days of NGF) for each group (early or late) were performed, and we found that NSE expression was not modified for any of the variables utilized (Fig. 4, upper panel), but TH expression was almost lost in the late group independently of NGF exposition—proliferation early vs. proliferation late, p = 0.0095; and differentiation early vs. differentiation late, p = 0.0238; according to Mann–Whitney's U test (Fig. 4, lower panel). | ||
| Fig. 3 Percentage of cells in the synthesis (S) phase for the early and late passage groups after 0, 3 and 6 days of NGF exposition. Cells were fixed prior and after 3 and 6 days stimulation of NGF, stained with propidium iodide and counted in a cytometer to determine cells in G0-G1, S and G2-M phases of the cell cycle. Only S phase are showed. The percentage of cells in the S phase decreased significantly with respect to day 0 (p < 0.001), but no differences were observed between days 3 and 6. Differences between early and late passages were found only in day 3 after NGF exposition (p < 0.05). Bars represent mean ± SEM. Data obtained from 3 independent experiments with culture duplicates. * = p < 0.05 (early vs. late), c = p < 0.001, compared to day 0, two-way ANOVA and Bonferroni post-test when significant values were obtained. | ||
Immunocytochemistry
When we evaluated the expression of tyrosine hydroxylase enzyme by immunocytochemistry in random fields, we found no cells positive for the enzyme in any of the groups; however, analyzing the entire coverslip to specifically find a marked cell, we found a few small clusters, but only in the early group, with no change with the addition of NGF (Fig. 4). The cells marked for TH appear more homogeneous in morphology (rounded and small somas, cell borders with high contrast and clusters with small number of cells) than the surrounding cells, and these clusters respond, developing neurites and establish contacts with other cells and neurites when stimulated with NGF (Fig. 5, panels g–i, and not shown) . We also evaluate tubulin ßIII expression in all early and late groups, with and without NGF, but the marker was not expressed in any of the conditions tested (not shown). | ||
| Fig. 4 Relative expression of neuron-specific enolase and tyrosine hydroxylase mRNA by quantitative real-time PCR. In the upper panel NSE expression is showed, with no change in the groups and conditions tested. The cells retain their neuronal phenotype. In the lower panel mRNA levels for TH fall significantly in the late group with respect to the early one independently of NGF exposure (p = 0.0095 for PE vs. PL, and 0.0238 for DE vs. DL, Mann–Whitney's U test). Statistical comparisons were made with ΔCt values. Graphics were derived from the formula 2-(ΔΔCt) for the mean fold-change expression. Ranges calculated with 2-(ΔΔCt ± SEMΔCt). PE = proliferation early, PL = proliferation late, DE = differentiation early, DL = differentiation late. The Y-axis scale is logarithmic to show the low levels of expression in late groups. Bars = mean ± ranges. Data obtained from n = 3–5, with PCR and culture duplicates. * = p < 0.05, ** = p < 0.01. | ||
Discussion
The purpose of in vitro assays with neuronal cell lines is to serve as primary models of screening for potential neurotoxics, in order to decide about the pertinence of further testing using more complex models and based on the relative uniformity and high throughput the cellular lines provide.4–6 Nevertheless, cells are living organisms that evolve depending on the environmental conditions, and even for a well characterized and widely used cell line such as PC12, many reports describe abnormal or mutant phenotypes since the late seventies, shortly after these cells became a useful research tool.12–15,23–27 The scientific literature clearly shows that the responses obtained from a heterogeneous, altered phenotype cell line lack uniformity and are not reproducible. Previous reports of viability to each of the compounds tested are reported in PC12 cells with different outcomes (Table 1). For arsenic, decreased viability to about 50% was obtained with 750 μg L−1 in 24 h,28 but we obtained this viability only in late passage cells with a slightly high arsenic concentration (1000 μg L−1). For lead, reports of viabilities around 80% are described for concentrations of 20–200 μg dL−1 in 24 h,29,30 and even for 3000 μg dL−1 in 72 h,29,31 but we obtained no decreased viability at the concentrations tested (5–1000 μg dL−1). For the mitochondrial toxin 3-nitropropionic acid, wide viability outcomes are reported too, from cells with high susceptibility, e.g. 60% viability with 20 μM in 24 h32 and 90% viability with the same 20 μM,33 to cells with high resistance with concentrations between 4000 and 8000 μM to obtain <60% viability in 24–48 h;34,35 in our study from concentrations of 1000–5000 μM and only in late passage cells we found a minimal decrease in viability to around 90%. Finally, for 6-OHDA, a widely used toxin for catecholaminergic cells, no less variation in results was found in the literature. Some authors used low doses (25 μM per 24 h and 20 μM per 6 h) for achieving 60–70% viabilities,36,37 others, concentrations between 75 and 150 μM for 24 h to obtain viabilities between 60 and 40%38–40 and even some obtained 50% viabilities with 150 μM in 15 minutes41 or 1000 μM in 12 h.42 Our results showed a viability around 50% with concentrations between 100 and 150 μM. Interestingly, in one of these works, a photomicrography of PC12 cells after 7 days of NGF treatment is shown, and the cells have a fibroblast-like morphology, flat appearance and scarcely appearing neurites.40 Another important internal source of variation arises when we compare results obtained in early passages to those obtained in old passages, as in this protocol, comparing passages 5 and 16 we found that late passages were more susceptible to 3-NPA and arsenite while early passages were more vulnerable to 6-OHDA.There are two interrelated hypotheses to explain change in the phenotype of PC12 cells. Some reports propose that in older passages there is a selection of cells that lose the functional expression of the high affinity NGF receptors conferring a more resistant phenotype to adverse conditions with every passage.14,15,43–45 The second hypothesis sustains that mutant PC12 cells express a mitogenic protein of the Wnt family that reset them into a more undifferentiated or primitive state in neural crest derived cells such as PC12.12,14,15,43–49 The cells we used had no developed neurites even after 2 weeks of NGF stimulation (not shown), and analyzing the synthesis phase (S) of the cell cycle we found that a residual proportion of cells remain in proliferation even after 6 days of NGF stimulation (Fig. 3) with late passage cells having a more proportion of cells than the earlier ones.
There have been reports of mutant PC12 cells lacking tyrosine hydroxylase and norepinephrine transporter expression since the late eighties,23,24 an important fact because PC12 are mainly catecholaminergic cells and also because expression of TH mRNA and enzyme is a characteristic of naive PC12 cells.13,50 According to this, our results show a lack of expression of tyrosine hydroxylase mRNA, but no changes in expression of neuron-specific enolase, which implies that the neuronal phenotype is not lost. When we evaluate the enzyme by immunocytochemistry, no positive cells were observed when random fields were analyzed, however, with a more detailed inspection we found isolated clusters of cells positive to TH enzyme but only in early groups and in very low numbers (around 10–15 clusters in the whole coverslip) independently of NGF exposition and even some of them developed short neurites after 6 days of NGF treatment. The morphology of these cells was more homogeneous than the surrounding negative cells, with a round, small nucleus and soma, non-flat phenotype and with evidence of physical contact between cells or prolongations derived from them (Fig. 5, lower panels). These results were unexpected, since the expression of the enzyme controlling catecholamine synthesis is distinctive of PC12 and other catecholaminergic cells.
 | ||
| Fig. 5 Immunocytochemistry to tyrosine hydroxylase enzyme after 0, 3 and 6 days of exposure to 50 ng mL−1 nerve growth factor in PC12 cells with altered phenotype. The first two rows images (panels a–f) were acquired from random field search and no positive cells were found. When the search was directed to find positive cells, occasionally clusters of cells positive for the enzyme were found but only and scarcely in the early group. The number of clusters did not change with NGF exposition, but cells appear to respond and develop neurites (panels g–i). Representative photomicrographs taken in a fluorescence microscope with 40× lens. Scale 50 μm. green = tyrosine hydroxylase, blue (Hoechst) = cell nucleus. | ||
Conclusions
With this work, we demonstrate the importance of evaluating the right identity of cell lines before any experiment, and common practices as sharing cell aliquots between laboratories needs to be done with reserves. Cells evolve and even cell lines appear to do so, therefore there would be a good practice to reevaluate cell phenotype with some regularity.Conflict of interest statement
The authors declare that there are no conflicts of interest.Funding
Consejo Nacional de Ciencia y Tecnología (student grant 372299), Fondos Concurrentes para Investigación UASLP (CR11-FRC-09-24.24), grants CONACYT Ciencia Básica 105937 and PIFI-2010-24MSV0011E-13.Acknowledgements
The authors thank Dr García-Sepúlveda for comments on the manuscript. Also they thank N López, ME Martínez, A Varela and YG Cataño for their technical support.References
- U. S. EPA (U. S. Environmental Protection Agency), The U.S. Environmental Protection Agency's Strategic Plan for Evaluating the Toxicity of Chemicals, 2009. [accessed: 08/01/12]. Available: http://www.epa.gov/spc/toxicitytesting/.
- U. S. GAO (U. S. Government Accounting Office), Chemical Regulation: Options Exists to Improve EPA's Ability to Assess Health Risks and Manage its Chemical Review Program. GAO-05-458, 2005. [accessed: 06/14/12]. Available: http://www.gao.gov/new.items/d05458.pdf.
- NRC (National Research Council), Toxicity Testing in the 21st Century: A Vision and a Strategy, 2007. [accessed: 08/01/12]. Available: http://books.nap.edu/openbook.php?record_id=11970.
- N. M. Radio and W. R. Mundy, Neurotoxicology, 2008, 29, 361–376 CrossRef CAS.
- K. S. Kang and J. E. Trosko, Toxicol. Sci., 2011, 120(Suppl 1), S269–S289 CrossRef CAS.
- E. J. Tokar, W. Qu and M. P. Waalkes, Toxicol. Sci., 2011, 120(Suppl 1), S192–S203 CrossRef CAS.
- N. M. Radio, J. M. Breier, T. J. Shafer and W. R. Mundy, Toxicol. Sci., 2008, 105, 106–118 CrossRef CAS.
- L. A. Greene and A. S. Tischler, Proc. Natl. Acad. Sci. U. S. A., 1976, 73, 2424–2428 CrossRef CAS.
- J. L. Connolly, L. A. Greene, R. R. Viscarello and W. D. Riley, J. Cell Biol., 1979, 82, 820–827 CrossRef CAS.
- J. D. Pollock, M. Krempin and B. Rudy, J. Neurosci., 1990, 10, 2626–2637 CAS.
- T. F. Martin and R. N. Grishanin, Methods Cell Biol., 2003, 71, 267–286 CrossRef CAS.
- M. A. Bothwell, A. L. Schechter and K. M. Vaughn, Cell, 1980, 21, 857–866 CrossRef CAS.
- D. N. Dixon, R. A. Loxley, A. Barron, S. Cleary and J. K. Phillips, In Vitro Cell. Dev. Biol., 2005, 41, 197–206 CAS.
- D. D. Eveleth and R. A. Bradshaw, J. Cell Biol., 1992, 117, 291–299 CrossRef CAS.
- S. H. Green, R. E. Rydel, J. L. Connolly and L. A. Greene, J. Cell Biol., 1986, 102, 830–843 CrossRef CAS.
- J. Xiao, Q. Zhou and Y. Liu, J. Neurosci. Res., 2002, 69, 104–109 CrossRef CAS.
- C. D. Toscano and T. R. Guilarte, Brain Res. Rev., 2005, 49, 529–554 CrossRef CAS.
- K. Jomova, Z. Jenisova, M. Feszterova, S. Baros, J. Liska, D. Hudecova, C. J. Rhodes and M. Valko, J. Appl. Toxicol., 2011, 31, 95–107 CAS.
- Z. Pang and J. W. Geddes, J. Neurosci., 1997, 17, 3064–3073 CAS.
- K. J. Livak and T. D. Schmittgen, Methods, 2001, 25, 402–408 CrossRef CAS.
- J. O'Brien, I. Wilson, T. Orton and F. Pognan, Eur. J. Biochem., 2000, 267, 5421–5426 CrossRef CAS.
- B. B. Rudkin, P. Lazarovici, B. Z. Levi, Y. Abe, K. Fujita and G. Guroff, EMBO J., 1989, 8, 3319–3325 CAS.
- J. K. Andersen, M. B. Zhang, X. H. Zhong, Y. Y. Rozenberg and B. D. Howard, J. Neurochem., 1990, 55, 559–567 CrossRef CAS.
- C. M. Bitler, M. B. Zhang and B. D. Howard, J. Neurochem., 1986, 47, 1286–1293 CrossRef CAS.
- Y. Shoji-Kasai, M. Morishima, R. Kuwahara, S. Kondo, M. Itakura and M. Takahashi, Biochim. Biophys. Acta, 2001, 1499, 180–190 CrossRef CAS.
- A. Suzuki and Y. Tsutomi, Toxicol. In Vitro, 2000, 14, 337–343 CrossRef CAS.
- R. Van Buskirk, T. Corcoran and J. A. Wagner, Mol. Cell Biol., 1985, 5, 1984–1992 CAS.
- S. Frankel, J. Concannon, K. Brusky, E. Pietrowicz, S. Giorgianni, W. D. Thompson and D. A. Currie, Neurotoxicology, 2009, 30, 529–537 CrossRef CAS.
- A. M. Sharifi, S. H. Mousavi, M. Bakhshayesh, F. K. Tehrani, M. Mahmoudian and S. Oryan, Toxicol. Lett., 2005, 160, 43–48 CrossRef CAS.
- J. Xu, L. D. Ji and L. H. Xu, Toxicol. Lett., 2006, 166, 160–167 CrossRef CAS.
- J. Xu, L. D. Ji and L. H. Xu, Environ. Toxicol., 2010, 25, 55–60 CAS.
- Z. Wei, S. Chigurupati, P. Bagsiyao, A. Henriquez and S. L. Chan, Neurotox. Res., 2009, 16, 14–29 CrossRef CAS.
- S. J. Lee, Y. C. Youn, E. S. Han and C. S. Lee, Neurochem. Res., 2005, 30, 1191–1200 CrossRef CAS.
- E. M. Kawamoto, M. Gleichmann, L. M. Yshii, S. Lima Lde, M. P. Mattson and C. Scavone, Braz. J. Med. Biol. Res., 2012, 45, 58–67 CrossRef CAS.
- B. S. Mandavilli, I. Boldogh and B. Van Houten, Brain. Res.: Mol. Brain Res., 2005, 133, 215–223 CrossRef CAS.
- S. Eminel, A. Klettner, L. Roemer, T. Herdegen and V. Waetzig, J. Biol. Chem., 2004, 279, 55385–55392 CrossRef CAS.
- K. S. Ha, K. M. Kim, Y. G. Kwon, S. K. Bai, W. D. Nam, Y. M. Yoo, P. K. Kim, H. T. Chung, T. R. Billiar and Y. M. Kim, FASEB J., 2003, 17, 1036–1047 CrossRef CAS.
- M. K. Takata, F. Yamaguchi, K. Nakanose, Y. Watanabe, N. Hatano, I. Tsukamoto, M. Nagata, K. Izumori and M. Tokuda, J. Biosci. Bioeng., 2005, 100, 511–516 CrossRef CAS.
- L. Tao, X. Li, L. Zhang, J. Tian, X. Li, X. Sun, X. Li, L. Jiang, X. Zhang and J. Chen, PLoS ONE, 2011, 6, e26055 CAS.
- N. Yamamoto, H. Sawada, Y. Izumi, T. Kume, H. Katsuki, S. Shimohama and A. Akaike, J. Biol. Chem., 2007, 282, 4364–4372 CrossRef CAS.
- K. Hanrott, L. Gudmunsen, M. J. O'Neill and S. Wonnacott, J. Biol. Chem., 2006, 281, 5373–5382 CrossRef CAS.
- M. Wang, Z. Zhang, L. C. Cheang, Z. Lin and S. M. Lee, Chin. Med., 2011, 6, 16 CrossRef.
- K. Fujita, P. Lazarovici and G. Guroff, Environ. Health Perspect., 1989, 80, 127–142 CrossRef CAS.
- M. T. Kasaian and K. E. Neet, Neurochem. Res., 1990, 15, 1167–1174 CrossRef CAS.
- B. A. Yankner and E. M. Shooter, Proc. Natl. Acad. Sci. U. S. A., 1979, 76, 1269–1273 CrossRef CAS.
- A. H. Chou and B. D. Howard, Oncogene, 2002, 21, 6348–6355 CrossRef CAS.
- A. H. Chou, S. Zheng, T. Itsukaichi and B. D. Howard, Brain. Res.: Mol. Brain Res., 2000, 77, 232–245 CrossRef CAS.
- G. M. Shackleford, K. Willert, J. Wang and H. E. Varmus, Neuron, 1993, 11, 865–875 CrossRef CAS.
- M. Ikeya, S. M. Lee, J. E. Johnson, A. P. McMahon and S. Takada, Nature, 1997, 389, 966–970 CrossRef CAS.
- K. S. Kim, D. H. Park, T. C. Wessel, B. Song, J. A. Wagner and T. H. Joh, Proc. Natl. Acad. Sci. U. S. A., 1993, 90, 3471–3475 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2013 |