HepG2/C3A 3D spheroids exhibit stable physiological functionality for at least 24 days after recovering from trypsinisation
Krzysztof Wrzesinski†a, Maria Chiara Magnone†b, Line Visby Hansenc, Marianne Ehrhorn Krused, Tobias Bergauere, Maria Bobadillab, Marcel Gublerb, Jacques Mizrahib, Kelan Zhangd, Christina M. Andreasenf, Kira Eyð Joensena, Signe Marie Andersena, Jacob Bastholm Olesena, Ove B. Schaffalitzky de Muckadellg and Stephen J. Fey*a
aDepartment of Biochemistry and Molecular Biology, University of Southern Denmark, Odense, Denmark. E-mail: sjf@bmb.sdu.dk; Fax: +45 6550 2467; Tel: +45 6550 2440
bpRED, Pharma Research and Early Development, DTA CVM, F. Hoffmann-La Roche, Basel, Switzerland. E-mail: maria_chiara.magnone@roche.com; Fax: +41 61 68 79141; Tel: +41 61 20 10500
cScience and Technology, Aarhus, Denmark. E-mail: linevh@orion.au.dk; Tel: +45 7455 2400
dDrugMode ApS, Southern Denmark Research Park, Odense, Denmark. E-mail: kelanzhang@hotmail.com; Fax: +45 6315 7801; Tel: +45 6315 7800
epRED, Pharma Research and Early Development, Translational Research Sciences (TRS), F. Hoffmann-LaRoche, Basel, Switzerland. E-mail: tobias.bergauer@roche.com
fDepartment of Orthopaedics and Traumatology, Odense University Hospital, Odense, Denmark. E-mail: christina.moeller.andreasen@ouh.regionsyddanmark.dk; Tel: +45 5160 5534
gDepartment of Medical Gastroenterology, Odense University Hospital, Odense, Denmark. E-mail: sdm@ouh.regionsyddanmark.dk; Fax: +45 6611 1328; Tel: +45 6541 2750
First published on 16th January 2013
Abstract
Primary human hepatocytes are widely used as an in vitro system for the assessment of drug metabolism and toxicity. Nevertheless a cell system with higher stability of physiological functions is required for the investigation of drugs’ mode of action, pathway analyses and biomarkers evaluations. We recently discovered that the human hepatocellular carcinoma cell line, HepG2/C3A, cultured as spheroids in a 3D system can recover their main functions after trypsinisation within about 18 days. The objective of this study was to investigate whether the spheroids’ metabolic functions remained stable after this recovery period. Therefore we evaluated physiological capabilities of the spheroids (cell survival, growth rate, glycogenesis, ATP, cholesterol and urea synthesis and drug metabolism) and the expression of key genes related to the main liver pathways in spheroids cultured for an additional 24 days after full recovery (day 18). Here we show that after the recovery period, the 3D spheroid culture can provide a metabolically competent homeostatic cell model which is in equilibrium with its culture environment for more than 3 weeks. Such a stable system could be used for the assessment of the drugs’ mode of action, for biomarkers evaluation and for any systems biology studies which require medium- to long-term stability of metabolic functions.
Introduction
The liver is a central organ for drug metabolism and therefore in vitro hepatocytes models (both primary cells and cell lines) are widely used for evaluating the safety and efficacy of compounds and their mode of action on specific pathways. These studies have illustrated that primary hepatocytes have limited stability in their physiological functions in vitro, and that hepatocyte cell lines do not fully reflect liver functions.1–3 An in vitro system is required that recapitulates liver functions in vitro and that maintains those functions for a sufficient amount of time for testing the mid- and long-term effects of compounds.In a recent study, we compared cells grown in a classical 2D cell culture and 3D spheroid formats in otherwise identical culture conditions. We reported that the 3D spheroid culture systems allow cells to recover from trypsinisation and re-establish their advanced functions (such as cellular organisation, cholesterol and urea synthesis and cytochrome P450 expression) within 15–18 days. During the recovery process, the cells in the spheroids are engaged in re-establishment of their key functions and their growth rate slows down. The cells undergo a clear transition approximately 18 days after culture initiation after which the cell activities stabilise. Before transition the amounts of ATP, urea, cholesterol and adenylate kinase all increase. After the transition, the amounts of adenylate kinase and cholesterol secreted fall while ATP and urea increase (at least up to day 21 at which timepoint that study was ended).4
In this study we investigate the question as to whether the cell functions recovered after the first 21 days in culture are maintained from day 21 to day 42. Few studies have followed the physiological capacity of immortal cells for six weeks post trypsinisation and investigated whether the 3D spheroid system is able to respond to physiological and pharmacological stimulations in a way that reproduces in vivo liver functions.
The parameters that have been investigated in this study were general cell viability (growth rate, ATP synthesis and adenylate kinase release), basal physiological capacity as illustrated by glucose metabolism, urea production, cholesterol synthesis and response to pharmacological treatment (inhibition of cholesterol production by lovastatin). We have also evaluated the expression of key genes involved in the liver functions under evaluation (e.g. glycogen and glucose metabolism, cholesterol synthesis, urea cycle, bile acids synthesis and transport) to confirm the stability of the molecular pathways behind the physiological functions.
All of these assays, without exception, demonstrate that, once the spheroids have passed through the transition (which occurs about 15–18 days after culture initiation), they exhibit a very stable functionality for at least a further 24 days. Thus 3D spheroids cultivated in the microgravity ProtoTissue™ bioreactor provide a biological system ‘at rest’ or in equilibrium where a minimum of background ‘noise’ from extraneous cellular processes is of value. The spheroid system provides a useful tool for drug discovery for understanding compounds’ mode of action and for the evaluation of pharmacodynamics and/or mechanistic biomarkers.
Materials and methods
For the sake of comparability, spheroid preparation and determination of growth, protein content, ATP, urea, cholesterol and glycogen were carried out by following exactly the same procedures as reported previously.4Standard culture conditions
The immortal human hepatocellular carcinoma cell line, HepG2/C3A (ATCC CRL-10741), was grown in standard tissue culture conditions (D-MEM (containing 1 g glucose L−1)).Spheroid culture conditions (3D culture)
Protein determination in spheroids
The amount of protein present in a sample was determined using the fluorescence-based ProStain Protein Quantification Kit (Active Motif Inc., cat. no. 15001). There were duplicate biological replicates and nine technical replicates.Growth rate and protein content estimation in spheroids
Spheroids were photographed on days 1, 3, 6, 8, 10, 13, 15, 17, 21, 25, 34 and 42 post culture initiation and the area of on average 181 spheroids measured individually at each time point and used to calculate the protein content of the spheroid. Briefly photomicrographs were taken using an Olympus IX81 motorized microscope and an Olympus DP71 camera. The ‘shadow’ area of spheroids was measured using the ‘Fitted Polygon Area’ function.6ATP assay for cell viability
Samples of the hepatocytes grown as spheroids (usually 6–2 spheroids per assay point depending on the age of the culture) were collected and cell viability was measured using the CellTiter-Glo luminescent cell viability assay (Promega, cat. no. G7571). The data were normalized with reference to a standard curve for ATP, to the untreated control and to the amount of cellular protein present.Adenylate kinase assay for cell viability
Adenylate kinase in spent media was measured using the bioluminescent cytotoxicity assay kit (Lonza Toxilight assay kit, cat. no. LT07-117). Media from spheroid cultures were collected in triplicate in connection with refreshing the growth media (typically at 48 h intervals).Glycogen synthesis in basal and insulin-stimulated conditions
Spheroids were serum starved for 20 h in DMEM with 5.5 mM glucose, 0.1% fatty acid free-BSA, 10 mM HEPES and 50 IU ml−1 penicillin–streptomycin. After starvation, the medium was completely aspirated, a small sample of spheroids was used for quality assessment and Trypan Blue staining to ensure that the viability was still >95%. The remaining spheroids were divided into 18 separate bioreactors (approximately 30 mg wet weight spheroids/bioreactor). Spheroids were treated with 0 or 100 nM Humulin® in either DMEM with 5.5 or 11.1 mM glucose containing 1.6 or 3.2 μCi ml−1D-[U-14C]-glucose respectively and cultivated for 2.5 h. Experiments were set up in quadruplicate, except for the blank control samples (where no radioactivity was added) that was set up in duplicate. After treatment, spheroids were washed three times in 37 °C PBS and lysed in 30% KOH by shaking. After 15 minutes, an equal volume of double distilled H2O was added. A small aliquot was withdrawn from each cell lysate and used for protein determination by the Bradford method.7Cell lysates were incubated at 95 °C for 1 h and then cooled in ice. 2 mg unlabeled glycogen and 1 ml 95% ethanol were added to each sample and they were left overnight at −20 °C to precipitate the glycogen. Samples were spun at 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 500g for 10 minutes and the supernatant was aspirated. Precipitates were solubilized by shaking in formic acid. 1 ml OptiPhase Hisafe 2 liquid scintillation cocktail was added and samples were mixed thoroughly. The amount of radioactive material in each sample was quantified using a Wallac 1450 microbeta scintillation counter. The glycogen content of the individual samples was related to their total protein content.
500g for 10 minutes and the supernatant was aspirated. Precipitates were solubilized by shaking in formic acid. 1 ml OptiPhase Hisafe 2 liquid scintillation cocktail was added and samples were mixed thoroughly. The amount of radioactive material in each sample was quantified using a Wallac 1450 microbeta scintillation counter. The glycogen content of the individual samples was related to their total protein content.
In a parallel experiment, the influence of the glucose concentration in the media was investigated. The above procedure was repeated except that the incubation was carried out in DMEM with 5.5, 11.1 or 25 mM glucose containing 1.6, 3.2 or 7.3 μCi ml−1D-[U-14C]-glucose respectively and cultivated for 2.5 h. No additional insulin was added.
Urea assay
Urea was measured using the Urea Assay Kit (Biovision, cat. no. K375-100) according to the manufacturer's instructions. Briefly, samples of growth media from duplicate cultures spheroids were centrifuged to remove any cellular debris and used for urea determination. A standard curve was prepared in the same way for each assay plate using a urea standard (Biovision, cat. no. 700623).Cholesterol assay
Cholesterol in the spent media was measured using the Amplex Red Cholesterol Assay Kit (Invitrogen, cat. no. A12216) according to the manufacturer's instructions with slight modifications. Positive, negative and standard curves were added to each reading plate.Gene expression
RNA was purified using the RNeasy Minikit cat. no. 74104 and RNase-Free DNase Set cat. no. 79254 both from Qiagen according to the manufacturer's instructions. Cells in the spheroids were disrupted by adding 350 μl ‘RLT buffer’ plus 350 μl 70% ethanol and vortexing. The sample was then transferred to the RNeasy column and centrifuged at 8000g for 15 s at room temperature. RNase free DNase 1 (80 μl) was added to the column and incubated at room temperature for 15 min. The column was then washed by adding 350 μl RLT buffer and centrifuged at 8000g for 15 s at room temperature. This was repeated with ‘RPE buffer’ (500 μl) twice where the second spin was prolonged to 2 min. Following this the RNeasy column was transferred to a new microtube and centrifuged at 11000g for 1 min. The RNeasy column was once again transferred to a new microtube and the RNA was eluted by adding 50 μl H2O and centrifuged at 8000g for 1 min. The elution wash was repeated once and the eluate was pooled. Absorbance was measured at 230, 260 and 280 nm in a spectrophotometer and the quality was approved if the ratios 260/280 and 260/230 were both greater than 1.8.Reverse transcription of RNA was accomplished using the SuperScript III First-Strand Synthesis Kit (Invitrogen) according to the manufacturer's protocol with a total RNA input of 100 ng per reaction. cDNA samples were then diluted 1:3 in TE buffer and 14 cycles of pre-amplification were carried out using 2× TaqMan PreAmp Master Mix (Applied Biosystems) and pooled Taqman Assays (Applied Biosystems) at a final concentration of 0.2× per assay. The following thermocycler program was used: 95 °C for 10 min, followed by 14 cycles at 95 °C for 15 s and 60 °C for 4 min. Pre-amplified cDNA products were diluted 1:5 in TE buffer. qPCR was performed using the 96.96 dynamic array (Fluidigm Corporation, CA, USA) following the manufacturer's protocol (Fluidigm Quick Reference Card, PN 68000130, Rev. B). Briefly, for each sample a 5 μl sample mix was prepared with 1× GE Sample Loading Reagent (Fluidigm), 1× Taqman Gene Expression Mastermix (Applied Biosystems) and diluted, pre-amplified cDNA. For the assay mix 1× Assay Loading Reagent (Fluidigm) was mixed with each of the Taqman Assays (final concentration: 10×), respectively. Priming of the Fluidigm array with a control line fluid and mixing of the sample and assay reagents were done with an IFC controller. qPCR was performed using a BioMark Instrument with the following cycling parameters: 95 °C for 10 min, followed by 40 cycles at 95 °C for 15 s and 60 °C for 1 min. Data were collected and analyzed with Real-Time PCR Analysis Software (Fluidigm Corporation, CA, USA).
Inhibition of cholesterol synthesis by lovastatin
Spheroids (20 mg total protein) were treated with final concentrations of 0, 1 or 10 μM lovastatin, 0.5% DMSO in DMEM for either 30 min or 18 h in 10 ml bioreactors.Cholesterol synthesis was measured by then adding 7 μCi ml−1 [1–14C]-sodium acetate (Perkin Elmer, cat. no.: NEC084H001MC) to triplicate 3D cultures in bioreactors and incubating for 4 h. The cholesterol content of the individual samples was related to their total protein content determined from a small sample of the NaOH spheroid lysate.7
Data and statistical analysis
In all assays, except for gene expression, data were normalized with reference to the amount of protein present, either determined directly where possible or by estimation when a direct determination was not possible. A single HepG2/C3A cell was assumed to contain 0.2 ng protein.For gene expression normalization was performed using geometric mean expression of housekeeping genes GUSB, H6PD and ALAS1: ΔCq = Cq gene of interest − Cq geomean Housekeepers. The Relative Quantification methodology (ΔCq) was used to calculate expression changes over time relative to day 21.
For the statistical analysis of the effect of glucose concentration and lovastatin treatment, the two-tailed Student's t-test was used. For analysis of variance of cholesterol synthesis at the different time points, one-way analysis of variance (ANOVA) with Tukey error protection was used. For analysis of variance of glycogen synthesis stimulated by insulin at the different time points two-way ANOVA was performed. Analyse-it Method Evaluation Tool version 2.12 by Analyse-it Software, Ltd was used to aid statistical analysis.
Results
The aim of this study was to determine whether hepatocytes cultured as 3D spheroids could stably maintain their functionality over 3 weeks after the recovery transition and could stably respond to pharmacological treatment and physiological stimulation. For this reason we examined a series of cellular functions specific to hepatocytes.Growth rate
Growth rate of the spheroids was evaluated by following the morphological changes and size increase until day 42. The averaged starting size for the spheroids was 200 μm on day 0, 900 μm on day 21 and 1400 μm on day 42 (Fig. 1A–D). To provide a more accurate evaluation of the growth rate, protein content was measured. As shown in Fig. 1E the doubling time increased from day 21 to day 42 showing that the cell growth rate was slowing down.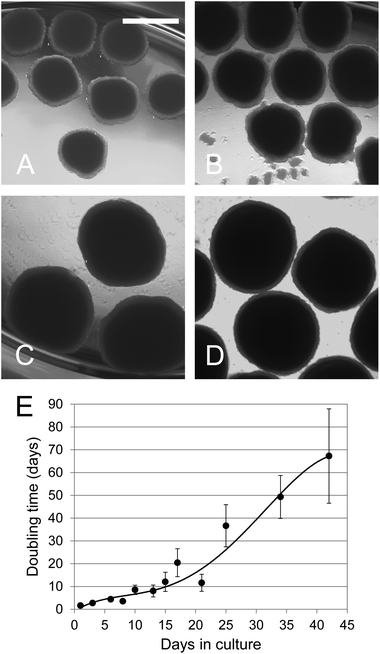 | ||
| Fig. 1 Growth of C3A spheroids during 42 days. (A–D) Photomicrographs of spheroid cultures at 21, 28, 35 and 42 days respectively. The bar in (A) indicates 1 mm. (E) Cell growth in 3D cultures. Spheroids were cultivated in the ProtoTissue™ bioreactor. Duplicate bioreactors were terminated at different times and the protein content of on average 170 spheroids was estimated at each time point for each bioreactor using the relationship between their shadow area and protein content. The growth rate was determined from the incremental increase in the total protein content of the bioreactor. The error bars indicating standard deviation are in some cases smaller than the symbol. | ||
Spheroid viability
ATP and adenylate kinase levels were followed to assess the viability of the spheroids and measurements normalized per microgram of protein to correct for increases in cell numbers.During the first 21 days the amount of ATP increased. Thereafter the ATP levels stabilised and remained constant (Fig. 2A). The data for adenylate kinase showed that the percentage of dead cells is also essentially constant (Fig. 2B). Both assays thus indicate that the viability of the cultures between day 21 and 42 remains constant, illustrating that the falling growth rate was not due to a reduction in viability.
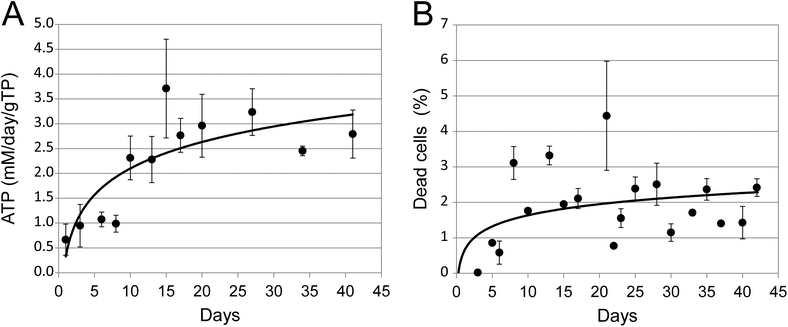 | ||
| Fig. 2 Viability of spheroids in long term cultures. (A) The total amount of ATP present in quadruplicate 3D cultures. Data were normalised to mM per day per gram of total cellular protein (gTP). (B) The percentage of dead cells present in quadruplicate 3D cultures, as estimated by adenylate kinase release. Error bars indicate standard deviation and are in some cases smaller than the symbol. | ||
Basal 3D spheroids functionality
To assess the functionality of the spheroids between day 21 and day 42, we selected three physiological functions of hepatocytes: glucose metabolism, cholesterol synthesis and urea production. These analyses were carried out at intervals during the 3 week study (typically at day 21, 28, 35 and 42 post culture initiation). All analyses were normalised to the amount of protein present in the samples. Measurements of cholesterol and urea metabolism were non-invasive and thus could be performed repeatedly on the same culture while the glucose metabolism assay required separate cultures for each data point.Glucose metabolism
The rate of glycogen synthesis was assayed at different concentrations of glucose in the medium. Spheroids were provided with radioactive glucose (at a constant specific activity) and glycogen synthesis was quantitated by measuring the amount of radioactive glucose incorporation into glycogen. The results (Fig. 3) showed that glycogen synthesis was roughly proportional to the increase in glucose concentrations in the media, with a steady increase from 5.5 to 25 mM.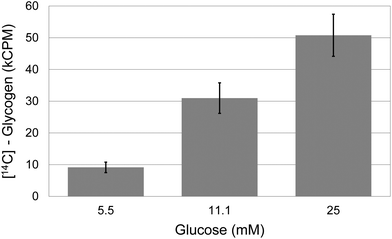 | ||
| Fig. 3 Glycogen synthesis in spheroids. Relationship between the glucose concentration in the growth medium and glyconeogenesis in quadruplicate samples of 21 day old spheroids. Error bars indicate standard deviation. | ||
Urea and cholesterol synthesis and secretion
Two important functions of the liver are the synthesis and secretion of urea and cholesterol. Spent media was assayed for urea or cholesterol secretion at multiple times during the study period. The amount of urea produced was somewhat variable but still appeared essentially constant at about 1.6 ± 0.35 mM per day per gTP (grams of total cellular protein) averaged over the observation period of 21 days (Fig. 4A). The gene expression level for arginase (Arg1), the enzyme catalysing the hydrolysis of arginine to ornithine and urea, was correspondingly stable over the 3 weeks period (Table 1). As can be seen from Fig. 4B, the amount of cholesterol secreted was reasonably constant at about 30 μg per day per gram of cellular protein. This is consistent with the observation that the gene expression levels of HMG-CoAR were stable over the 3 weeks (Table 1).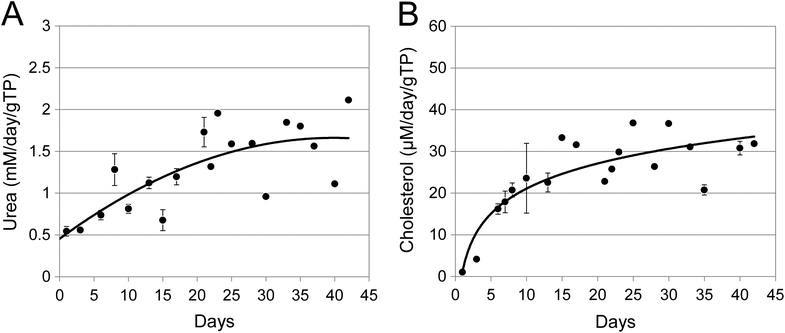 | ||
| Fig. 4 Urea and cholesterol production in long term spheroid cultures. The growth media was collected from duplicate ProtoTissue™ bioreactors, clarified by centrifugation and the amount of urea (A) or cholesterol (B) present quantitated at the times shown. Data were normalized to mM or μM per day per gram of total cellular protein (gTP). The data from day 0 to day 21 for both urea and cholesterol were previously presented in ref. 4. Error bars indicate standard deviation and are in some cases smaller than the symbol. | ||
| Gene name | Biological function | Normalized median expression (at day 21) | Expression categorya | Relative normalized expression % (compared to day 21 = 100%) | ||
|---|---|---|---|---|---|---|
| Day 28 | Day 35 | Day 42 | ||||
| a For convenience they have been grouped into three expression groups: high: ΔCq (−16 ≤ x < −6); medium: ΔCq (−6 ≤ x < 6); low: ΔCq (6 ≤ x < 16). | ||||||
| SLC2A2 (GLUT2) GS | Glucose import and glycogen synthesis | 1.2 | Medium | 90 | 98 | 95 |
| High | 91 | 85 | 80 | |||
| FBP1 | Gluconeogenesis | 4.8 | Medium | 81 | 75 | 92 |
| G6PC | −2.5 | Medium | 98 | 98 | 98 | |
| PCK2 | −0.6 | Medium | 96 | 94 | 97 | |
| ARG1 | Urea production | 7.0 | Low | 87 | 93 | 90 |
| HMGCR SLCO1B1 (OATP1B1) | Cholesterol biosynthesis | −1.8 | Medium | 96 | 91 | 97 |
| Lovastatin transport | −15.9 | High | 91 | 98 | 90 | |
| SLCO1A2 | Bile acid import/export and synthesis | −15.9 | High | 95 | 98 | 97 |
| CYP7A1 | 6.6 | Low | 88 | 91 | 91 | |
| SLC10A1 | 12.0 | Low | 84 | 75 | 63 | |
| CYP3A4 | Drug metabolism | 10.2 | Low | 68 | 59 | 61 |
| CYP1A1 | Paracetamol detoxification | 5.4 | Medium | 84 | 90 | 77 |
| CYP1A2 | 4.6 | Medium | 95 | 89 | 90 | |
| ALB | Liver specific genes | −9.3 | High | 95 | 96 | 97 |
| F7 | 0.9 | Medium | 94 | 97 | 89 | |
| FABP1 | −5.6 | High | 94 | 98 | 89 | |
| TF | −7.0 | High | 92 | 94 | 96 | |
It is important to note that the steady state rates of synthesis for both urea and cholesterol were significantly above the synthesis rates seen during the first few days of culture.
Gene expression
Hepatocytes in the liver are the primary site for the synthesis of many of the most abundant proteins in the blood. Gene expression analysis performed here confirmed that RNAs for several of these proteins (albumin, factor VII, the fatty acid binding protein and transferrin) were all expressed at moderate or high levels. In addition, the expression of several genes related to key pathways in hepatocytes functionality was evaluated over the 3 weeks period (Table 1). All genes showed detectable expression levels and maintained a stable pattern from day 21 to day 42 (data normalized to the expression level on day 21).3D spheroids functionality under physiological and pharmacological stimulations
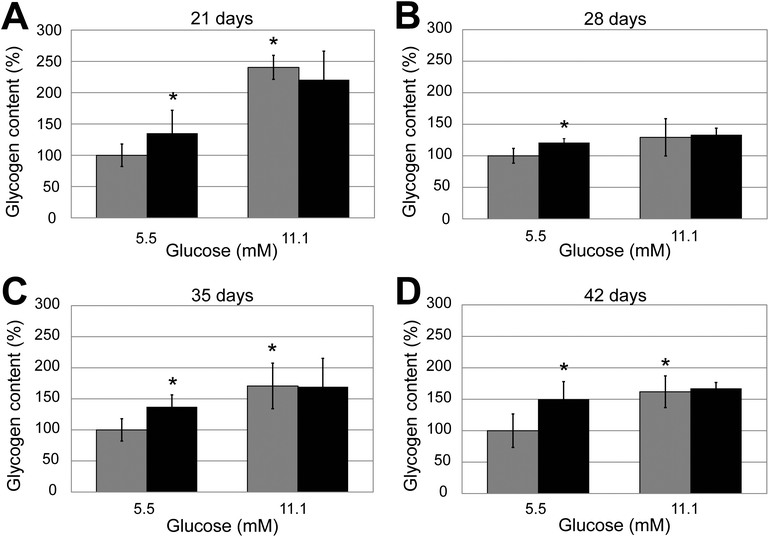 | ||
| Fig. 5 Effect of insulin on glycogen synthesis in long term spheroid cultures. Quadruplicate spheroid cultures of different ages (A: 21 days; B: 28 days; C: 35 days and D: 42 days) were grown in the presence of either 5.5 or 11 mM glucose and treated or mock treated with 100 nM insulin. Results have been normalised to the level of glycogen synthesis in the control mock-treated spheroids grown in 5.5 mM glucose. Error bars indicate standard deviation. | ||
A comparison of the increase in glycogen synthesis comparing spheroids grown in 11.1 compared to 5.5 mM glucose showed that the difference was statistically significant (Student's t-test) for the 21, 35 and 42 day cultures (p = 0.03, 0.01 and 0.01 respectively, indicated in the figure by the * over the black bars) but not at 28 days (p = 0.12).
| Spheroid age (days) | Treatment (h) | Lovastatin | Student's t-test | |||
|---|---|---|---|---|---|---|
| 0 μM | 10 μM | |||||
| CPM | SD | CPM | SD | p | ||
| 21 | 4.5 | 38281 | 3068 | 462 | 113 | 0.002 |
| 42 | 4.5 | 37557 | 7646 | 7452 | 11939 | 0.020 |
| Spheroid age (days) | Treatment (h) | Lovastatin | Student's t-test | |||
|---|---|---|---|---|---|---|
| 0 μM | 1 μM | |||||
| CPM | SD | CPM | SD | p | ||
| 35 | 22 | 56077 | 16810 | 110636 | 19185 | 0.020 |
Discussion
Human hepatocytes cultures are commonly used in drug development for a range of functional assessments (ADME, metabolic functions, biomarker and pathways analysis). Nevertheless it is known that primary hepatocytes in culture do not retain some of the key functions they exert in vivo using the classical cell culture conditions.The main difference between cells cultured using the classical culture conditions (in the bottom of microtitre plates, flasks etc., i.e. ‘flat or 2D culture’ conditions) and spheroids (true 3D culture) is the fact that the cells can grow undisturbed in a 3D environment (i.e. without suffering damage by trypsinisation), receive an active supply of nutrients as the growth media ‘flows past’ and they can develop tight interactions with other cells and recover functionality typical of normal hepatocytes. This allows the cells to develop several ultrastructural and physiological features that do not normally have time to develop when the same cells are grown in otherwise the same conditions using classical cell and tissue culture procedures. These features include tight junctions, microvillae, bile canaliculae-like tubules, glycogen granules, urea and cholesterol secretion. Judged by their morphologic appearance, growth rate, the production of ATP, adenylate kinase, cholesterol and urea, the spheroids need at least 18 days to recover.4 These observations would be fully compatible with the recent report that sandwich-cultured rat hepatocytes need 6 days to establish cell polarity and bile canaliculi8 because in the spheroid system presented here, the hepatocytes first have to synthesise their own extracellular matrix (which is otherwise provided in the sandwich-culture models) before they do the same.
The outstanding question examined in this report concerns the functional stability of hepatocyte spheroids after the recovery process and their reactivity to physiological and pharmacological stimulations. In particular, we have investigated basal 3D spheroid activities (cell growth and expression of liver-specific genes) and four of the core functions performed by the liver: glycogen synthesis and response to insulin stimulation; urea production, cholesterol production and its inhibition by lovastatin. In addition, we have measured the expression of some key genes related to hepatocyte specific functions. These results have shown that the features developed by the spheroids are stable for at least 21 days once they have been established (i.e. spheroids are metabolically stable from day 21 to 42 in culture). This period of stability is probably much longer because spheroids have been cultivated in our laboratories without trypsinisation for up to 302 days (at which time point the culture still looked perfectly healthy).
Cell growth and gene expression
The investigation of the basic functions of the 3D spheroid system showed that between 21 and 42 days of culture the cells are able to proliferate and maintain their viability. This is particularly relevant because of the growing modality of the cultures which may limit oxygen and nutrient availability to the cells in the inner part of the spheroids and impact the validity of the functional assessments.In addition, gene expression analysis showed that the cells in the 3D spheroids express a wide set of liver-specific proteins, thus indicating that they are in a fully differentiated state.
Finally, 3D spheroids also express genes of key pathways related to liver functions, which make them amenable for exploratory investigations such as biomarker discovery and drug mode of action studies.
Glycogen synthesis
One of the primary functions of the liver is to assist in the regulation of blood sugar by converting glucose to glycogen (glycogenesis) at times when the blood sugar level is high and convert it back again when blood sugar levels are low. The anabolic synthesis is regulated in part by the hormone insulin which is secreted into the blood stream from the pancreas upon food intake. Glucose uptake into the liver is facilitated by SLC2A2 (GLUT2, a passive, high capacity low-affinity transporter) shown by gene expression to be expressed at expectable, stable levels throughout the 21 day study period.9 Glucose is then phosphorylated by the enzyme hexokinase, effectively trapping the glucose-6-phosphate inside the cell. In the liver, glucokinase has a low affinity for glucose (Km of about 10 mM), and is not inhibited by glucose-6-phosphate. This allows hepatocytes to accumulate large amounts of glucose 6-phosphate when blood glucose concentrations are high.10 In our spheroid cultures we indeed observed an increased glycogen synthesis depending on the concentration of glucose in the medium (from 5.5 to 25 mM). Once in the cells, glucose can enter a number of metabolic pathways including glycogen synthesis. We observed a stable pattern of expression of glycogen synthase from day 21 to 42. In addition, we observed a stable increase in glycogen synthesis in response to insulin at 5.5 mM glucose concentration in the medium. Insulin binding regulates the glycogen production via the activation of protein kinase β. This kinase indirectly induces the dephosphorylation and activation of glycogen synthase (increasing the rate of glycogen synthesis) and dephosphorylates and inactivates glycogen phosphatase (decreasing the rate of glycogen breakdown).11 Interestingly, an increase in glycogen synthesis was not observed at 11.1 mM glucose concentration in the medium. It is tempting to speculate that the non-response to insulin in conditions of high extracellular glucose mimics a condition of relative insulin resistance as the one observed in type 2 diabetes. However additional parameters (e.g. phosphorylation of insulin receptor and downstream factors) should be evaluated to gather a more complete picture.In addition to the reactivity of the system to insulin in terms of glycogen synthesis, we evaluated the expression of key genes in the gluconeogenesis pathway, such as glucose 6-phosphatase, fructose biphosphatase and phosphoenolpyruvate carboxykinase, and we observed a similar level of expression from day 21 to 42, indicating the maintenance of gluconeogenic pathway over 3 weeks (Table 1).
Overall, our data indicate that the spheroids maintain their functionality in glucose and glycogen metabolism from day 21 to 42. Our data are partially in contrast with a study by Dabos et al., where they found significant changes in a number of parameters (including glucose, lactate, pyruvate, amino acids, urea and ethanol production) during a 21 day spheroid culture of primary porcine hepatocytes. Interestingly they described a progressive switch in the metabolism from anaerobic (and active gluconeogenesis) to aerobic (with restricted gluconeogenesis) during days 7–10.12 The observed differences may reflect intrinsic differences in growth potential between the decline of primary cells and immortal cell lines in culture.
Urea
The urea cycle is the main pathway allowing the detoxification from ammonia and it is one of the main functions of the liver. One key enzyme of the urea cycle is arginase 1 (Arg1) that converts arginine in ornithine and urea. In vivo, urea production is in the region of 0.74 mM urea per day per gTP (grams of total protein)4 and both primary hepatocytes and HepG2/C3A cells have been shown to produce similar amounts (0.55 mM per day per gTP13,14).One key feature of hepatocytes cell lines grown in 2D systems, such as HepG2 and HepG2/C3A cells, is that they have lost the capability to detoxify ammonia, in that they have a non-functional urea cycle (HepG2 cells do not express Arg115), and the urea that they do produce (0.160 mM per day per gTP) is produced via Arg2 (HepG2/C3A cells13,16). The level of urea production of HepG2/C3A cells was shown to be sensitive to glucose levels, being highest (1.7 mM per day per gTP) in a glucose-free media.14 In our 3D spheroid system, urea production between day 21 and 42 is in the range of 1.6 ± 0.35 mM per day per gTP. This system stably expresses Arg1 at low levels, thus suggesting that ammonia detoxification may be possible.
Cholesterol
Another of the essential functions of the liver is to produce cholesterol. The liver synthesizes about 25% of the body's daily requirement,17 and this corresponds to a daily production of about 5.5 μM per gram of hepatocyte protein.4 Cholesterol is used in part to maintain cell membrane fluidity and in part as an essential precursor of a number of important molecules such as steroid hormones, fat soluble proteins and bile acids. Cholesterol is synthesised from acetate by several steps, taking place in the endoplasmic reticulum and the mitochondria, in the mevalonate pathway. The first part of the reaction pathway occurs in the cytosol, but the irreversible and rate limiting step from 3-hydroxy-3-methylglutaryl-CoA (HMG CoA) to mevalonate is catalysed by the HMG-CoA reductase (HMG-CoAR). Because of this, compounds affecting HMG-CoAR activity are widely used in the clinic to regulate the synthesis of cholesterol.17 The cholesterol lowering group of drugs, the ‘statins’, resemble the HMG part of HMG-CoA and have a much higher affinity for the active site of the HMG-CoAR, enabling them to act as competitive inhibitors.18 Furthermore, the mevalonate pathway is controlled by feedback inhibition of HMG-CoAR and HMG-CoA synthase by the end product, cholesterol. This regulation is important for the hepatocyte to balance the internal and external sources of cholesterol in order to sustain mevalonate synthesis and at the same time avoid steroid accumulation.19Cholesterol synthesis was not stable and reached a peak at around day 10 in Hep2 cells cultured for 21 days on alginate spheroids.20 In our spheroid system, the cholesterol levels appear constant between day 21 and day 42. Consistently, HMG-CoAR gene expression levels do not change throughout the three week period.
Out of the 12 transporters (SLCO1A2, SLCO2B1, SLCO1B1, SLCO3A1, SLCO4A1, SLCO1B3, SLC22A7, SLC22A8, SLC22A1, SLC10A1, SLC15A1, and SLC15A2) investigated in HepG2 cells, Libra found 11 expressed. Of these, the gene expression of SLCO1B1, SLCO3A1, and SLCO1B3 was greatly repressed, while the expression of SLCO2B1, SLC22A7, and SLC22A8 was either maintained or increased in HepG2 in comparison to their expression in human liver.21 Currently it is not clear whether the high expression of SLCO1A2 and SLCO1B1 seen here is a specific difference between the HepG2 cells and HepG2/C3A or whether the spheroid culture has induced their expression. Several other transporters have also been shown to be expressed and active in HepG2 and HepG2/C3A cell lines. These include P-gp (MDR1 or ATPB1), and MRP2 (cMOAT),22–24 and several amino acid transporters (SLC7A11, SLC1A4, and SLC3A2).25
Interestingly the increasing amounts of cholesterol seen following the recovery period would be expected to induce an increase in SCL10A1 and SCL22A1 (but not of SCLO1A2 (OATP1A2) or CYP3A4).26 This would be a logical preparation for bile synthesis and secretion. The results observed here: low expression of SCL10A1, CYP3A4 and high expression of SCLO1A2 therefore present an incomplete picture.
It is interesting to note that the same HepG2/C3A cells, which in 2D systems show several limitations (e.g. low cholesterol production, no urea cycle, reduced cytochromes function), recover the functionality typical of human liver in the 3D spheroid system.
Technical limitations of 3D spheroids
There are some technical limitations in working with our 3D spheroid system. First of all the classical microtitre plates format used with 2D systems is not applicable to the spheroids, thus implying a lower throughput of the experimental setting. Second, the 3D spheroid system is less amenable to microscopy and high content imaging as compared to 2D systems, which makes the direct visualisation of cells or organelles more complicated.Finally, a general limitation shared between our 3D spheroid system and other 3D and 2D systems that are composed of primary hepatocytes or immortalized cell lines is that they do not capture the interactions with other relevant liver cell types, such as Kupffer cells, stellate cells and endothelial cells, which are functional partners of hepatocytes in the liver.
Nevertheless, the 3D spheroid system shows a number of advantages that largely outweighs the limitations. First of all, this culturing system allows HepG2/C3A cells to recover some key functions that are typical of the liver and that are lost in 2D systems. Whether this is due to the resuming of the differentiation program or to the simple fact that cells can recover undisturbed for a longer time is not clear at this point in time. Certainly, using HepG2/C3A cells provides a practically unlimited amount of identical starting material, which ensures a greater biological homogeneity as compared to primary hepatocytes.
Conclusions
Spheroids constructed from the human HepG2/C3A hepatocellular carcinoma cell line show a number of characteristics on the ultrastructural, expression and functional levels, which suggests that they mimic the function of the human liver and that these attributes are reproducible and stable over at least the 3 weeks used as the study period.In addition, the 3D spheroid system allows the cells to be functionally stable for several weeks (in a pilot study spheroids were cultured for up to 302 days), thus enabling long-term studies.
The low biological variability and the long-term stability make this system particularly useful for the investigations of the compounds mode of action, toxicology and for biomarker discovery. Further developments (e.g. deep focus confocal microscopy) and novel handling procedures will pave the way for the introduction of 3D culture into the mainstream of biology.
Acknowledgements
Part of this study was generously supported by F. Hoffmann-La Roche and by MC2 Biotek A/S.Notes and references
- S. Wilkening, F. Stahl and A. Bader, Drug Metab. Dispos., 2003, 31, 1035–1042 CrossRef CAS.
- S. N. Hart, Y. Li, K. Nakamoto, E. A. Subileau, D. Steen and X. B. Zhong, Drug Metab. Dispos., 2010, 38, 988–994 CrossRef CAS.
- M. Lubberstedt, U. Muller-Vieira, M. Mayer, K. M. Biemel, F. Knospel, D. Knobeloch, A. K. Nussler, J. C. Gerlach and K. Zeilinger, J. Pharmacol. Toxicol. Methods, 2011, 63, 59–68 CrossRef.
- K. Wrzesinski and S. J. Fey, Toxicol. Res., 2013 10.1039/c2tx20060k.
- K. Wrzesinski, Molecular Markers Associated with Hepatotoxicity: Development of In Vitro Test System Based on Human Cells, Vdm Verlag, Saarbrücken, Germany, 2009 Search PubMed.
- S. J. Fey and K. Wrzesinski, Toxicol. Sci., 2012, 127, 403–411 CrossRef CAS.
- M. M. Bradford, Anal. Biochem., 1976, 72, 248–254 CrossRef CAS.
- D. Fu, Y. Wakabayashi, J. Lippincott-Schwartz and I. M. Arias, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 1403–1408 CrossRef CAS.
- F. Q. Zhao and A. F. Keating, Curr. Genomics, 2007, 8, 113–128 CrossRef CAS.
- M. Larion and B. G. Miller, Arch. Biochem. Biophys., 2012, 519, 103–111 CrossRef CAS.
- M. Peak, A.-H. Molham and L. Agius, Biochem. J., 1992, 282, 797–805 CAS.
- K. J. Dabos, L. J. Nelson, T. J. Bradnock, J. A. Parkinson, I. H. Sadler, P. C. Hayes and J. N. Plevris, Biochim. Biophys. Acta, Gen. Subj., 2001, 1526, 119–130 CrossRef CAS.
- D. Mavri-Damelin, L. H. Damelin, S. Eaton, M. Rees, C. Selden and H. J. Hodgson, Biotechnol. Bioeng., 2008, 99, 644–651 CrossRef CAS.
- V. V. Iyer, H. Yang, M. G. Ierapetritou and C. M. Roth, Biotechnol. Bioeng., 2010, 107, 347–356 CrossRef CAS.
- D. Mavri-Damelin, S. Eaton, L. H. Damelin, M. Rees, H. J. Hodgson and C. Selden, Int. J. Biochem. Cell Biol., 2007, 39, 555–564 CrossRef CAS.
- C. Filippi, S. A. Keatch, D. Rangar, L. J. Nelson, P. C. Hayes and J. N. Plevris, J. Hepatol., 2004, 41, 599–605 CrossRef CAS.
- D. W. Russell, Cardiovasc. Drugs Ther., 1992, 6, 103–110 CrossRef CAS.
- J. A. Tobert, Nat. Rev. Drug Discovery, 2003, 2, 517–526 CrossRef CAS.
- J. L. Goldstein and M. S. Brown, Nature, 1990, 343, 425–430 CrossRef CAS.
- L. H. Damelin, S. Coward, S. F. Choudhury, S. A. Chalmers, I. J. Cox, N. J. Robertson, G. Revial, M. Miles, R. Tootle, H. J. Hodgson and C. Selden, Arch. Biochem. Biophys., 2004, 432, 167–177 CrossRef CAS.
- A. Libra, C. Fernetti, V. Lorusso, M. Visigalli, P. L. Anelli, F. Staud, C. Tiribelli and L. Pascolo, J. Pharmacol. Exp. Ther., 2006, 319, 809–817 CrossRef CAS.
- K. A. Wojtal, E. de Vries, D. Hoekstra and S. C. van Ijzendoorn, Mol. Biol. Cell, 2006, 17, 3638–3650 CrossRef CAS.
- J. M. Prot, C. Aninat, L. Griscom, F. Razan, C. Brochot, C. G. Guillouzo, C. Legallais, A. Corlu and E. Leclerc, Biotechnol. Bioeng., 2011, 108, 1704–1715 CrossRef CAS.
- A. Nakano, D. Tsuji, H. Miki, Q. Cui, S. M. El Sayed, A. Ikegame, A. Oda, H. Amou, S. Nakamura, T. Harada, S. Fujii, K. Kagawa, K. Takeuchi, A. Sakai, S. Ozaki, K. Okano, T. Nakamura, K. Itoh, T. Matsumoto and M. Abe, PLoS One, 2011, 6, e27222 CAS.
- J. I. Lee, J. E. Dominy Jr., A. K. Sikalidis, L. L. Hirschberger, W. Wang and M. H. Stipanuk, Physiol. Genomics, 2008, 33, 218–229 CrossRef CAS.
- V. Dias and V. Ribeiro, Fundam. Clin. Pharmacol., 2007, 21, 445–450 CrossRef CAS.
Footnote |
| † These authors equally contributed to the study. |
| This journal is © The Royal Society of Chemistry 2013 |