 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Bis(carbazolyl) derivatives of pyrene and tetrahydropyrene: synthesis, structures, optical properties, electrochemistry, and electroluminescence†
Bilal R. Kaafarani*a, Ala'a O. El-Balloulia, Roman Trattnigb, Alexandr Fonaric, Stefan Saxb, Brigitte Wexd, Chad Riskoe, Rony S. Khnayzera, Stephen Barlowe, Digambara Patraa, Tatiana V. Timofeevac, Emil J. W. Listbf, Jean-Luc Brédase and Seth R. Mardere
aDepartment of Chemistry, American University of Beirut, Beirut 1107-2020, Lebanon. E-mail: bilal.kaafarani@aub.edu.lb; Tel: +961-3151451
bNanoTecCenter Weiz Forschungsgesellschaft mbH, Franz-Pichler-Straße 32, A-8160 Weiz, Austria
cDepartment of Biology & Chemistry, New Mexico Highlands University, Las Vegas, NM 87701, USA
dDepartment of Natural Sciences, Lebanese American University, Byblos, Lebanon
eSchool of Chemistry and Biochemistry and Center for Organic Photonics and Electronics, Georgia Institute of Technology, Atlanta, Georgia 30332, USA
fInstitut für Festkörperphysik, Technische Universität Graz, Petersgasse 16, A-8010 Graz, Austria
First published on 8th January 2013
Abstract
Tetrahydropyrene and pyrene have been functionalized in their 2,7-positions with carbazole and 3,6-di-tert-butylcarbazole groups, and the properties of these new compounds are compared to analogous carbazole and 3,6-di-tert-butylcarbazole derivatives of benzene and biphenyl using X-ray crystallography, UV-vis absorption and fluorescence spectroscopy, electrochemistry, and quantum-chemical calculations. The absorption spectra are similar to those of their biphenyl-bridged analogues, although TD-DFT calculations indicate a different description of the excited states in the pyrene case, with the lowest observed absorption no longer corresponding to the S0 → S1 transition. The 3,6-di-tert-butylcarbazole compounds show reversible electrochemical oxidations; the benzene, biphenyl, tetrahydropyrene, or pyrene bridging groups have little impact on the first oxidation potential. Bilayer organic light-emitting diodes incorporating the tetrahydropyrene and pyrene derivatives as emitters show deep-blue electroluminescence.
Introduction
Organic electroluminescent devices are of interest for use in flat panel displays and for lighting.1–3 Full-color displays require the use of red, green, and blue emitters; relative to red and green emitters, blue emitters are not as well optimized in terms of their efficiency, color purity, and stability.4–7 Efficient charge injection from electrodes into the adjacent organic layers, balanced charge transport, confinement of hole–electron recombination and the resultant excitons in the desired emissive layer, and high quantum efficiencies of luminescence are all important factors in achieving efficient devices, which should ideally exhibit low drive voltages and high current and power efficiencies.8 Efficient blue-emitting devices are challenging to fabricate because the large optical and transport gap required for the emitter means that efficient hole and/or electron injection can be hard to achieve at moderate drive voltages, while care also has to be taken to avoid the possibility of exciton energy transfer from the emissive layer to neighboring materials. Blue OLEDs can be obtained by using either fluorescent or phosphorescent materials. Although the latter approach can potentially result in much higher quantum efficiency, successful implementation typically involves blending of a heavy-metal phosphorescent “guest” into an organic “host”, both the singlet and triplet excited-state energy of which must be larger than that of the “guest”, while triplet energy transfer to neighboring layers must be prevented, thus leading to even more challenging issues regarding injection and exciton confinement.Polyaromatic molecules, such as diphenylanthracene,9 have been studied as potential fluorescent blue emitters. Pyrene has a remarkably long fluorescence lifetime,10 yet also exhibits high photoluminescence (PL) efficiency. In addition, it has a strong tendency to form excimers,11,12 while pyrene excitation spectra are sensitive to microenvironment changes.13 Although pyrene has high charge-carrier mobility and a lower ionization potential than many other efficient blue emitters, for example polyfluorenes, facilitating hole injection, pyrene itself is generally unsuitable for use in OLEDs due to its tendency to crystallize.5 Accordingly, a number of functionalized pyrenes have been developed as blue emitters.14 For example, solution-processable blue-emissive layers in OLEDs have been obtained by incorporating the pyrene core in dendrimers or polymers.15–17
Carbazole-based materials have been widely used as donors in a variety of organic electronic devices and, in general, exhibit hole-transport properties, although hybrids with various acceptor moieties such as oxadiazoles,18–20 and even 4,4′-di(carbazol-9-yl)biphenyl21 (CBP, compound 2a in Fig. 1), are ambipolar to varying extents. In OLEDs, carbazole-based materials have been used as hole-transport materials,22–24 as emitters,19,22,25 and as hosts for other fluorescent26 or phosphorescent emitters.9,20,27–29 Although carbazoles typically exhibit irreversible electrochemistry due to dimerization and polymerization of the corresponding radical cations at their 3,6-positions,30–32 blocking of those positions can suppress these reactions; moreover, 3,6-di(tert-butyl) substitution of the carbazole moieties of CBP (to give 2b, Fig. 1) has also been reported to result in a significantly higher glass-transition temperature.9
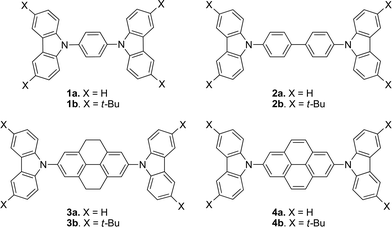 | ||
| Fig. 1 Compounds studied in this work. | ||
Given the fluorescent properties of pyrene and the transport properties of carbazoles, combining these two motifs in a single molecule may offer an interesting combination of physical properties and, perhaps, a route to new electroluminescent materials. Indeed, several such examples with a variety of architectures have been reported; however, these materials have generally presented quite extensive conjugation resulting in green emission.33–37 Here, we report the synthesis, characterization, and electroluminescent properties of molecules in which two carbazole or 3,6-di-tert-butylcarbazole moieties are attached through the nitrogen atom to the 2,7 positions of pyrene (4a and 4b, Fig. 1), which, in contrast to previously reported carbazole–pyrene materials, show blue fluorescence. We compare the structures, optical and electrochemical properties, orbital structure, and reorganization energies of 4a,b to those of analogous compounds in which the pyrene is replaced with tetrahydropyrene (3a,b), biphenyl (2a,b), or benzene (1a,b).
Experimental
General
The following compounds were synthesized according to literature procedures: 4,5,9,10-tetrahydropyrene, 6,38 2,7-dibromo-4,5,9,10-tetrahydropyrene, 7,39 2,7-dibromopyrene, 8,40 and 3,6-di-tert-butyl-9H-carbazole, 9b.411,4-Bis(carbazol-9-yl)benzene (1a)
Following a general literature procedure,42 a mixture of carbazole (1.00 g, 5.99 mmol), 1,4-dibromobenzene (0.70 g, 2.9 mmol), copper powder (0.38 g, 5.9 mmol), K2CO3 (2.48 g, 17.9 mmol) and 18-crown-6 (0.79 g, 2.9 mmol) were suspended in 1,2-dichlorobenzene (30 mL) and purged with argon for 30 min. The reaction mixture was heated to reflux for 7 d under an argon atmosphere. The crude mixture was filtered and the residue was washed with CHCl3 (3 × 10 mL). The solid was precipitated by adding cold ethanol (ca. 100 mL) to the brown filtrate. The brown solid was then recrystallized from toluene–ethanol to obtain 1a as light brown solid (0.37 g, 34%), mp (determined by DSC) 325.9 °C (Lit. 310;42 32743). 1H NMR (300 MHz, CDCl3): δ 8.19 (d, J = 7.8 Hz, 4H), 7.82 (s, 4H), 7.57 (d, J = 8.2 Hz, 4H), 7.47 (td, J = 7.3, 1.2 Hz, 4H), 7.33 (td, J = 7.3, 1.2 Hz, 4H). 13C{1H} NMR (75.5 MHz, CDCl3): δ 140.75, 136.67, 128.38, 126.14, 123.57, 120.47, 120.29, 109.77. Anal calcd for: C30H20N2: C, 88.21; H, 4.93; N, 6.86. Found: C, 88.00; H, 4.95; N, 6.73%.1,4-Bis(3,6-di-tert-butylcarbazol-9-yl)benzene (1b)
The title compound was synthesized using essentially the same approach as 1a. A mixture of 9b (1.00 g, 3.6 mmol), 1,4-dibromobenzene (0.42 g, 1.79 mmol), copper powder (0.23 g, 3.6 mmol), K2CO3 (1.5 g, 10.8 mmol) and 18-crown-6 (0.47 g, 1.79 mmol) were suspended in 1,2-dichlorobenzene (30 mL) and purged with argon for 30 min. The reaction was refluxed for 7 d under an argon atmosphere. The crude mixture was filtered and the residue was washed with CHCl3 (3 × 10 mL). The filtrate was evaporated under reduced pressure and the residue was purified by column chromatography using hexane–CH2Cl2 (20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) as eluent. The brown solid was then recrystallized from toluene–ethanol to obtain 1b as white solid (0.55 g, 48%), mp (determined by DSC) 373 °C (no literature melting point has been reported for this compound). 1H NMR (300 MHz, CDCl3): δ 8.18 (d, J = 0.9 Hz, 4H), 7.77 (s, 4H), 7.55–7.52 (dd, J = 8.7, 0.9 Hz, 4H), 7.51–7.50 (d, J = 8.7 Hz, 4H), 1.49 (s, 36H). 13C{1H} NMR (75.5 MHz, CDCl3): δ 143.12, 139.12, 136.66, 128.22, 127.8, 123.74, 123.48, 109.20, 34.77, 32.02. Anal calcd for: C46H52N2: C, 87.29; H, 8.28; N, 4.43. Found: C, 87.01; H, 8.40; N, 4.47%.
:1) as eluent. The brown solid was then recrystallized from toluene–ethanol to obtain 1b as white solid (0.55 g, 48%), mp (determined by DSC) 373 °C (no literature melting point has been reported for this compound). 1H NMR (300 MHz, CDCl3): δ 8.18 (d, J = 0.9 Hz, 4H), 7.77 (s, 4H), 7.55–7.52 (dd, J = 8.7, 0.9 Hz, 4H), 7.51–7.50 (d, J = 8.7 Hz, 4H), 1.49 (s, 36H). 13C{1H} NMR (75.5 MHz, CDCl3): δ 143.12, 139.12, 136.66, 128.22, 127.8, 123.74, 123.48, 109.20, 34.77, 32.02. Anal calcd for: C46H52N2: C, 87.29; H, 8.28; N, 4.43. Found: C, 87.01; H, 8.40; N, 4.47%.4,4′-Bis(carbazol-9-yl)biphenyl (2a)
The title compound was synthesized according to a general literature procedure.42 A mixture of carbazole (2.50 g, 15.0 mmol), 1,8-diiodobiphenyl (2.00 g, 4.93 mmol), copper powder (0.94 g, 15 mmol), K2CO3 (5.45 g, 39.4 mmol) and 18-crown-6 (1.30 g, 4.92 mmol) were suspended in 1,2-dichlorobenzene (50 mL) and purged with argon for 30 min. The reaction mixture was refluxed for 7 d under argon atmosphere. The crude mixture was filtered and the filtrate was cooled in fridge; the solid formed was collected and recrystallized twice from ethyl acetate to obtain 2a as light brown needles (1.3 g, 47%), mp (determined by DSC) 282.2 °C (Lit. 290;42 28144). 1H NMR (300 MHz, CDCl3): δ 8.19 (d, J = 7.2 Hz, 4H), 7.94 (m overlapping, 4H), 7.74 (m overlapping, 4H), 7.54 (d, J = 7.2 Hz, 4H), 7.48–7.43 (td, J = 6.9, 1.2 Hz, 4H), 7.35–7.31 (td, J = 6.9, 1.2 Hz, 4H). 13C{1H} NMR (75.5 MHz, CDCl3): δ 140.80, 139.29, 137.24, 128.53, 127.49, 126.2, 123.48, 120.38, 120.07, 109.82. Anal calcd for C36H24N2: C, 89.23; H, 4.99; N, 5.78. Found: C, 89.07; H, 4.80; N, 5.79%.4,4′-Bis(di-tert-butylcarbazol-9-yl)biphenyl (2b)
A mixture of 3,6-di-tert-butyl-9H-carbazole 9b (2.47 g, 8.84 mmol), 1,8-diiodobiphenyl (1.20 g, 2.95 mmol), copper powder (0.56 g, 8.8 mmol), K2CO3 (3.26 g, 0.0236 mol) and 18-crown-6 (0.78 g, 3.0 mmol) were suspended in 1,2-dichlorobenzene (30 mL) and purged with argon for 30 min. The reaction was refluxed for 7 d under argon atmosphere. The crude mixture was filtered and the filtrate was left in the fridge for 2 d; the resulting solid was collected and recrystallized from ethyl acetate to obtain 2b as white solid (0.80 g, 30%), mp (determined by DSC) 438.0 °C (no literature melting point has been reported for this compound). 1H NMR (300 MHz, CDCl3): δ 8.17 (d, J = 1.5 Hz, 4H), 7.91–7.89 (d, J = 8.4 Hz, 4H), 7.71 (d, J = 8.4 Hz, 4H), 7.52–7.49 (dd, J = 1.5 Hz, J = 8.7 Hz, 4H), 7.46 (d, J = 8.7 Hz, 4H). 13C{1H} NMR (75.5 MHz, CDCl3): δ 142.98, 139.16, 138.83, 137.65, 128.37, 127.05, 123.68, 123.46, 116.31, 109.28, 34.76, 32.05. Anal calcd for C52H56N2: C, 88.09; H, 7.96; N, 3.95. Found: C, 87.93; H, 7.97; N, 3.96%.2,7-Bis(carbazol-9-yl)-4,5,9,10-tetrahydropyrene (3a)
A 10 mL microwave tube was charged with carbazole (289 mg, 1.73 mmol), 2,7-dibromo-4,5,9,10-tetrahydropyrene 7 (300 mg, 0.824 mmol), and a magnetic stir bar. In a glove box under nitrogen atmosphere, tris(dibenzylideneacetone)dipalladium(0) (60 mg, 0.066 mmol), tri-tert-butylphosphine (165 mg, 0.568 mmol), sodium tert-butoxide (353 mg, 3.67 mmol), and dry toluene (6 mL) were added. The tube was sealed under nitrogen and the reaction was conducted in the microwave under standard control conditions (power: 180 W; max temp: 125 °C; run time: 2 min; hold time: 60 min; pressure: 120 psi). The reaction mixture was then poured into ice-water (100 mL) and extracted with chloroform (3 × 50 mL); the extracts were successively washed with water and brine. The solution was dried over anhydrous MgSO4 and was run through a short bed of silica. The solvent was removed under reduced pressure and the resulting solid was recrystallized from toluene–ethanol to yield 3a as white solid (393 mg, 55%), mp (determined by DSC) 394.2 °C. 1H NMR (300 MHz, CDCl3): δ 8.19–8.16 (d, J = 7.8 Hz, 4H), 7.54–7.51 (d, J = 8.1 Hz, 4H), 7.54–7.45 (td, J = 7.2, 1.2 Hz, 4H), 7.34 (s, 4H), 7.33–7.29 (td, J = 7.2, 1.2 Hz, 4H), 3.08 (s, 8H). 13C{1H} NMR (75.5 MHz, CDCl3): δ 140.91, 137.05, 136.50, 129.46, 125.89, 124.50, 123.36, 120.32, 119.86, 110.00, 28.34. HRMS-EI (m/z): [M]+: calcd for C40H28N2, 536.2252; found, 536.2234. Anal calcd for C40H28N2: C, 89.52; H, 5.26; N, 5.22. Found: C, 89.37; H, 5.23; N, 5.22%.2,7-Bis(3,6-di-tert-butylcarbazol-9-yl)-4,5,9,10-tetrahydropyrene (3b)
A 10 mL microwave tube was charged with 3,6-di-tert-butyl-9H-carbazole 9b (484 mg, 1.73 mmol), 2,7-dibromo-4,5,9,10-tetrahydropyrene 7 (300 mg, 0.824 mmol), and a magnetic stir bar. In a glove box under nitrogen atmosphere, tris(dibenzylideneacetone)dipalladium(0) (60 mg, 0.066 mmol), tri-tert-butylphosphine (165 mg, 0.568 mmol), sodium tert-butoxide (353 mg, 3.67 mmol), and dry toluene (6 mL) were added. The tube was sealed under nitrogen and the reaction was conducted in the microwave under standard control conditions (power: 180 W; max temp: 125 °C; run time: 2 min; hold time: 60 min; pressure: 120 psi). The reaction mixture was then poured into ice-water (100 mL) and extracted with chloroform (3 × 50 mL); the extracts were successively washed with water and brine. The solution was dried over anhydrous MgSO4 and was run through a short bed of silica. The solvent was removed under reduced pressure and the resulting solid was recrystallized from toluene–ethanol to yield 3b as white solid (405 mg, 65%). 1H NMR (300 MHz, CDCl3): δ 8.16 (d, J = 0.9 Hz, 4H), 7.52–7.48 (dd, J = 8.7, 1.8 Hz, 4H), 7.47–7.43 (d, J = 9.0 Hz, 4H), 7.32 (s, 4H), 3.05 (s, 8H), 1.49 (s, 36H). 13C{1H} NMR (75.5 MHz, CDCl3): δ 142.74, 139.27, 136.84, 129.10, 129.03, 124.08, 123.54, 123.32, 116.21, 109.41, 34.75, 32.04, 28.37. HRMS-EI (m/z): [M]+: calcd for C56H60N2, 760.4757; found, 760.4723. Anal calcd for C56H60N2: C, 88.37; H, 7.95; N, 3.68. Found: C, 88.18; H, 7.80; N, 3.67%.2,7-Bis(carbazol-9-yl)pyrene (4a)
A 10 mL microwave tube was charged with carbazole (196 mg, 1.17 mmol), 2,7-dibromopyrene 8 (200 mg, 0.555 mmol), and a magnetic stir bar. In a glove box under nitrogen atmosphere, tris(dibenzylideneacetone)dipalladium(0) (40 mg, 0.044 mmol), tri-tert-butylphosphine (110 mg, 0.379 mmol), sodium tert-butoxide (235 mg, 2.44 mmol), and dry toluene (6 mL) were added. The tube was sealed under nitrogen and the reaction was conducted in the microwave under power control conditions (power: 180 W; max temp: 125 °C; run time: 2 min; hold time: 60 min; pressure: 120 psi). The reaction mixture was then poured into ice-water (100 mL) and extracted with chloroform (3 × 50 mL); the extract were then washed with water and brine, dried over anhydrous MgSO4, and run through a short bed of silica. The solvent was removed under reduced pressure and the resulting solid was recrystallized twice from toluene–ethanol to yield 4a as white solid (185 mg, 63%), mp (determined by DSC) 364.6 °C. 1H NMR (300 MHz, CDCl3): δ 8.46 (s, 4H), 8.26–8.23 (m overlapping, 4H), 8.23 (s overlapping, 4H), 7.58–7.55 (d, J = 7.8 Hz, 4H), 7.49–7.44 (td, J = 6.9, 1.2 Hz, 4H), 7.39–7.34 (td, J = 7.8, 1.2 Hz, 4H). 13C{1H} NMR (75.5 MHz, CDCl3): δ 141.41, 135.76, 132.57, 128.14, 126.14, 123.76, 123.57, 120.48, 120.26, 109.63, 105.00. HRMS-EI (m/z): [M]+: calcd for C40H24N2, 532.1939; found, 532.1936. Anal calcd for C40H24N2: C, 90.20; H, 4.54; N, 5.26. Found: C, 89.84; H, 4.47; N, 5.27%.2,7-Bis(3,6-di-tert-butylcarbazol-9-yl)pyrene (4b)
A 10 mL microwave tube was charged with 3,6-di-tert-butyl-9H-carbazole 9b (326 mg, 1.17 mmol), 2,7-dibromopyrene 8 (200 mg, 0.555 mmol), and a magnetic stir bar. In a glove box under nitrogen atmosphere, tris(dibenzylideneacetone)dipalladium(0) (40 mg, 0.044 mmol), tri-tert-butylphosphine (110 mg, 0.379 mmol), sodium tert-butoxide (235 mg, 2.44 mmol), and dry toluene (6 mL) were added. The tube was sealed under nitrogen and the reaction was conducted in the microwave under power control conditions (power: 180 W; max temp: 125 °C; run time: 2 min; hold time: 60 min; pressure: 120 psi). The reaction mixture was then poured into ice-water (100 mL) and extracted with chloroform (3 × 50 mL); the extracts were washed with water and brine, dried over anhydrous MgSO4, and run through a short bed of silica. The solvent was removed under reduced pressure and the resulting solid was recrystallized from toluene–ethanol to yield 4b as white solid (339 mg, 81%). 1H NMR (300 MHz, CDCl3): δ 8.43 (s, 4H), 8.23 (s, 4H), 8.19 (s, 4H), 7.52 (s, 4H), 7.51 (s, 4H), 1.51 (s, 36H). 13C{1H} NMR (75.5 MHz, CDCl3): δ 143.19, 139.74, 136.17, 132.46, 128.05, 123.78, 123.56, 123.37, 116.40, 109.02, 105.00, 34.81, 32.04. HRMS-EI (m/z): [M]+: calcd for C56H56N2, 756.4444; found, 756.4426. Anal calcd for C56H56N2: C, 88.84; H, 7.46; N, 3.70. Found: C, 88.68; H, 7.59; N, 3.63%.Photophysical studies
The photophysical studies of compounds 1–4 were performed using 1–5 μM solutions of each compound prepared in different solvents. The procedure involved the measurement of 1–2 mg of each compound in a vial for the preparation of a stock solution (1 mM) in DCM. Dilutions were done in another vial to obtain 100 μM solutions from which 40–200 μL were taken, placed in different vials, and left to dry. Different solvents were then added to each vial (4 mL) to obtain 1–5 μM solutions. A JASCO V-570 UV-NIR spectrophotometer was used for measurements of the absorption spectra, and the fluorescence measurements were done using a Jobin-Yvon-Horiba Fluorolog III spectrofluorimeter. The excitation source was a 100 W xenon lamp and the slit width was fixed at 5 nm for all measurements, while the excitations were done at 330 or 320 nm. In order to find the fluorescence lifetime of compounds (1–4), a Jobin-Yvon-Horiba FluorologIII spectrofluorimeter was used with a pulsed diode laser (282 nm). The decay data were analyzed using Data Analysis software to fit decays to single or double exponential with χ2 < 3.X-Ray structure determination
X-ray diffraction experiments were carried out with a Bruker SMART APEX II CCD diffractometer, using Mo Kα radiation (λ = 0.71073 Å) at 100 K; details of the solution and refinement and given in Table S1 (ESI†), while cif files are available from the Cambridge Crystallographic Data Center where the crystal structures of 1a, 2b, 3a, and 4b have been deposited and allocated the deposition numbers CCDC 899423, 899424, 899425, and 899426, respectively. The raw data frames were integrated with the SAINT+ program using narrow-frame algorithm.45 Absorption corrections were applied using the semi-empirical method of the SADABS program.46 The structures were solved by direct methods and refined using the Bruker SHELXTL programs suite47 by full-matrix least-squares methods on F2 with SHELXL-97 in anisotropic approximation for all non-hydrogen atoms. Compound 1a was found to crystallize in the orthorhombic space group Pbca, with an entire molecule in the asymmetric unit, whereas crystals of 2b, 3a, and 4b belong to the monoclinic space group P21/c with half of the molecule in the asymmetric unit. In 2b disorder was modeled for the bridging six-membered ring over two positions with partial occupancies of 0.53(3) and 0.47(3). In 3a disorder was modeled for the Csp3 atoms in the tetrahydropyrene core over two positions with partial occupancies of 0.63(8) and 0.37(8). All H atoms were placed in idealized positions and refined with constrained C–H distances and Uiso(H) values set to 1.2Ueq or 1.5Ueq (for methyl group) of the attached C atom.Organic light-emitting devices and electroluminescence-measurements
Devices were fabricated using a standard sandwich assembly to investigate these new materials as the active layer in OLEDs: indium tin oxide (ITO)/poly(3,4-ethylenedioxythiophene)–polystyrenesulfonic acid (Baytron P VPAI 4083) (PEDOT:PSS)/3a, 3b, 4a, 4b/1,3,5-tris(1-phenyl-1H-2-benzimidazole) (TPBi)/Ca/Al. ITO-covered glass substrates were mechanically cleaned by the use of acetone and isopropyl alcohol. Afterwards the substrates were subjected to various supersonic treatments in deionized water, toluene, and isopropanol. A dry cleaning step in oxygen plasma finished the cleaning procedure. On top of the ITO-substrates a layer of PEDOT:PSS was applied via spin-coating under ambient conditions and dried under dynamic vacuum according to the specifications. Layers of 3a and 4a with a thickness of 80 nm were evaporated at an evaporation rate of 0.5 Å s−1 from a resistively heated crucible under dynamic vacuum in an evaporation chamber having an initial base pressure <1.0 × 10−6 mbar. Film thicknesses were monitored using a quartz-crystal microbalance. Compounds 3b and 4b were spin-coated from 5 mg mL−1 THF solutions in inert atmosphere and dried at 80 °C for 2 h in vacuum. The resulting layer thickness amounted to 75 nm. The cathode materials (TPBi, Ca, Al) were deposited in an evaporation chamber under high vacuum (p < 1.0 × 10−6 mbar). TPBi (10 nm) was evaporated uniformly onto the substrate as an electron transport layer, Ca (10 nm) and Al (100 nm) were deposited through a shadow mask thus forming multiple devices with a device-area of 9 mm2 on a single substrate.Electroluminescence (EL) spectra were acquired using an ORIEL spectrometer with an attached calibrated charge-coupled device (CCD) camera. Current–luminance–voltage (I–L–V) characteristics were recorded in a customized setup using a Keithley 2612A source measure unit for recording the I–V characteristics while the luminance was measured by a Keithley 6485 Picoammeter using a photodiode calibrated by a Konica-Minolta LS-100 Luminancemeter.
Quantum-chemical studies
The ground states and radical-cation states of 1b–4b were calculated at the density functional theory (DFT) level by employing the global hybrid B3LYP functional48–51 in conjunction with a 6-31G(d,p) basis set.52,53 The low-lying singlet excited states were evaluated using the neutral ground-state geometries via time-dependent density functional theory (TDDFT) with the B3LYP functional and the same basis set. All calculations were performed using the Gaussian09 (Revision A.02) software suite.54Results and discussion
Synthesis
Compounds 1–4 were synthesized from Ullmann or Buchwald–Hartwig coupling of the appropriate carbazole to the dibromo/diiodo derivatives of the bridging groups. The bridging groups required for the synthesis of compounds 3 and 4 were synthesized from pyrene, 5, as shown in Scheme 1, according to literature procedures: pyrene was first activated using RANEY® nickel and then hydrogenated at 40 psi for 3 days in the presence of Pd/C to yield 4,5,9,10-tetrahydropyrene, 6.38 2,7-Dibromo-4,5,9,10-tetrahydropyrene, 7, was obtained through the bromination of 6 using bromine in acetic acid.39 Oxidation of 7 using bromine and CS2 at room temperature yielded 2,7-dibromopyrene, 8.40 Compound 8 cannot be obtained from direct bromination of 5, which gives 1,3,6,8-tetrabromopyrene55 although recently a transition-metal catalyzed C–H activation approach to the direct 2,7-diborylation of 5 has been reported.56 3,6-Di-tert-butyl-9H-carbazole, 9b, was synthesized from 9H-carbazole, 9a, using a previously described ZnCl2-mediated Friedel–Crafts alkylation with 2-chloro-2-methylpropane in nitromethane (Scheme 2).41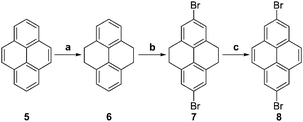 | ||
| Scheme 1 Synthesis of 2,7-dibromo-4,5,9,10-tetrahydropyrene and 2,7-dibromopyrene. (a) (1) RANEY® nickel; (2) Pd/C, H2 (40 psi); (b) AcOH, Br2, NaOH; (c) CS2, Br2. | ||
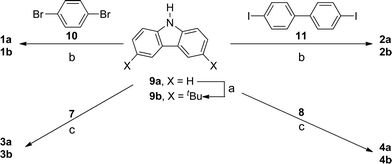 | ||
| Scheme 2 Synthesis of bis(carbazolyl) derivatives of benzene, biphenyl, tetrahydropyrene, and pyrene. (a) tBuCl, nitromethane, ZnCl2; (b) Cu, K2CO3, 18-crown-6, 1,2-dichlorobenzene (reflux, 1 week); (c) Pd2(dba)3, PtBu3, NaOtBu, toluene, 120 psi, 1 h (microwave). | ||
The well-known compounds 1a and 2a were obtained essentially as described in the literature,42 using Ullmann condensation reactions of 9a with 1,4-dibromobenzene, 10, and 4,4′-diiodobiphenyl, 11, respectively (Scheme 2). Their tert-butyl-substituted analogues, 1b and 2b, have been reported: 1b has been obtained from the Ullmann reaction of 9b and 1,4-diiodobenzene (although no experimental details and characterizing data were reported),57 while 2b was obtained by Friedel–Crafts alkylation of 2a.9 We synthesized 1b and 2b from 9b and the appropriate dibromoarene/diiodoarene bridging groups using the same Ullmann conditions used for 1a and 2a.
The Ullmann approach requires stoichiometric quantities of copper reagent and, typically, high temperatures (>200 °C) and long reaction times, and often gives only moderate yields; an alternative is the Buchwald–Hartwig palladium-catalyzed C–N coupling reaction.58,59 We obtained 3 and 4 in moderate to good yield from reaction of the appropriate dibromopyrene and carbazole derivatives (Scheme 2), using the tris(dibenzylideneacetone)dipalladium(0)/tri-tert-butylphosphine catalyst system, as described for N-arylation of carbazole in ref. 60, but using microwave irradiation to accelerate the reaction (with a maximum temperature of 125 °C, run time of 2 min, hold time of 60 min, pressure of 120 psi, and power of 180 W).
Crystal and molecular structures
Single crystals of 1,4-bis(carbazol-9-yl)benzene (1a), 4,4′-bis(3,6-di-tert-butylcarbazol-9-yl)biphenyl (2b), 2,7-bis(carbazol-9-yl)-4,5,9,10-tetrahydropyrene (3a), and 2,7-bis(3,6-di-tert-butylcarbazol-9-yl)pyrene (4b) were obtained by crystallization from dichloromethane–ethyl acetate solution and their structures were determined using X-ray diffraction; the structure of 2a has previously been reported,61 while suitable crystals were not obtained for the remaining materials. Details of the solutions and refinements are listed in Table S1† and the molecular structures are shown in Fig. 2. The molecules are held together in the crystal by van der Waals interactions and no short intermolecular contacts or close π-stacking interactions were found (see ESI† for packing diagrams). Density functional theory (DFT) optimizations of the ground-state structures of 1b, 2b, 3b, and 4b were carried out for direct comparison; selected DFT (B3LYP/6-31G(d,p))-calculated geometric parameters are given in Table S1† and compared with the crystallographic parameters.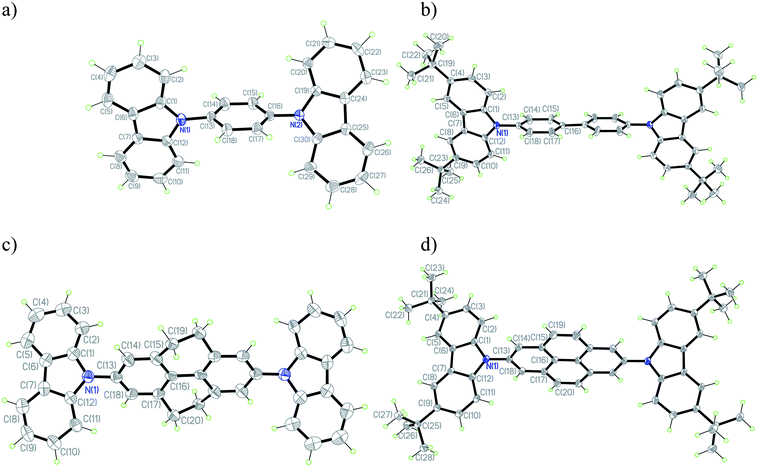 | ||
| Fig. 2 ORTEP plots of 1a (a), 2b (b), 3a (c), and 4b (d) with the numbering scheme in projection on the best plane. In the cases of 2b and 3a, where conformational disorder was found, only the major conformation is shown. Thermal ellipsoids are shown with 50% probability level. Hydrogen atoms are drawn as circles of arbitrary small radii for clarity. | ||
In the “gas-phase” DFT calculations, the biphenyl unit of 2b is characterized by a twist angle of 37°, the CH2CH2 “bridges” of 3b lead to a lower twist of 17°, while only in the pyrene derivative 4b do the “CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH” bridges lead to complete planarization of the biphenyl portion of the bridge. Molecules of 2b, 3a, and 4b are located on crystallographic inversion centers in their crystals, meaning that the biphenyl portions of the bridging moieties are apparently planar, as is the case in the previously reported structure of 2a,61 although this is likely an artifact of disorder, as in the case of biphenyl itself,62 precluding a meaningful comparison of experimental and DFT bridge geometries. In both the crystallographically determined and DFT structures, the carbazole moieties are significantly twisted from coplanarity with the adjacent bridging group, consistent with the expected effects of steric interactions and with other structures of N-aryl carbazole derivatives; however, in the calculated structures, the angles between these two planes are all close to 53°, while there is considerable variation in the angles found in the crystal structures (see Table S2†). Although the nitrogen atoms in the DFT structures are all fully planarized, the coordination geometries of the carbazole nitrogen atoms in all four crystal structures are not perfectly planar: the extent of pyramidalization is, however, well within the range of values that have been observed for other N-aryl carbazoles.63–66 These geometric differences may indicate relatively shallow potential energy surfaces with respect to these distortions so that minor crystal packing effects can lead to variations.
CH” bridges lead to complete planarization of the biphenyl portion of the bridge. Molecules of 2b, 3a, and 4b are located on crystallographic inversion centers in their crystals, meaning that the biphenyl portions of the bridging moieties are apparently planar, as is the case in the previously reported structure of 2a,61 although this is likely an artifact of disorder, as in the case of biphenyl itself,62 precluding a meaningful comparison of experimental and DFT bridge geometries. In both the crystallographically determined and DFT structures, the carbazole moieties are significantly twisted from coplanarity with the adjacent bridging group, consistent with the expected effects of steric interactions and with other structures of N-aryl carbazole derivatives; however, in the calculated structures, the angles between these two planes are all close to 53°, while there is considerable variation in the angles found in the crystal structures (see Table S2†). Although the nitrogen atoms in the DFT structures are all fully planarized, the coordination geometries of the carbazole nitrogen atoms in all four crystal structures are not perfectly planar: the extent of pyramidalization is, however, well within the range of values that have been observed for other N-aryl carbazoles.63–66 These geometric differences may indicate relatively shallow potential energy surfaces with respect to these distortions so that minor crystal packing effects can lead to variations.
On the other hand, there is good general agreement between crystallographically determined and calculated bond lengths, suggesting that DFT describes the electronic structure of these molecules well (see below). For example, the bridging N(carbazole)–C(aryl bridge) bonds are in the range of 1.422(4)–1.428(4) Å (1.417–1.419 Å, DFT), agreeing well with bond lengths reported in literature.63–67 The C–C bond lengths of the DFT and X-ray structures follow similar patterns. Notably, the calculations indicate that the central C–C bond of the biphenyl portion of the bridge shortens from biphenyl (1.483 Å) to tetrahydropyrene (1.473 Å) to pyrene (1.424 Å), consistent (allowing for the low precision of the 2b structure) with the trends in the corresponding crystallographic bond lengths (1.489(2), 1.464(12), 1.470(4), and 1.422(3) Å for 2a,612b, 3a, and 4b, respectively). While the small difference in the C–C bond lengths of the tetrahydropyrene and biphenyl bridges likely reflects constraints imposed by the CH2CH2 groups, the much shorter central bond in the pyrene system indicates that there is a significant change in electronic structure of the bridge (see below for a discussion of the molecular orbitals) and that the simplistic representation of pyrene as a biphenyl moiety planarized by CHCH groups is inadequate.
Absorption and fluorescence studies
Fig. 3a and b show the absorption and fluorescence spectra respectively of the carbazole and 3,6-di-tert-butylcarbazole derivatives (1–4) in DMF (see ESI† for spectra acquired in different solvents). In all cases, the absorption spectra showed a common sharp peak at around 295 nm and broader weaker features in the range 310–340 nm. In the case of 2a, these have been described as carbazole-localized transitions and transitions between more extensively delocalized π and π* orbitals, respectively68 although carbazole itself also shows a weak transition at ca. 330 nm in addition to a strong absorption at ca. 290 nm.69 The absorption maxima, emission maxima, and Stokes shift of each compound are summarized in Table 1. 3,6-tert-Butyl substitution of the carbazole moieties results in small red-shifts in absorption (ca. 0–80 meV) and fluorescence (ca. 70–135 meV) maxima compared to their unsubstituted analogues, consistent with a previous comparison of 2a and 2b.9 The pyrene derivatives have the lowest energy emission maxima (429 and 450 nm for 4a and 4b, respectively) and exhibit the greatest Stokes shifts (between the lowest energy observed absorption maximum and the fluorescence maximum) of any of the compounds examined, although another weaker emission feature is present at ca. 380–390 nm; as discussed below, this is consistent with the lowest energy absorption maximum not corresponding to S0 → S1 in the pyrene compounds.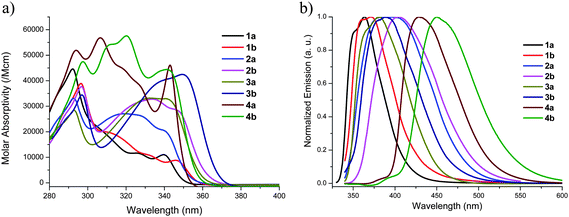 | ||
| Fig. 3 Absorption (a) and normalized emission (b) spectra of compounds 1–4 in DMF. | ||
| Compound | λmaxabs (nm) (ε (M−1 cm−1)) | λmaxem (nm) | Stokes shift (cm−1) | Φfb | τfl/nsc |
|---|---|---|---|---|---|
| a Optical data in other solvents are presented in Tables S5 and S6 in the ESI.b Fluorescence quantum yield.c Fluorescence lifetime.d Biexponential fit; the relative contribution of the two components is also indicated. | |||||
| 1a | 340 (11000) | 363 | 1864 | 0.46 | 6.56 |
| 1b | 346 (8800) | 371 | 1948 | 0.24 | 13.2 (53%), 5.73 (47%)d |
| 2a | 340 (20500) | 387 | 3572 | 0.42 | 2.50 |
| 2b | 347 (28400) | 404 | 4066 | 0.15 | 3.05 |
| 3a | 341 (33000) | 381 | 3079 | 0.49 | 1.95 |
| 3b | 349 (42500) | 389 | 2946 | 0.69 | 2.03 (56%), 0.12 (44%)d |
| 4a | 343 (46000) | 429 | 5844 | 0.44 | 2.78 (1%), 24.7 (99%)d |
| 4b | 341 (44400) | 450 | 7103 | 0.42 | 18.9 |
Table 2 provides the vertical transition energies (and wavelengths), transition dipoles, and electronic configurations of the lowest-lying excited states with appreciable oscillator strength as determined at the TDDFT B3LYP/6-31G(d,p) level of theory. The calculated maxima, though slightly red-shifted with respect to experiment,70 agree well with the empirical trends. For 1b through 3b, the first vertical transition with appreciable oscillator strength is the S0 → S1 transition; in each case these are well-described as HOMO → LUMO excitations (although in 1b, the HOMO → LUMO+2 excitation also contributes to the transition). Depictions of the molecular orbitals involved in the transitions are located in the ESI;† in the case of 1b, the HOMO and LUMO are fully delocalized across both carbazole units and the central phenylene bridge. In the case of 2b and 3b, the HOMO has a similar delocalized character, while the LUMO is mainly localized on the central biphenyl portion with only modest extension on the carbazole (consistent with previous reports of the frontier orbitals of 2a and 2b9). Thus, the lowest-lying transitions are indeed well-described as π–π* transitions, as previously suggested for 2a,68 although they have significant carbazole-to-bridge charge-transfer character.
| Compound | Evert (eV) | λvert (nm) | μge (Debye) | Electronic configuration | |
|---|---|---|---|---|---|
| 1b | S0 → S1 | 3.78 | 328 | 6.07 | HOMO → LUMO (69%); HOMO → LUMO+2 (29%) |
| 2b | S0 → S1 | 3.46 | 359 | 7.24 | HOMO → LUMO (98%) |
| 3b | S0 → S1 | 3.33 | 372 | 7.55 | HOMO → LUMO (98%) |
| 4b | S0 → S1 | 2.78 | 446 | 0.87 | HOMO → LUMO (98%) |
| S0 → S5 | 3.50 | 354 | 2.62 | HOMO → LUMO+1 (75%); HOMO-4 → LUMO (23%) | |
| S0 → S7 | 3.73 | 332 | 9.69 | HOMO-4 → LUMO (64%); HOMO → LUMO+1 (22%); HOMO-7 → LUMO+1 (5%); HOMO → LUMO+3 (3%) |
The picture differs for 4b, where the S0 → S1 transition, which largely corresponds to excitation from a carbazole-localized HOMO to a pyrene-localized LUMO (Fig. 4), has negligible transition dipole. This pyrene-localized LUMO has very little wavefunction distribution, as expected, on the 2- and 7-positions that connect the carbazole end groups to the pyrene center – in direct contrast with the phenyl (1b) and biphenyl(-like) bridges (2b and 3b) of the other systems – therefore limiting the wavefunction overlap of the ground and excited states. Consistent with the small transition dipole moments linking these states, the fluorescence lifetimes of 4a and 4b are longer than those observed for the other compounds in most solvents (see Table 2 and ESI†). Two higher-lying transitions (S0 → S5 and S0 → S7) with mixed HOMO → LUMO+1 and HOMO-4 → LUMO character are the first transitions with any appreciable transition dipole; they presumably correspond to the lowest-energy experimentally observed absorption maximum. Assuming the main observed emission peak to be the S0 → S1 emission, the large Stokes shifts seen for 4a and 4b are, therefore, easily understood (the small emission peak at ca. 370–380 nm perhaps occurring from one of the higher lying states with more significant coupling to the ground state). The LUMO+1 is mainly pyrene-localized, closely resembling the LUMO+1 of an isolated pyrene,56,71–80 while the HOMO-4 is located on the pyrene unit and corresponds with the HOMO of pyrene itself (see Fig. 4). The most important contributor to S0 → S5 is the HOMO → LUMO+1 excitation, indicating that this transition has significant quadrupolar carbazole-to-pyrene charge-transfer character and some similarity to S0 → S1 for the other compounds. The HOMO-4 → LUMO excitation is the most important contributor to the S0 → S7 transition; this transition is primarily a pyrene-localized transition, closely corresponding to the S0 → S2 transition of pyrene.56
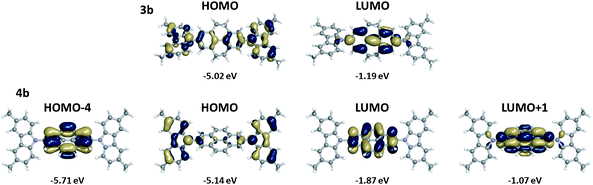 | ||
| Fig. 4 Illustrations of the frontier molecular orbitals and molecular orbital energies of 3b and 4b evaluated at the B3LYP/6-31 G(d,p) level. | ||
Fluorescence quantum yields of 1–4 were determined in different solvents (Table S5†). It was observed that solvent polarity affected the overall fluorescence quantum yield. In general, higher fluorescence quantum yields were observed in non-polar solvents (i.e. cyclohexane and hexane) and medium-polarity solvents (i.e. DCM and THF), while lower fluorescence quantum yields were recorded in polar solvents such as acetonitrile, methanol and ethanol. Solvent dependence was also manifested in the fluorescence lifetime (τf).81 Data for the eight carbazole derivatives were fit to mono- or biexponential decays. Generally, there was no clear trend among the lifetime values in different solvents. However, the lowest lifetime (τ1 or τavg) was obtained in hexane for compounds 1a, 2a, 2b, 3a, 3b, 4a, 4b, while 1b was an exception with the lowest τavg value being found in ethanol (see ESI†). Importantly, the recorded lifetimes for compounds 1(a,b), 2(a,b), 3(a,b) were less than 10 ns, while the pyrene-based compounds (4a and 4b) exhibited higher lifetimes in most solvents with values up to 22 ns. Nonetheless, these lifetimes fall considerably short of those found for pyrene itself (ca. 400 ns).82 It should be noted that despite the small calculated S0 → S1 transition dipoles and the long fluorescence lifetimes for 4a and 4b, the solution fluorescence quantum yields are still moderately large (up to 56%, see Table S5†) and comparable to those found for 1–3. The observed biexponential decays for some compounds (1a only in methanol, 1b (except in acetonitrile), 3b and 4b in all the solvents) may be attributed to various origins of the radiative transitions such as intramolecular twisting in the excited state or charge transfer states.83 The radiative (kr) and non-radiative (knr) rate constants of 1–4 in various solvents are summarized in Table S6.†
Redox properties
The solution electrochemistry of 1a–4a and 1b–4b was investigated using cyclic voltammetry (CV) using 0.1 M nBu4NPF6 in dichloromethane as electrolyte. Compounds 1a–4a all undergo irreversible oxidation, consistent with previous reports for other carbazole derivatives without 3,6-substitution, including 2a;61 this irreversibility is attributed to coupling of carbazole radical cations through those positions, in which the spin density is relatively high.30–32 In contrast, 1b–4b all undergo two sequential reversible oxidation processes indicating that these coupling reactions are effectively suppressed; this qualitative difference is shown for the example of 4avs.4b in Fig. 5, while the other CVs are shown in the ESI.† Although irreversible oxidations are observed for many carbazoles with useful solid-state hole-transport properties, such as PVK84 and 2a,61 reversible electrochemistry might be expected to lead to more stable device behavior due to fewer decomposition reactions of the charge-carrying species.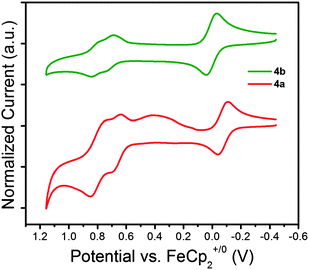 | ||
| Fig. 5 Cyclic Voltammograms (50 mV s−1) for 1,4-bis(carbazol-9-yl)pyrene (4a) and 1,4-bis(3,6-di-tert-butylcarbazol-9-yl)pyrene (4b) with ferrocene as an internal standard in CH2Cl2–0.1 M nBu4NPF6. | ||
The first oxidation potentials, E+/01/2, of the reversibly oxidized tert-butyl derivatives 1b–4b materials vary by only ca. 0.1 V within the series; DFT-calculated values of ionization potential (IP) also show relatively minor variation. For 2b, 3b, and 4b, the minor variations in the DFT ΔSCF estimates of gas-phase vertical and adiabatic IPs, or of the IPs according to Koopmans' theorem, correlate very well with those in E+/01/2; the correlation is less good for 1b; as in the previously reported (more pronounced) discrepancy between trends in gas-phase IP values and electrochemical potentials for bis(dimethylamino)benzene, biphenyl, and tolane derivatives, this is likely to be due to a greater solvation effect on the electrochemical potentials for the species with the smaller bridge.85 The similarity of redox potentials and calculated IPs within the series is consistent with the largely carbazole-localized nature of the HOMO in all cases, while some of the variation can be attributed to the varying bridge contributions; in particular, 4b is significantly less readily ionized than 3b owing to the destabilizing interaction of the carbazole and bridge-based orbitals in the latter system, while there is barely any pyrene contribution to the HOMO of 4b.
In the case of the reversibly oxidized 3,6-di(tert-butyl)carbazole derivatives, 1b–4b, differential pulse voltammetry (DPV)86 was also used to better separate the two oxidation features and better determine the separation ΔE1/2 = E2+/+1/2 − E+/01/2. As shown in Fig. 6 and Table 3, there is considerable variation in the separation between the two oxidations. The largest separation is seen for 1b; this is expected given the shorter bridge between the two carbazole redox centers (for example, greater values of ΔE1/2 are seen for bis(di-p-anisylamino)benzene and bis(dimethylamino)benzene than for their respective biphenyl-bridged analogues),85,87 which gives rise to increased delocalization, and, in the case of a class-II monocation, increased electrostatic interactions, stabilizing the monocation. The variation between the remaining compounds shows no clear trend; however, values of ΔE1/2 are known to depend on a number of additional factors,88 some of which are perhaps in competition with one another in the present series.
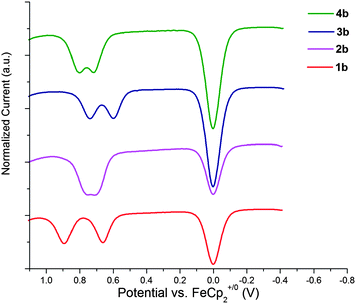 | ||
| Fig. 6 Oxidative differential pulse voltammograms (20 mV s−1; 50 mV pulse width) for bis(3,6-di-tert-butylcarbazol-9-yl) derivatives in CH2Cl2–0.1 M nBu4NPF6; the feature at 0 V corresponds to oxidation of the ferrocene internal standard. | ||
| Cpd | Experiment | Theory | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| E+/01/2a (V) | E+2/+1/2a (V) | ΔE1/2 (mV) | λonsetb (nm) | Eoptgb (eV) | IPsc (eV) | EAsd (eV) | IPverte (eV) | IPadie (eV) | IPKTf (eV) | EAKTf (eV) | IPKT − EAKT (eV) | |
| a Potentials obtained from the DPV measurements.b Onset of optical absorption.c Estimated solid-state IP obtained according to IP = eE+/01/2 + 4.8 eV.d Estimated of the solid-state IP obtained according to EA = IP − Eoptg; see text for caveats.e ΔSCF values of vertical and adiabatic ionization energies for isolated molecules.f DFT gas-phase values estimated using Koopmans' theorem, i.e. IPKT = −EHOMO and EAKT = −ELUMO. | ||||||||||||
| 1b | +0.66 | +0.89 | 237 | 358 | 3.46 | 5.5 | 2.0 | 6.21 | 6.13 | 5.12 | 0.73 | 4.39 |
| 2b | +0.71 | +0.75 | 76 | 363 | 3.42 | 5.5 | 2.1 | 6.10 | 6.05 | 5.13 | 1.15 | 3.97 |
| 3b | +0.60 | +0.74 | 148 | 370 | 3.35 | 5.4 | 2.1 | 5.99 | 5.92 | 5.02 | 1.19 | 3.83 |
| 4b | +0.72 | +0.80 | 104 | 358 | 3.46 | 5.5 | 2.1 | 6.11 | 6.06 | 5.14 | 1.87 | 3.27 |
The solid-state IPs of the compounds were estimated to be in the range 5.4–5.6 eV according to the empirically established approximate relation, IPs = eE+/01/2 + 4.8 eV, where e is the electronic charge and where the potential is relative to ferrocenium/ferrocene. The value of 5.5 eV estimated for 2b is close to a value of 5.7 eV directly measured by UV photoelectron spectroscopy for 2b (and also for 2a).9,89 Estimates of the electron affinity (EA) in the solid state, obtained using the crude approximation, EAs = IPs − Eoptg, where Eoptg is an estimate of the energy of the first singlet excited state, are also given in Table 3. In general, this approach will lead to overestimates of EAs due to the effects of exciton binding energy. Moreover, as discussed in the previous section, the onset of strong absorption correspond to the S0 → S1 transition for 1b–3b, but S0 → S5 for 4b, meaning that this approach is unlikely to even afford meaningful trends in EAs in the present series. Indeed the estimated EAs values are all similar, while the DFT estimate of the EA obtained from Koopmans’ theorem for 4b is much larger than that of 2b or 3b (Table 3). The trends in EAKT reflect the fact that the π-system of the bridge increases in size from 1b to 2b/3b and then, again, to 4b; in addition, the LUMO in 4b is strongly pyrene localized and less destabilized by contributions from the carbazole portions of the molecule than that of 1b–3b.
The DFT geometries of 1b˙+–4b˙+ were examined to gain further insight into the redox properties of these species. In principle, cations such as these might be either class-II (unsymmetrical with charge largely localized around one of the nitrogen atoms) or III (delocalized) mixed-valence species.90 Electron self-interaction errors in DFT often leads to artificially delocalized solutions;91 therefore, while delocalized class-III structures are obtained from the DFT calculations and this result is similar to what is experimentally established for bis(diarylamino)biphenyl radical cations,92 it is possible that some or all of these radical cations may in fact exhibit localized structures, in which case adiabatic IPs, geometric changes, and reorganization energies will differ somewhat from those given in Table S4.† The largest geometric changes occur in the vicinity of the carbazole–bridge linkages. The N(carbazole)–C(aryl bridge) bonds shorten by 0.01–0.02 Å, with the bond length change being largest for the smallest example, 1b. The induced strain of the alkyl linkage in the bridge of 3b induces a slightly larger change to the N–C bond versus the unconstrained biphenyl bridge of 2b, while the N–C bond change is smallest for 4b, where there is relatively little N–C(bridge) antibonding character in the HOMO. A considerable reduction in the torsion angle between the carbazole units and the neighboring aryl rings of the bridge also occurs on oxidation, again with the largest change occurring for 1b (11° reduction), an intermediate change for 3b (9°), and the smallest for 2b and 4b (ca. 6°). The twist angle between the two benzene rings of the biphenyl ring of 2b also decreases (by 7°) on oxidation, but does not significantly change in the case of 3b (where the constraints of the alkyl bridges lead to a lower twist in the neutral geometry, as discussed above). The planarization of the biphenyl bridge of 2b˙+ is reminiscent of previously reported computational and crystallographic studies of bis(diarylamino)biphenyl derivatives and their radical cations.92,93
The varying degrees of geometric modifications on oxidation lead to notable differences in the neutral/radical cation intramolecular reorganization energy, which, according to Marcus theory,94 is related to the barrier to hole transport. The smaller 1b, which undergoes the largest changes to the geometric structure on oxidation, has the largest intramolecular reorganization energy (176 meV). The alkyl bond constraints in 3b, interestingly, lead to a considerably larger intramolecular reorganization energy (150 meV) than that found in 2b (112 meV). This difference is similar to that between bis(diarylamino)biphenyl and fluorene derivatives.95,96 The intramolecular reorganization energy of 4b (90 meV) is the smallest and is comparable with that of the high hole-mobility material pentacene (98 meV).97 All of these intramolecular organization energies are smaller than those estimated for many prototypical hole-transport materials, including TPD (290 meV)95 and sexithiophene (300 meV).98
Organic light-emitting devices
To investigate the electroluminescent (EL) properties of compounds 3a, 3b, 4a and 4b, OLEDs were assembled in a standard sandwich geometry: ITO/PEDOT:PSS/[carbazole derivative]/TPBi/Ca/Al; 3a and 4a were deposited by evaporation, while 3b and 4b were spin-cast. 1,3,5-Tris(1-phenyl-1H-benzo[d]imidazol-2-yl)benzene (TPBi) is widely used as a hole-blocking/electron-transporting material,8 and in the current devices was deposited by evaporation to facilitate electron injection into the active layer. All devices give deep blue EL, with maxima in the range 422–435 nm, suggesting that emission occurs primarily from the carbazole-material layer; TPBi films show a fluorescence maximum at 383 nm.99Fig. 7 depicts the current density–voltage–luminance (J–V–L) characteristics of devices using 3a and 3b, respectively, as light-emitting layers. The tetrahydropyrene-based OLEDs gave maximum luminance values of 138 cd m−2 at 9 V for 3a and 361 cd m−2 at 10.8 V for 3b. The onset voltages of 5.3 and 5.5 V for 3a and 3b, respectively, are rather high and indicate either imbalanced charge injection or transport within the devices. Consequently the maximum device efficiencies remained low (0.02 and 0.07 cd A−1 for 3a and 3b, respectively). Devices using 3a as the light-emitting layer show an EL peak maximum at 422 nm and a weak shoulder around 375 nm. The spectra correspond to Commission Internationale de l'Eclairage 1931 (CIE1931) coordinates of x = 0.159 and y = 0.046, corresponding to a deep blue emission. For 3b devices, a slightly narrower EL emission peak was observed in a similar region to that seen for 3a devices (EL maximum at 425 nm), but a weak and slightly structured emission is also seen in the red spectral region at around 620 nm; similar EL features in PVK films have been attributed to electrophosphorescent emission.100 The overall spectrum corresponds to CIE1931 x = 0.193 and y = 0.078, which is slightly red-shifted compared to the EL-emission of 3a, but still located in the deep-blue region of the visible spectrum.
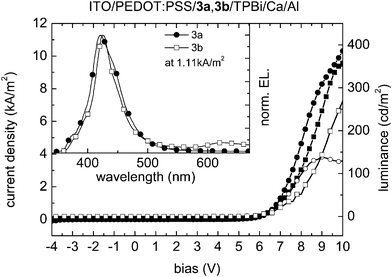 | ||
| Fig. 7 Current density (3a: line with filled circles; 3b: line with filled squares)/luminance (3a: line with open circles; 3b: line with open squares) as a function of the bias voltage of ITO/PEDOT:PSS/3a, 3b/TPBi/Ca/Al devices. Inset: electroluminescence emission spectra at a current density of 1.11 kA m−2. | ||
Fig. 8 shows the J–V–L characteristics of analogous devices in which 4a and 4b are used as emitters. Maximum luminance values of 1076 cd m−2 were obtained for the 4a device at 7.0 V and 316 cd m−2 at 7.6 V for 4b, respectively. The onset voltages of 4.0 V (4a) and 4.2 V (4b) are somewhat smaller than when tetrahydropyrene-bridged molecules 3a and 3b were used as the active layer, perhaps due to more facile electron injection into the pyrene molecules (see values of EAKT in Table 3). However, the overall device efficiencies of 0.16 cd A−1 for 4a and 0.04 cd A−1 for 4b still remain low and can again be attributed to an unbalanced charge-carrier transport in the active materials. The EL-spectra in the inset of Fig. 8 show a broad and slightly structured EL emission peak for compound 4a in the range 410–570 nm with its maximum at 435 nm and a distinct shoulder around 455 nm. The spectrum corresponds to CIE1931 coordinates x = 0.151 and y = 0.092 (deep blue). For compound 4b a narrow and structureless molecular emission peak between 410 nm and 525 nm, peaking at 435 nm, can be observed, resulting in x = 0.156 and y = 0.044 (CIE1931), at the far blue end of the visible spectrum. None of the reported devices exhibited any sign of excimer emission.
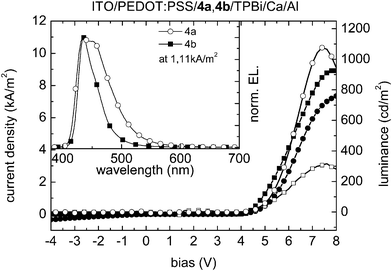 | ||
| Fig. 8 Current density (4a: line with filled circles; 4b: line with filled squares)/luminance (4a: line with open circles; 4b: line with open squares) as a function of the bias voltage of ITO/PEDOT:PSS/4a,4b/TPBi/Ca/Al devices. Inset: electroluminescence emission spectra at a current density of 1.11 kA m−2. | ||
The device work demonstrates that the tetrahydropyrene and pyrene-bridged compounds are in principle suitable emitting materials for deep-blue OLEDs. While the brightnesses and efficiencies are low compared to state-of-the-art multilayer electrophosphorescent devices employing iridium-based emitters,101,102 some of present devices compare reasonably well to blue electrofluorescent devices with comparably simple device architectures, such as devices recently reported based on dendronized or polymerized pyrene derivatives;15–17 for example a single-layer blue-turquoise device based on a polypyrene was reported to exhibit a peak luminance of 300 cd m−2 at 8 V and a maximum efficiency of ca. 0.3 cd A−1. It is anticipated that more efficient OLEDs may be obtained by introduction of additional transport layers on the anode side to ensure a more balanced charge injection.103
Conclusion
Tetrahydropyrene derivatives 3a,b show similar absorption and emission spectra to their biphenyl analogues, 2a and 2b. The pyrene derivatives 4a,b show qualitatively different behavior: DFT calculations indicate significant differences in the key frontier orbitals and that the lowest-lying strong absorption is no longer S0 → S1, the emission is considerably red-shifted relative to that of the other compounds, and the fluorescence lifetimes are generally longer. All of the tert-butyl-substituted compounds studied have reversible electrochemistry, the first oxidation potential showing only minor variations with the bridging group. DFT calculations indicate significant variation in the reorganization energy for hole-transfer between biphenyl, tetrahydropyrene, and pyrene derivatives, with the smallest values being found for the pyrene materials. Both tetrahydropyrene and pyrene derivatives have been used as blue fluorescent emitters in simple OLED architectures; lower turn-on voltages for the pyrene materials may be related to more facile electron injection from the electron-transporting layer. These molecules may also have applications as hole-transporting materials and perhaps as hosts for green phosphors in OLEDs.Acknowledgements
This work was supported by the Petroleum Research Foundation of the American Chemical Society (grant #: 47343-B10), Solvay S.A., the Science and Technology Center Program of the National Science Foundation (DMR-0120967), and NSF PREM program (DMR-0934212). B. R. K. thanks AUB for faculty development funds.References
- C. W. Tang and S. A. VanSlyke, Appl. Phys. Lett., 1987, 51, 913–915 CrossRef CAS.
- K. Müllen and U. Scherf, Organic Light Emitting Devices. Synthesis, Properties and Applications, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, 2006 Search PubMed.
- L. Xiao, Z. Chen, B. Qu, J. Luo, S. Kong, Q. Gong and J. Kido, Adv. Mater., 2011, 23, 926–952 CrossRef CAS.
- Y.-H. Kim, S. J. Lee, S.-Y. Jung, K.-N. Byeon, J.-S. Kim, S. C. Shin and S.-K. Kwon, Bull. Korean Chem. Soc., 2007, 28, 443–446 CrossRef CAS.
- C. Tang, F. Liu, Y.-J. Xia, L.-H. Xie, A. Wei, S.-B. Li, Q.-L. Fan and W. Huang, J. Mater. Chem., 2006, 16, 4074–4080 RSC.
- R. M. Adhikari, R. Mondal, B. K. Shah and D. C. Neckers, J. Org. Chem., 2007, 72, 4727–4732 CrossRef CAS.
- B. K. Shah, D. C. Neckers, J. Shi, E. W. Forsythe and D. Morton, J. Phys. Chem. A, 2005, 109, 7677–7681 CrossRef CAS.
- Y. Shirota and H. Kageyama, Chem. Rev., 2007, 107, 953–1010 CrossRef CAS.
- M.-H. Ho, B. Balaganesan, T.-Y. Chu, T.-M. Chen and C. H. Chen, Thin Solid Films, 2008, 517, 943–947 CrossRef CAS.
- J. B. Birks, Photophysics of Aromatic Molecules, Wiley-Interscience, London, 1970 Search PubMed.
- T. Förster and K. Kasper, Z. Phys. Chem., 1954, 1, 275–277 CrossRef.
- J. B. Birks, D. J. Dyson and I. H. Munro, Proc. R. Soc. London, Ser. A, 1963, 275, 575–588 CrossRef CAS.
- K. Kalyanasundaram and J. K. Thomas, J. Am. Chem. Soc., 1977, 99, 2039–2044 CrossRef CAS.
- C.-H. Yang, T.-F. Guo and I. W. Sun, J. Lumin., 2007, 124, 93–98 CrossRef CAS.
- T. Qin, W. Wiedemair, S. Nau, R. Trattnig, S. Sax, S. Winkler, A. Vollmer, N. Koch, M. Baumgarten, E. J. W. List and K. Müllen, J. Am. Chem. Soc., 2011, 133, 1301–1303 CrossRef CAS.
- R. Trattnig, T. M. Figueira-Duarte, D. Lorbach, W. Wiedemair, S. Sax, S. Winkler, A. Vollmer, N. Koch, M. Manca, M. A. Loi, M. Baumgarten, E. J. W. List and K. Müllen, Opt. Express, 2011, 19, A1281–A1293 CrossRef CAS.
- T. M. Figueira-Duarte, P. G. D. Rosso, R. Trattnig, S. Sax, E. J. W. List and K. Müllen, Adv. Mater., 2010, 22, 990–993 CrossRef CAS.
- Y. Tao, Q. Wang, L. Ao, C. Zhong, J. Qin, C. Yang and D. Ma, J. Mater. Chem., 2010, 20, 1759–1765 RSC.
- L. Duan, J. Qiao, Y. Sun and Y. Qiu, Adv. Mater., 2011, 23, 1137–1144 CrossRef CAS.
- Y. Zhang, C. Zuniga, S.-J. Kim, D. Cai, S. Barlow, S. Salman, V. Coropceanu, J.-L. Brédas, B. Kippelen and S. R. Marder, Chem. Mater., 2011, 4002–4015 CrossRef CAS.
- N. Matsusue, Y. Suzuki and H. Naito, Jpn. J. Appl. Phys., Part 1, 2005, 44, 3691–3694 CrossRef CAS.
- J. Kido, K. Hongawa, K. Okuyama and K. Nagai, Appl. Phys. Lett., 1993, 63, 2627–2629 CrossRef CAS.
- Q. Zhang, Y. F. Hu, Y. X. Cheng, G. P. Su, D. G. Ma, L. X. Wang, X. B. Jing and F. S. Wang, Synth. Met., 2003, 137, 1111–1112 CrossRef CAS.
- S.-J. Kim, J. Leroy, C. Zuniga, Y. Zhang, L. Zhu, J. S. Sears, S. Barlow, J.-L. Brédas, S. R. Marder and B. Kippelen, Org. Electron., 2011, 12, 1314–1318 CrossRef CAS.
- G. Kremser, O. T. Hofmann, S. Sax, S. Kappaun, E. J. W. List, E. Zojer and C. Slugovc, Monatsh. Chem., 2008, 139, 223–231 CrossRef CAS.
- Y. Wang, Y. Hua, X. Wu, L. Zhang, Q. Hou, F. Guan, N. Zhang, S. Yin and X. Cheng, Org. Electron., 2008, 9, 692–698 CrossRef CAS.
- Y. Tao, Q. Wang, C. Yang, Q. Wang, Z. Zhang, T. Zou, J. Qin and D. Ma, Angew. Chem., Int. Ed., 2008, 47, 8104–8107 CrossRef CAS.
- Z. Zhao, X. Xu, H. Wang, P. Lu, G. Yu and Y. Liu, J. Org. Chem., 2008, 73, 594–602 CrossRef CAS.
- C. Adachi, M. A. Baldo and S. R. Forrest, J. Appl. Phys., 2000, 87, 8049–8055 CrossRef CAS.
- J. F. Ambrose and R. F. Nelson, J. Electrochem. Soc., 1968, 115, 1159–1164 CrossRef CAS.
- J. F. Ambrose, L. L. Carpenter and R. F. Nelson, J. Electrochem. Soc., 1975, 122, 876–894 CrossRef CAS.
- G. Inzelt, J. Solid State Electrochem., 2003, 7, 503–510 CrossRef CAS.
- T. Yamada, F. Suzuki, A. Goto, T. Sato, K. Tanaka and H. Kaji, Org. Electron., 2011, 12, 169–178 CrossRef CAS.
- K. R. J. Thomas, J. T. Lin, Y.-T. Tao and C.-W. Ko, Adv. Mater., 2000, 12, 1949–1951 CrossRef CAS.
- Y. Xing, X. Xu, P. Zhang, W. Tian, G. Yu, P. Lu, Y. Liu and D. Zhu, Chem. Phys. Lett., 2005, 408, 169–173 CrossRef CAS.
- Z. Zhao, X. Xu, F. Wang, G. Yu, P. Lu, Y. Liu and D. Zhu, Synth. Met., 2006, 156, 209–214 CrossRef CAS.
- H.-C. Ting, Y.-M. Chen, H.-W. You, W.-Y. Hung, S.-H. Lin, A. Chaskar, S.-H. Chou, Y. Chi, R.-H. Liu and K.-T. Wong, J. Mater. Chem., 2012, 22, 8399–8407 RSC.
- D. M. Connor, S. D. Allen, D. M. Collard, C. L. Liotta and D. A. Schiraldi, J. Org. Chem., 1999, 64, 6888–6890 CrossRef CAS.
- D. Rausch and C. Lambert, Org. Lett., 2006, 8, 5037–5040 CrossRef CAS.
- H. Lee and R. G. Harvey, J. Org. Chem., 1986, 51, 2847–2848 CrossRef CAS.
- Y. Liu, M. Nishiura, Y. Wang and Z. Hou, J. Am. Chem. Soc., 2006, 128, 5592–5593 CrossRef CAS.
- B. E. Koene, D. E. Loy and M. E. Thompson, Chem. Mater., 1998, 10, 2235–2250 CrossRef CAS.
- Q. Zhang, J. Chen, Y. Cheng, L. Wang, D. Ma, X. Jing and F. Wang, J. Mater. Chem., 2004, 14, 895–900 RSC.
- R. Anemian, Y. Morel, P. L. Baldeck, B. Paci, K. Kretsch, J.-M. Nunzi and C. Andraud, J. Mater. Chem., 2003, 13, 2157–2163 RSC.
- SAINT+; 6.2a, Bruker Analytical X-ray System, Inc., Madison, WI, 2001 Search PubMed.
- SADABS, Bruker Analytical X-ray System, Inc., Madison, WI, 1999 Search PubMed.
- SHELXTL; 6.10, Bruker Analytical X-ray System, Inc., Madison, WI, 1997 Search PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS.
- S. H. Vosko, L. Wilk and M. Nusair, Can. J. Phys., 1980, 58, 1200–1211 CrossRef CAS.
- P. J. Stephens, F. J. Devlin, C. F. Chabalowski and M. J. Frisch, J. Phys. Chem., 1994, 98, 11623–11627 CrossRef CAS.
- P. C. Hariharan and J. A. Pople, Theor. Chim. Acta, 1973, 28, 213–222 CrossRef CAS.
- M. M. Francl, W. J. Pietro, W. J. Hehre, J. S. Binkley, M. S. Gordon, D. J. Defrees and J. A. Pople, J. Chem. Phys., 1982, 77, 3654–3665 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision A.02, Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
- H. Volmmann, H. Becher, M. Corell and H. Streeck, Justus Liebigs Ann. Chem., 1937, 531, 1–159 CrossRef.
- A. G. Crawford, A. D. Dwyer, Z. Liu, A. Steffen, A. Beeby, L.-O. Palsson, D. J. Tozer and T. B. Marder, J. Am. Chem. Soc., 2011, 133, 13349–13362 CrossRef CAS.
- A. Hameurlaine and W. Dehaen, Tetrahedron Lett., 2003, 44, 957–959 CrossRef CAS.
- M. S. Driver and J. F. Hartwig, J. Am. Chem. Soc., 1996, 118, 7217–7218 CrossRef CAS.
- J. P. Wolfe, S. Wagaw and S. L. Buchwald, J. Am. Chem. Soc., 1996, 118, 7215–7216 CrossRef CAS.
- H. Nakashima, S. Kawakami, S. Shitagaki and S. Seo, US Pat. 2007/0096639, 2007.
- P. J. Low, M. A. J. Paterson, D. S. Yufit, J. A. K. Howard, J. C. Cherryman, D. R. Tackley, R. Brook and B. Brown, J. Mater. Chem., 2005, 15, 2304–2315 RSC.
- Although biphenyl was formerly considered as adopting a planar conformation in the crystal (J. Trotter, Acta Crystallogr., 1961, 14, 1135; A. Hargreaves and A. H. Rizvi, Acta Crystallogr., 1962, 15, 365), this was subsequently found to be an artefact of disorder, with a phase transition being observed to give an ordered phase below 40 K with the twist angle of ca. 10° in that phase (H. Cailleau, J. L. Baudour and C. M. E. Zeyen, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1979, 35, 426); moreover, when found in general positions within other structures, biphenyl molecules are also found to be twisted (e.g., D. Naae, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1979, 35, 2765; A. Pénicaud, O. Y. Carréon, A. Perrier, D. J. Watkin and C. Coulon, J. Mater. Chem. 2002, 12, 913; R. E. Douthwaite, A. Taylor and A. C. Whitwood, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2005, 61, o328).
- W. Li, J. Qiao, L. Duan, L. Wang and Y. Qiu, Tetrahedron, 2007, 63, 10161–10168 CrossRef CAS.
- M. A. Reddy, A. Thomas, B. Sridha, V. J. Rao and K. Bhanuprakash, Tetrahedron Lett., 2011, 52, 6942–6947 CrossRef CAS.
- W. Wu and J. Tong, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2011, 67, o1919 Search PubMed.
- S. Saha and S. Samanta, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1999, 55, 1299–1300 Search PubMed.
- F. H. Allen, O. Kennard, D. G. Watson, L. Brammer, A. G. Orpen and R. Taylor, J. Chem. Soc., Perkin Trans. 2, 1987, S1–S19 RSC.
- V. Jankus, C. Winscom and A. P. Monkman, J. Chem. Phys., 2009, 130, 074501 CrossRef.
- I. B. Berlman, Handbook of Fluorescence Spectra of Aromatic Molecules, Academic Press, New York, 2nd edn, 1971 Search PubMed.
- The effects of electron self-interaction errors in the general DFT formalism can often lead to red-shifted absorption spectra for conjugated systems.
- J. V. Goodpaster, J. F. Harrison and V. L. McGuffin, J. Phys. Chem. A, 1998, 102, 3372–3381 CrossRef CAS.
- Y. Bito, N. Shida and T. Toru, Chem. Phys. Lett., 2000, 328, 310–315 CrossRef CAS.
- M. Parac and S. Grimme, Chem. Phys., 2003, 292, 11–21 CrossRef CAS.
- S. Grimme and M. Parac, ChemPhysChem, 2003, 4, 292–295 CrossRef CAS.
- B.-C. Wang, J.-C. Chang, H.-C. Tso, H.-F. Hsu and C.-Y. Cheng, J. Mol. Struct.: THEOCHEM, 2003, 629, 11–20 CrossRef CAS.
- M. Dierksen and S. Grimme, J. Chem. Phys., 2004, 120, 3544–3554 CrossRef CAS.
- Y. H. Park and B.-S. Cheong, Curr. Appl. Phys., 2006, 6, 700–705 CrossRef.
- I. S. K. Kerkines, I. D. Petsalakis, G. Theodorakopoulos and W. Klopper, J. Chem. Phys., 2009, 131, 224315 CrossRef.
- O. K. Bazyl, G. V. Maier, T. N. Kopylova and V. I. Danilova, Zh. Prikl. Spektrosk., 1982, 37, 80–86 CAS.
- S. Grimme and F. Neese, J. Chem. Phys., 2007, 127, 154116 CrossRef.
- A. Sharma and S. Schulman, Introduction to Fluorescence Spectroscopy, Wiley-Interscience, USA, 1999 Search PubMed.
- J. L. Kropp, W. R. Dawson and M. W. Windsor, J. Phys. Chem., 1969, 73, 1747–1752 CrossRef CAS.
- S. Krotkus, K. Kazlauskas, A. Miasojedovas, A. Gruodis, A. Tomkeviciene, J. V. Grazulevicius and S. Jursenas, J. Phys. Chem. C, 2012, 116, 7561–7572 CAS.
- J. D. Anderson, E. M. McDonald, P. A. Lee, M. L. Anderson, E. L. Ritchie, H. K. Hall, T. Hopkins, E. A. Nash, J. Wang, A. Padias, S. Thayumanavan, S. Barlow, S. R. Marder, G. Jabbour, S. Shaheen, B. Kippelen, N. Peyghambarian, R. M. Wightman and N. R. Armstrong, J. Am. Chem. Soc., 1998, 120, 9646–9655 CrossRef CAS.
- C. Risko, V. Coropceanu, S. Barlow, V. Geskin, K. Schmidt, N. E. Gruhn, S. R. Marder and J.-L. Brédas, J. Phys. Chem. C, 2008, 112, 7959–7967 CAS.
- F. Scholz, Electroanalytical Methods: Guide to Experiments and Applications, Springer, New York, 2002 Search PubMed.
- C. Lambert and G. J. Nöll, J. Am. Chem. Soc., 1999, 121, 8434–8442 CrossRef CAS.
- J. E. Sutton and H. Taube, Inorg. Chem., 1981, 20, 3125–3134 CrossRef CAS.
- However, a rather different UV-PES value of 6.25 eV has also been reported for 2a (A. Kahn, N. Koch and W. Gao, J. Polym. Sci., Part B: Polym. Phys., 2003, 41, 2529).
- J. Hankache and O. S. Wenger, Chem. Rev., 2011, 111, 5138–5178 CrossRef CAS.
- T. Bally and W. T. Borden, Reviews in Computational Chemistry, Wiley-VCH, New York, 1999 Search PubMed.
- P. J. Low, M. A. J. Paterson, H. Puschmann, A. E. Goeta, J. A. K. Howard, C. Lambert, J. C. Cherryman, D. R. Tackley, S. Leeming and B. Brown, Chem.–Eur. J., 2004, 10, 83–91 CrossRef CAS.
- V. Coropceanu, M. Malagoli, J. M. André and J.-L. Brédas, J. Am. Chem. Soc., 2002, 124, 10519–10530 CrossRef CAS.
- R. A. Marcus, Rev. Mod. Phys., 1993, 65, 599–610 CrossRef CAS.
- M. Malagoli and J.-L. Brédas, Chem. Phys. Lett., 2000, 327, 13–17 CrossRef CAS.
- R. D. Hreha, C. P. George, A. Haldi, B. Domercq, M. Malagoli, S. Barlow, J.-L. Brédas, B. Kippelen and S. R. Marder, Adv. Funct. Mater., 2003, 13, 967–973 CrossRef CAS , and references cited therein.
- V. Coropceanu, M. Malagoli, D. A. da Silva Filho, N. E. Gruhn, T. G. Bill and J.-L. Brédas, Phys. Rev. Lett., 2002, 89, 275503 CrossRef CAS.
- D. A. da Silva Filho, V. Coropceanu, D. Fichou, N. E. Gruhn, T. G. Bill, J. Gierschner, J. Cornil and J.-L. Brédas, Philos. Trans. R. Soc., A, 2007, 365, 1435–1452 CrossRef CAS.
- Z. Wang, P. Lu, S. Chen, Z. Gao, F. Shen, W. Zhang, Y. Xu, H. S. Kwok and Y. Ma, J. Mater. Chem., 2011, 21, 5451–5456 RSC.
- L. Qian, D. Bera and P. H. Holloway, Appl. Phys. Lett., 2007, 90, 103511 CrossRef.
- Y.-C. Zhu, L. Zhou, H.-Y. Li, Q.-L. Xu, M.-Y. Teng, Y.-X. Zheng, J.-L. Zuo, H.-J. Zhang and X.-Z. You, Adv. Mater., 2011, 23, 4041–4046 CrossRef CAS.
- C. Ulbricht, B. Beyer, C. Friebe, A. Winter and U. S. Schubert, Adv. Mater., 2009, 21, 4418–4441 CrossRef CAS.
- Y. Shen, A. R. Hosseini, M. H. Wong and G. G. Malliaras, ChemPhysChem, 2004, 5, 16–25 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Packing diagrams, select bond angles and dihedral angles of the ground state and radical-cation states, absorption and emission data in different solvents, frontier molecular orbitals, CV plots, DSC plots, 1H NMR spectra, and cif files. CCDC 899423–899426. For ESI and crystallographic data in CIF or other electronic format See DOI: 10.1039/c2tc00474g |
| This journal is © The Royal Society of Chemistry 2013 |