Engineering of a O6-alkylguanine-DNA alkyltransferase chimera and repair of O4-alkyl thymidine adducts and O6-alkylene-2′-deoxyguanosine cross-linked DNA†
Francis P.
McManus
and
Christopher J.
Wilds
*
Department of Chemistry and Biochemistry Concordia University, 7141 Sherbrooke Street West, Montreal, Canada. E-mail: Chris.Wilds@concordia.ca; Fax: +1-514-848-2424 ext. 5798; Tel: +1-514-848-2424 ext. 5798
First published on 24th January 2013
Abstract
A soluble human O6-alkylguanine-DNA alkyltransferase (hAGT) chimera was engineered containing the active site of OGT (residues 139–159) and an additional S134P mutation. The resulting hAGT chimera not only retained hAGT's ability to repair bulky O6-alkylene-2′-deoxyguanosine interstrand cross-linked DNA damage but also displayed enhanced repair of various O4-alkyl thymidine adducts.
O 4-Methyl-thymidine (O4MeT) in the genome that results from exposure to tobacco derived N-nitrosamines or chemotherapeutic agents is highly mutagenic because of its ability to form altered hydrogen bonding patterns.1 The stable O4MeT
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :dG pair resulting from a single round of replication, causes a dT to dC transition mutation, which is especially detrimental to the cell if this event occurs on proto-oncogenes or tumour suppressor genes.2
:dG pair resulting from a single round of replication, causes a dT to dC transition mutation, which is especially detrimental to the cell if this event occurs on proto-oncogenes or tumour suppressor genes.2
O
6-Methyl-2′-deoxyguanosine (O6MeG), another well known mutagenic nucleobase modification, generates dG:dC to dA:dT transition.3 Unlike its thymidine counter-part the O6MeG:dT intermediate (formed after a single round of replication) is a substrate for mismatch repair and direct repair in humans.4 The O4MeT:dG mismatch persists in the cell since it is neither a good substrate for mismatch repair nor direct repair, contributing to the mutagenic potential of O4MeT.5O4-Alkyl thymidine, which is believed to be a minor lesion, has been found at similar levels as O6MeG in the liver tissue of healthy volunteers, demonstrating its importance and prominence in the absence of exogenous alkylating agents.6
O 6-Alkylguanine-DNA alkyltransferases (AGT) are part of the direct repair pathway and are responsible for the removal of both O6-alkyl dG and O4-alkyl dT adducts.7 The repair occurs in a single step process where the active site Cys thiolate anion performs a nucleophilic attack on the α-carbon of the adduct located on the exocyclic oxygen.8 In the process the native DNA is restored and the AGT protein is irreversibly alkylated where it eventually undergoes degradation by the ubiquitin mediated pathway.9 AGT homologues show vast substrate differences. hAGT has been studied extensively and is believed to possess the greatest substrate range for O6-alkyl dG adducts. Among some of the “bulkier” substrates that have been shown to undergo repair by this protein are O6-2′-deoxyguanosine-alkylene-O6-2′-deoxyguanosine interstrand cross-links (ICL), which are DNA lesions that covalently link complementary DNA strands obstructing DNA unwinding and segregation, in the process inhibiting cell prolifieration due to physical obstruction.10
Escherichia coli OGT is believed to be the most efficient AGT at eliminating O4-alkyl dT.11 This is contrary to hAGT, which is extremely poor at removing even O4MeT. hAGT recognizes and binds O4MeT but cannot remove them efficiently, physically shielding the lesion from the nucleotide excision repair (NER) machinery and increasing the lesion's toxicity.12
The Loeb and Pegg groups have dedicated some effort to generate hAGT variants capable of increased repair of O4MeT. The Loeb laboratory employed a random sequence mutagenesis approach to generate multiple variants followed by a functional complementation assay to obtain a variant with 8 point mutations (C150Y, S152R, A154S, V155G, N157T, V164M, E166Q, and A170T).13 This variant showed a rate of repair of O4MeT that was roughly 11.5 times greater than that of hAGT.14 A horizontal gene transfer from OGT to hAGT was invoked by the Pegg group to generate their hAGT–OGT chimera. Their most promising construct harboured 8 mutations (V149I, C150G, S151R, S152N, A154T, V155M, G156T and N157G), which was achieved by substituting the hAGT amino acids by their respective residues in OGT. Unfortunately, incorporating numerous successive mutations generated a chimeric protein that was not soluble, requiring refolding procedures to be adopted. Their chimera displayed valuable properties when expressed in alkyltransferase and NER deficient E. coli cells, where reduced levels of mutations at dT:dA sites were observed upon cell exposure to methylating, ethylating and propylating agents when compared to similar cells expressing wild-type hAGT.15
In the present work, we extended the portion of OGT residues in the chimera towards the c-terminal but kept Pro 140 from hAGT to generate a soluble construct.16 The chimeras ability to repair alkylation adducts and interstrand cross-links at both the O4 atom of thymidine and O6 atom of 2′-deoxyguanosine was investigated using the substrates shown in Fig. 1, which were synthesized and characterized as previously described.
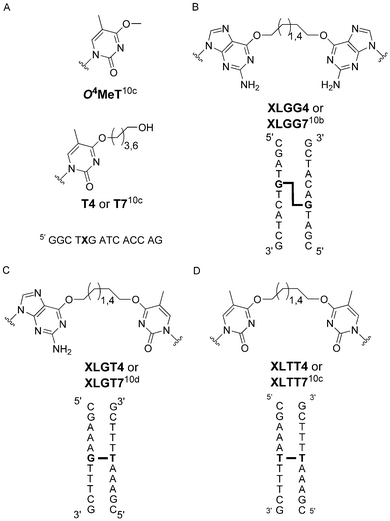 | ||
| Fig. 1 Structures of (a) O4-alkyl-thymidine mono-adducts and DNA sequence where X contains the modified residue, (b) O6-2′-deoxyguanosine-alkylene-O6-2′-deoxyguanosine interstrand cross-link and duplex where G-G is the ICL, (c) O6-2′-deoxyguanosine-alkylene-O4-thymidine interstrand cross-link and duplex where G-T is the ICL and (d) O4-thymidine-alkylene-O4-thymidine interstrand cross-link and duplex where T-T is the ICL. | ||
A slight variation of inverted PCR was employed to introduce residues 139–159 from OGT into the hAGT scaffold, while retaining Pro 140 (see Table 1 for sequences), which is known to confer hAGT with the ability to repair larger adducts such as ICL and O6-benzyl guanine.10a,b,17 The protein was expressed and purified as previously reported for hAGT.10b The protein was obtained in >90% purity as assessed by SDS-PAGE and its mass was in accordance with the calculated value, indicating no post-translational modification of the foreign protein (see ESI, Fig. 2 and 3†). The successive mutations introduced into the chimera did not substantially affect the secondary structure of the overall protein as assessed by far-UV circular dichroism, which revealed a similar signature to hAGT. The alterations destabilized the protein with respect to hAGT reducing the Tm by 7.5 °C. Characterization of the chimera by intrinsic fluorescence showed a blue shift in tryptophan emission indicating a slight change in tertiary structure. This change in fluorescence is most likely a result of a variation in the local environment of Trp 65 to a less polar position. Trp 65 is located in the N-terminal domain of the protein but packs next to the altered region and is perhaps affected by the V139I alteration.18 C145S and R135G variants of the chimera protein were created by site-directed mutagenesis, purified and characterized (see ESI, Fig. 2–6†).
| Protein | Amino acid sequence (139–159)a |
|---|---|
| a Amino acid numbering based on hAGT sequence. b Most active chimera designed in ref. 15. | |
| hAGT | VPILIPCHRVVCSSGAVGNYS |
| OGT | ISIVVPCHRVIGRNGTMTGYA |
| hAGT-03b | VPILIPCHRVIGRNGTMTGYS |
| Chimera | ![[I with combining low line]](https://www.rsc.org/images/entities/char_0049_0332.gif) P P![[V with combining low line]](https://www.rsc.org/images/entities/char_0056_0332.gif) PCHRVIGRNGTMTGY PCHRVIGRNGTMTGY![[A with combining low line]](https://www.rsc.org/images/entities/char_0041_0332.gif) |
The P140S variant (introducing Ser 140 found in OGT) of the chimera created an insoluble construct when expressed under the pQE30 vector. This protein was also expressed using pET15B and purified under denaturing conditions in order to obtain a higher protein yield to be used in refolding attempts. Previously published refolding protocols for hAGT did not yield an appreciable amount of properly folded protein.19 Dialysis mediated refolding of the P140S chimera variant was greatly aided by high salt, Tris and L-arginine. Unfortunately, the refolded protein had an improper conformation as assessed by far-UV circular dichroism and intrinsic fluorescence and was unable to repair O6MeG and O4MeT (data not shown).
The chimeras ability to repair O6MeG in a 14 bp oligonucleotide (5′ GGC TTX ATC ACC AG, where X is O6MeG) appeared to be unaffected given the repair of O6MeG occurred in less than 15 s at room temperature, as was the case with hAGT (data not shown). The alterations improved the AGTs ability to remove O4MeT in a similar oligonucleotide (5′ GGC TXG ATC ACC AG, where X is O4MeT). As observed in Fig. 2, the chimera repaired O4MeT 30 times faster than hAGT, which is comparable to results reported by the Pegg group for their insoluble constructs, where their most proficient variant had a 47-fold increase over hAGT.15 Repair of O4MeT by OGT required less than 15 s making it at least 100-fold more proficient than the chimera (data not shown).
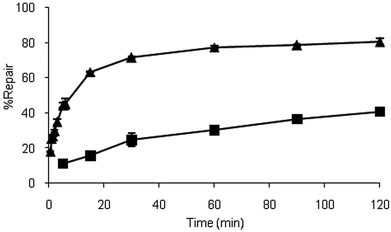 | ||
| Fig. 2 Time course dealkylation of O4MeT nucleoside at room temperature by hAGT (■) and the chimera (▲). | ||
Molecular modelling and molecular dynamics were employed to comprehend the reason for the chimeras increased ability to remove O4MeT over its human equivalent. Modelling of the chimera was performed using the hAGT-O6MeG co-crystal (PDB ID: 1t38) as template and placement of the C145 side chain was guided by the hAGT crystal structures 1yfh, 1t39 and 1eh6. O6MeG was mutated to O4MeT and the placement of O4MeT verified with PDB ID: 1yfh, as reference. Conjugate gradient minimization, simulated annealing and torsion angle dynamics were conducted using Crystallography & NMR System as done previously by our group.10c,20
The modelling data suggest the increased repair of O4MeT by the chimera may be due to changes in the shape of the loop that better accommodates the modified nucleoside as observed in Fig. 3. The N157G and S159A alterations in the chimera also make the loop more flexible resulting in a reduction in distance between the Cys 145 thiolate anion in the chimera to the methyl adduct of O4MeT by 0.65 Å relative to the native hAGT, which may be responsible for the enhanced repair observed.
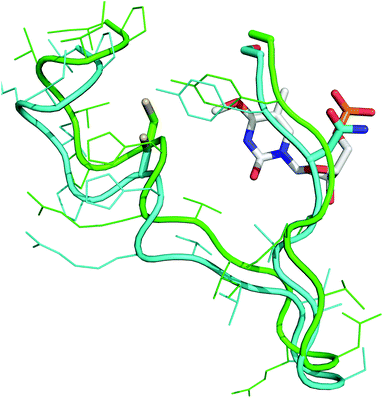 | ||
| Fig. 3 O 4MeT (white) in the active site of hAGT (cyan/grey) and the chimera (green/black). The side chains of Cys 145, Asn 157 and Ser/Ala 159 are represented as sticks. | ||
hAGT has two unfavourable interactions; Asn 157 which sterically clashes with the O4MeT sugar moiety and Ser 159 that is in close proximity with the C5 methyl of O4MeT hindering the flexibility of the loop and consequently inhibiting any penetration of O4MeT into the protein's active site.
The pre-minimized model of the chimera in complex with O4MeT revealed a potential clash between Arg 135 and the C5 methyl of the modified nucleobase. This residue is replaced by a Gly in OGT and Ala in Ada-C, suggesting that small amino acids are usually located at this position in AGTs capable of repairing O4MeT with appreciable ease. For this reason the R135G variant of the chimera was generated in hope of creating a more active variant. The repair capabilities of the chimera were compared to its R135G variant using a small library of O4-alkyl thymidine adducts. Overnight incubation at 37 °C of the AGT with the various O4-alkylated DNA revealed the R135G alteration was detrimental to the chimera. The R135G variant maintained the ability to repair O4MeT, but lost its capability to repair substrates T4 and T7 (see ESI, Fig. 7†). Repair of T4 and T7 by the original chimera was moderate, showing only 40% repair after an overnight incubation. These results are still promising since its human counterpart is unable to react with these substrates to any detectable extent.10c
hAGT is the only AGT shown to date to repair ICL DNA and several independent alterations in its amino acid sequence are known to abolish this ability. These alterations (aside from those involved in the proteins activity) include: P140A, P140K, V148L and Y158H, all of which are located in the portion of hAGT that is altered to form the chimera.10a,b To verify the effect of the alterations on the proteins ability to process ICL DNA time course repair assays with various ICL containing oligonucleotides were conducted. The time course assays revealed that the chimera is similar to hAGT and can repair ICL DNA involving the O6 atom of guanine. The repair of cross-linked substrates XLGG4 and XLGG7, where the O6 atoms of 2′-deoxyguanosine are attached by an alkylene linker, were carried out at virtually identical rates by hAGT and the chimera. The chimera, much like hAGT, repaired XLGG7 more rapidly and extensively than it did XLGG4, 50% repair in 3 h for XLGG7 as opposed to 30% repair in 8 h for XLGG4 (see ESI, Fig. 8†).
Similar repair trends were observed for the XLGT series, where the O6 atom of 2′-deoxyguanosine and O4 atom of thymidine are attached by an alkylene linker. XLGT7 was repaired with comparable rates by both AGTs whereas the repair of XLGT4 was virtually non-existent existent (see ESI, Fig. 9†). ESI-MS analysis of the repair reaction revealed the chimera selectively repairs the cross-link at the O6 atom of guanine. The repair event produces a complex comprised of the chimera covalently attached through its Cys 145 to the O4 atom of the cross-linked thymidine via the seven methylene linkage (see ESI, Fig. 10 and 11†). The same repair specificity was reported by our group for hAGT.10d
In light of the positive results with mono-adducts T4 and T7 repair assays with cross-linked DNA, where the O4-atoms of thymidine were attached by an alkylene linker (XLTT4 and XLTT7), were undertaken with the chimera. Unfortunately, the chimera was unable to repair XLTT4 nor XLTT7 (see ESI, Fig. 12†). As was the case with hAGT and OGT, XLTT4 and XLTT7 ICL DNA evaded repair by the chimera.10c The chimera bound XLTT4 and XLTT7 with low μM Kd as determined by electromobility shift assays indicating good binding of the ICL DNA by the protein. The chimera bound the XLTT DNA series more tightly than it did the XLGT, which the protein is able to partially repair, suggesting the absence of XLTT repair is not due to recognition of the substrate by the protein (see Table 2).
| DNA | K d (μM) | Protein:DNA stoichiometry |
|---|---|---|
a Binding assays were carried out using 0.5 nM dsDNA in 10 mM Tris-HCl (pH 7.6), 100 mM NaCl, 1 mM DTT, 10 μg mL−1 BSA and 2.5% glycerol at 21 °C.
b DNA sequence is 5′-CGAAA![[T with combining low line]](https://www.rsc.org/images/entities/char_0054_0332.gif) TTTCG/3′-GCTTTAAAGC.
c DNA sequence is 5′-CGAAA TTTCG/3′-GCTTTAAAGC.
c DNA sequence is 5′-CGAAA![[G with combining low line]](https://www.rsc.org/images/entities/char_0047_0332.gif) TTTCG/3′-GCTTT TTTCG/3′-GCTTT![[C with combining low line]](https://www.rsc.org/images/entities/char_0043_0332.gif) AAAGC. AAAGC.
|
||
| Control (T-A)b | 37.57 ± 6.48 | 1.91 ± 0.09 |
| XLTT4 | 2.56 ± 0.42 | 1.86 ± 0.16 |
| XLTT7 | 1.99 ± 0.12 | 1.80 ± 0.16 |
| Control (G-C)c | 3.96 ± 0.82 | 1.89 ± 0.18 |
| XLGT4 | 6.10 ± 0.10 | 2.12 ± 0.20 |
| XLGT7 | 3.88 ± 0.47 | 1.99 ± 0.13 |
The chimera interacts with dsDNA in a virtually identical fashion to hAGT, as indicated by our binding experiments. Like hAGT, the chimera displays a preference for DNA with a dG that is more than 2 nucleotides from the 5′ ends, as observed between the T-A and G-C controls. The chimera bound all DNA species with comparable dissociation constants and stoichiometry as those reported for hAGT using identical substrates.10c,d hAGT is believed to bind every 4 nucleotides where an observed stoichiometry of 2:1 hAGT:DNA has been documented for both 11-mer dsDNA and ssDNA, consistent with our findings with the chimera.21
The control DNA (11 bp) used in the binding assays had a Tm that was measured to be approximately 40 °C. For the ICL DNA, the Tm was found to be approximately 10 °C higher. In order to ensure duplex formation for the binding assays, experiments were performed at 21 °C.
Binding experiments with the R135G chimera variant showed no association of DNA in the tested range (0 to 30 μM) establishing the role of Arg 135 as a binding residue. The R135G alteration in the chimera appears to cause a decrease in binding affinity between the protein and DNA, limiting the proteins ability to repair bulkier adducts (such as T4 and T7) since they require a longer time to react with the proteins thiolate anion as a result of the distance between the reacting groups. The role of Arg 135 in the chimera is equivalent to its function in hAGT, where it is speculated that this residue is 1 of only 3 basic amino acids involved in the ionic interaction between the protein and dsDNA as well as being 1 of 2 residues involved in the interaction with ssDNA.22
Conclusions
We successfully generated a hAGT–OGT chimera that can be purified under native conditions. The chimera, containing a portion of the hAGT active site changed to the respective amino acids in OGT, displays some properties from OGT since it can repair O4MeT 30 times faster than hAGT. In addition, the chimera can remove larger adducts at the O4 atom of thymine, such as those found in T4 and T7, but does so poorly. The chimera maintained the ability to repair cross-links involving the O6 atom of guanine, a property that was previously thought to be unique to hAGT (see Table 3 for repair summary). The alterations in the primary construct were shown to have no effect on DNA binding affinity or stoichiometry, suggesting the modified residues are not involved in substrate recognition.| Lesion | hAGT | OGT | Chimera |
|---|---|---|---|
| a 10 pmol AGT with 2 pmol DNA at 37 °C. b 60 pmol AGT with 2 pmol DNA at 37 °C. (+++) >80% repair within 1 minute, (++) >80% repair within 1 h, (+) >10% repair within 16 h and (−) <10% repair within 16 h. | |||
| O 4MeTa(10c) | + | +++ | ++ |
| T4a(10c) | − | ++ | + |
| T7a(10c) | − | + | + |
| XLGG4b(10a) | + | − | + |
| XLGG7b(10a) | + | − | + |
| XLGT4b(10d) | − | − | − |
| XLGT7b(10d) | + | − | + |
| XLTT4b(10c) | − | − | − |
| XLTT7b(10c) | − | − | − |
Such a chimera may play a role in combinational chemotherapeutic regimens due to its added ability to repair undesired, highly mutagenic, O4-alkyl thymine adducts that are generated during cancer treatment and which are known to persist in mammalian cells.23 Since the chimera is based mostly on the hAGT scaffold many of the biological properties of the protein are maintained. For example, the presence of the 125-KAAR sequence responsible for targeting and maintaining the protein in the nucleus and the presence of Met 134, which is required for the steric clash that allows hAGT to be ubiquitinated and degraded is retained; the presence of the terminal regions of hAGT that impart the protein with the ability to interact with DNA with a high inter-protein cooperativity, which is required for concentrating the protein to segments of DNA that are undergoing transcription is also preserved.8,24
Acknowledgements
We are grateful to Dr Anthony E. Pegg (Penn. State University) for the plasmid encoding the wild-type hAGT gene. This research was supported by grants from the Natural Sciences and Engineering Research Council of Canada (NSERC), the Canada Foundation for Innovation (CFI) and the Canada Research Chair (CRC) program. FPM is the recipient of a postgraduate fellowship from NSERC.Notes and references
- (a) S. S. Hecht, Chem. Res. Toxicol., 1998, 11, 559–603 CrossRef CAS; (b) P. D. Lawley, D. J. Orr, S. A. Shah, P. B. Farmer and M. Jarman, Biochem. J., 1973, 135, 193–201 CAS; (c) B. Singer, Cancer Res., 1986, 46, 4879–4885 CAS; (d) M. W. Kalnik, M. Kouchakdjian, B. F. Li, P. F. Swann and D. J. Patel, Biochemistry, 1988, 27, 108–115 CrossRef CAS.
- B. D. Preston, B. Singer and L. A. Loeb, Proc. Natl. Acad. Sci. U. S. A., 1986, 83, 8501–8505 CrossRef CAS.
- E. L. Loechler, C. L. Green and J. M. Essigmann, Proc. Natl. Acad. Sci. U. S. A., 1984, 81, 6271–6275 CrossRef CAS.
- (a) D. Toorchen and M. D. Topal, Carcinogenesis, 1983, 4, 1591–1597 CrossRef CAS; (b) D. R. Duckett, J. T. Drummond, A. I. Murchie, J. T. Reardon, A. Sancar, D. M. Lilley and P. Modrich, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 6443–6447 CrossRef CAS; (c) G. Koike, H. Maki, H. Takeya, H. Hayakawa and M. Sekiguchi, J. Biol. Chem., 1990, 265, 14754–14762 CAS.
- (a) L. Samson, S. Han, J. C. Marquis and L. J. Rasmussen, Carcinogenesis, 1997, 18, 919–924 CrossRef CAS; (b) D. R. Duckett, J. T. Drummond, A. I. Murchie, J. T. Reardon, A. Sancar, D. M. Lilley and P. Modrich, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 6443–6447 CrossRef CAS.
- H. Kang, C. Konishi, T. Kuroki and N. Huh, Carcinogenesis, 1995, 16, 1277–1280 CrossRef CAS.
- (a) A. E. Pegg, Chem. Res. Toxicol., 2011, 24, 618–639 CrossRef CAS; (b) S. L. Gerson, Nat. Rev. Cancer, 2004, 4, 296–307 CrossRef CAS.
- D. S. Daniels, T. T. Woo, K. X. Luu, D. M. Noll, N. D. Clarke and A. E. Pegg, et al. , Nat. Struct. Mol. Biol., 2004, 11, 714–720 CAS.
- (a) K. S. Srivenugopal, X. H. Yuan, H. S. Friedman and F. Ali-Osman, Biochemistry, 1996, 35, 1328–1334 CrossRef CAS; (b) M. Xu-Welliver and A. E. Pegg, Carcinogenesis, 2002, 23, 823–830 CrossRef CAS.
- (a) Q. Fang, A. M. Noronha, S. P. Murphy, C. J. Wilds, J. L. Tubbs, J. A. Tainer, G. Chowdhury, F. P. Guengerich and A. E. Pegg, Biochemistry, 2008, 47, 10892–10903 CrossRef CAS; (b) F. P. McManus, Q. Fang, J. D. Booth, A. M. Noronha, A. E. Pegg and C. J. Wilds, Org. Biomol. Chem., 2010, 8, 4414–4426 RSC; (c) F. P. McManus, D. K. O'Flaherty, A. M. Noronha and C. J. Wilds, Org. Biomol. Chem., 2012, 10, 7078–7090 RSC; (d) F. P. McManus, A. Khaira, A. M. Noronha and C. J. Wilds, Bioconjugate Chem., 2013 DOI:10.1021/bc300553u.
- (a) H. Takinowaki, Y. Matsuda, T. Yoshida, Y. Kobayashi and T. Ohkubo, Protein Sci., 2006, 15, 487–497 CrossRef CAS; (b) S. R. Paalman, C. Sung and N. D. Clarke, Biochemistry, 1997, 36, 11118–11124 CrossRef CAS; (c) M. Sassanfar, M. K. Dosanjh, J. M. Essigmann and L. Samson, J. Biol. Chem., 1991, 266, 2767–2771 CAS.
- L. Samson, S. Han, J. C. Marquis and L. J. Rasmussen, Carcinogenesis, 1997, 18, 919–924 CrossRef CAS.
- L. P. Encell, M. M. Coates and L. A. Loeb, Cancer Res., 1998, 58, 1013–1020 CAS.
- (a) L. P. Encell and L. A. Loeb, Biochemistry, 1999, 38, 12097–12103 CrossRef CAS; (b) L. P. Encell and L. A. Loeb, Carcinogenesis, 2000, 21, 1397–1402 CrossRef CAS.
- Q. Fang, S. Kanugula, J. L. Tubbs, J. A. Tainer and A. E. Pegg, J. Biol. Chem., 2010, 285, 8185–8195 CrossRef CAS.
- G. Koike, H. Maki, H. Takeya, H. Hayakawa and M. Sekiguchi, J. Biol. Chem., 1990, 265, 14754–14762 CAS.
- (a) M. Crone, K. Goodtzova, S. Edara and A. E. Pegg, Cancer Res., 1994, 54, 6221–6227 Search PubMed; (b) M. Crone and A. E. Pegg, Cancer Res., 1993, 53, 4750–4753 Search PubMed.
- J. E. Wibley, A. E. Pegg and P. C. Moody, Nucleic Acids Res., 2000, 28, 393–401 CrossRef CAS.
- Q. Fang, S. Kanugula and A. E. Pegg, Biochemistry, 2005, 44, 15396–15405 CrossRef CAS.
- A. T. Brunger, P. D. Adams, G. M. Clore, W. L. DeLano, P. Gros and R. W. Grosse-Kunstleve, et al. , Acta Crystallogr., Sect. D: Biol. Crystallogr., 1998, 52, 30–42 Search PubMed.
- (a) M. Melikishvili, J. J. Rasimas, A. E. Pegg and M. G. Fried, Biochemistry, 2008, 47, 13754–13763 CrossRef CAS; (b) J. J. Rasimas, S. R. Kar, A. E. Pegg and M. G. Fried, J. Biol. Chem., 2007, 282, 3357–3366 CrossRef CAS.
- C. A. Adams, M. Melikishvili, D. W. Rodgers, J. J. Rasimas, A. E. Pegg and M. G. Fried, J. Mol. Biol., 2009, 389, 248–263 CrossRef CAS.
- N. Huh and M. F. Rajewsky, Carcinogenesis, 1986, 7, 435–439 CrossRef CAS.
- (a) A. Lim and B. F. Li, EMBO J., 1996, 15, 4050–4060 CAS; (b) R. B. Ali, A. K. Teo, H. K. Oh, L. S. Chuang, T. C. Ayi and B. F. Li, Mol. Cell. Biol., 1998, 18, 1660–1669 CAS; (c) I. Tessmer, M. Melikishvili and M. G. Fried, Nucleic Acids Res., 2012, 17, 8296–8308 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Chimera engineering and biophysical characterization, ICL repair and binding data. See DOI: 10.1039/c2tx20075a |
| This journal is © The Royal Society of Chemistry 2013 |