Synthesis of graphene by low-temperature exfoliation and reduction of graphite oxide under ambient atmosphere†
Bin
Shen
,
Dingding
Lu
,
Wentao
Zhai
* and
Wenge
Zheng
*
Ningbo Key Lab of Polymer Materials, Ningbo Institute of Material Technology and Engineering, Chinese Academy of Sciences, Ningbo, Zhejiang province 315201, China. E-mail: wtzhai@nimte.ac.cn; wgzheng@nimte.ac.cn; Fax: +86 0574 8668 5186; Tel: +86 0574 8668 5256
First published on 3rd October 2012
Abstract
We firstly report a facile approach to produce few-layered graphene sheets by low-temperature (130 °C) exfoliation and reduction of graphite oxide under ambient atmosphere with the aid of HCl. The obtained graphene materials exhibited high BET specific surface area (∼500 m2 g−1) and excellent electrical conductivity (∼1200 S m−1).
Graphene, a new class of two-dimensional (2D) nanomaterial consisting of a single layer of sp2 networks of carbon atoms, possesses extraordinary electrical, mechanical, and thermal properties.1 These unique features offer great promise for many applications including nanoelectronics, polymer composites, solar cells, as well as batteries.2–6 Graphene has been prepared by several approaches such as micromechanical exfoliation of graphite, chemical vapor deposition, epitaxial growth, and the exfoliation and reduction of graphite oxide (GO).7–10 However, the low productivity of the first three methods makes them unsuitable for large-scale applications and the exfoliation and reduction of GO becomes the most promising route for the bulk production of graphene.
Among different approaches for the exfoliation and reduction, thermal exfoliation of GO by rapid heating treatment at high temperature is the most promising one for mass production of graphene sheets.10,11 As reported by McAllister et al.,10 high temperature (1050 °C) and rapid heating (>2000 °C min−1) were the key parameters for full exfoliation of GO. However, the thermal treatment at such a high temperature with the fast heating rate involves huge energy consumption and difficulties in operation. It is necessary to develop an approach for thermal exfoliation of GO at a greatly reduced temperature. A few methods have been explored to obtain few-layered graphene sheets by thermally exfoliating GO under vacuum at a significantly low temperature (135–250 °C).12,13 The vacuum has been thought to exert an outward force on the expanding graphene layers and help accelerate the expansion of graphene layers. Moreover, thermal exfoliation and reduction of GO in a hydrogen (H2) atmosphere at ∼200 °C has also been reported, and the high exfoliation efficiency is due to the violent reaction between hydrogen and functional groups on GO.14 However, these methods require a special atmosphere, such as ultra-high vacuum, H2, which is still one of the big obstacles for the bulk production of graphene.
In this study, we explore a facile and fast approach to produce few-layered graphene sheets by low-temperature exfoliation and reduction of GO under ambient atmosphere (without high-vacuum or any other special atmosphere). The exfoliation temperature of GO was as low as 130 °C and the exfoliation process was carried out in air within ∼8 min. To the best of our knowledge, this is the first example of successful low-temperature exfoliation and reduction of GO under ambient atmosphere, which opens up new opportunities for the bulk production of graphene and the development of graphene composite materials.
As we know, the oxygen-containing groups of GO can decompose to produce gases like H2O and CO2 in the thermal treatment. For a successful exfoliation process, the pressure generated from evolved gases that cause rapid expansion should exceed the van der Waals forces holding GO sheets together.15 If GO is heated slowly at low temperature, gas evolution and expansion occur slowly enough that the lateral diffusion can relieve the generated pressure.15 Therefore, it is insufficient to yield high enough pressure to overcome the van der Waals forces without the aid of high-vacuum. The above discussion hints us that if the inner pressure can be reinforced at low temperature, a fast expansion–exfoliation of GO sheets may be achieved under ambient atmosphere and few-layered graphene can be produced at such a low temperature. We realized this process by introducing a volatile substance (HCl) into the inter-lamellar region of GO during the washing process. The volatile HCl could vaporize and expand due to the external heating, as well as the strong internal heating during the GO decomposition at a low temperature,11 supplying an extra pressure on expanding the GO sheets, which helps accelerate the expansion of GO and results in an effective exfoliation of GO under ambient atmosphere (as schematically represented in Fig. 1).
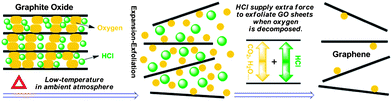 | ||
| Fig. 1 Schematic representation of low-temperature exfoliation of GO sheets under ambient atmosphere with the assistance of volatile HCl. | ||
Herein, GO was prepared by using the Staudenmaier method,14,16 and the detailed preparation process is described in the ESI.† In order to increase the filtrate efficiency, the GO slurry was filtered and washed with 10% HCl solution instead of deionized water to remove the sulfuric ions. Then, it was completely dried at 80 °C without washing with water to remove HCl. Therefore, some HCl could be kept in the inter-lamellar region of GO after drying. In order to confirm this supposal, the GO slurry with a concentration of 2 mg ml−1 was prepared and the pH was tested to be ∼2, which suggested the existence of HCl in the dried GO. Moreover, the XPS spectrum of GO (Fig. S1†) further confirmed the existence of Cl element of HCl in the GO sample. Finally, the obtained GO was placed in a pre-heated beaker (heated in an oven) at 130 °C (the initial decomposition temperature of oxygen-containing groups of GO as shown in Fig. S2†) under ambient atmosphere. About 8 min later, explosive expansion of GO accompanied by flash flame was observed (Fig. S3†). The volume of the GO sample expanded for a few hundred times in the whole process and few-layered graphene was obtained at the gram scale. The obtained sample was denoted as G-130 and the absence of the Cl element in its XPS spectrum (Fig. S1†) was indicative of the vaporization of HCl during the exfoliation. For comparison, the residual HCl in GO was intentionally washed off with water until the pH was neutral. As expected, the prepared GO could not be exfoliated at 130 °C under ambient atmosphere, which confirmed that the residual HCl played a positive role in promoting the exfoliation of GO.
The exfoliation degree of GO and the morphologies of the obtained G-130 were characterized by X-ray diffraction (XRD) and microscopic observations. Fig. 2a shows XRD patterns of pristine graphite, GO and resulting G-130. Compared with pristine graphite, which is characterized by a sharp peak at 26.6° (002) with a typical interlayer spacing of 0.33 nm, the XRD pattern of GO shows a typical peak located at 12.7° (002), corresponding to an interlayer spacing of 0.77 nm. This indicates that graphite was totally transformed into GO and most oxygen was bonded to the planar surface of graphite after oxidization. After the low-temperature heat-treatment under ambient atmosphere, the obvious characteristic peak around 12° disappeared, indicating that the periodic layered structure of GO sheets was exfoliated randomly. However, a new peak at 24.0° appears in the XRD pattern of G-130, and its intensity is very weak, as shown in the inset of Fig. 2a. This result indicates that aggregation persisted to some extent in G-130, which was supposed to arise from the strong van der Waals interactions between the G-130 sheets.
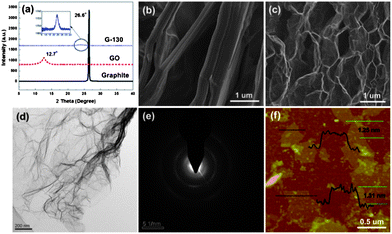 | ||
| Fig. 2 (a) XRD patterns of pristine graphite, GO and G-130 powder; (b and c) SEM images of GO and G-130 powder; (d and e) TEM images of G-130 sheets and their SAED; and (f) a typical tapping mode AFM image of G-130 sheets deposited on a mica substrate. | ||
The low-temperature exfoliation was also verified by scanning electron microscopy (SEM) observation. The representative SEM image of G-130 (Fig. 2c), compared to that of parent GO (Fig. 2b), showed that GO sheets were efficiently exfoliated to ultrathin graphene sheets with only a few parts connected with each other (Fig. 2c). This morphology is very similar to that of graphene prepared by high-temperature exfoliation at 1050 °C or low-temperature exfoliation under high-vacuum.10,12,13Fig. 2d presents a transmission electron microscopy (TEM) image of G-130. It is seen that G-130 resembles a transparent ultrathin film with a few thin ripples within the plane. As reported by researchers, the ripples are intrinsic properties of thin graphene sheets, due to the extra thermodynamic stability of the 2D membranes arising from microscopic crumpling.17 In addition, Fig. 2e shows the selected area electron diffraction (SAED) pattern of G-130. It is composed of diffraction rings and well-defined diffraction spots, confirming the well-crystallized single-layer graphene structure, though it is not as perfect as pristine monolayered graphene. This is reasonable because some defects still remained in the restored graphitic structure resulting from the removal of oxygen functionalities.
The surface area of the as-prepared G-130 was determined by BET surface area measurements. From the linear region of the graph (Fig. 3a) and using the BET equation, the BET specific surface area was determined to be ∼500 m2 g−1, which is lower than that of graphene obtained at 1050 °C (∼700 m2 g−1) and higher than that of graphene obtained at 200 °C under vacuum (∼400 m2 g−1).4,13 This was about ∼5.3 times lower than the ideal specific surface area (2630 m2 g−1) of a single graphene sheet,18 which indicated that each G-130 platelet was comprised of ∼5 to 6 individual graphene sheets on average. We further characterized the G-130 sheets by using AFM after fully sonicating these sheets (300 W, 15 min) with the aid of surfactant (sodium dodecylsulfate, SDS). Fig. 2f presents a typical AFM image, where the thickness of graphene sheets varies from 1.2 to 1.3 nm according to cross-sectional analysis. Statistics about the distribution of the thickness revealed that over 70% of the observed individual sheets were of the thickness below 1.3 nm. Considering the presence of SDS surrounding the G-130 sheets,13,19 as well as the “bumps” resulting from the residual oxygen functionalities,11,12 most of these sheets were believed to be single-layered. It indicates that these as-prepared G-130 sheets could be readily exfoliated to isolated sheets with the help of sonication.
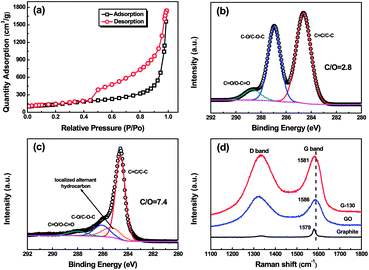 | ||
| Fig. 3 (a) Nitrogen adsorption and desorption isotherms of G-130; (b and c) C 1s XPS spectra of GO and G-130; and (d) Raman spectra of pristine graphite, GO and G-130 with a laser of 633 nm. | ||
In addition to the low-temperature exfoliation, GO sheets could also be reduced during the thermal treatment process. The XPS curves of GO and G-130 reveal that G-130 has a higher C/O atomic ratio (7.4) than GO (2.8), suggesting partial reduction during the exfoliation process (Fig. S1†). Fig. 3b and c show the C 1s spectra of GO and G-130. The binding energies at ∼284.6 eV, ∼285.3 eV, ∼286.5 eV and ∼288.3 eV are assigned to the unoxidized graphite carbon skeleton (C–C/C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C), localized alternant hydrocarbon,20,21 hydroxyl or epoxide group (C–OH/C–O–C) and carboxyl group (CO/O–CO), respectively. As listed in Table S1,† the relative atomic percentages of C–OH/C–O–C and CO/O–CO groups decrease to 14.2% and 6.5% for G-130 sheets, in comparison to 38.1% and 7.1% for GO sheets. The thermal reduction was further confirmed by Raman spectra. As shown in Fig. 3d, the G-band of G-130 red-shifts to 1581 cm−1, which is close to the value of pristine graphite (1579 cm−1), indicating the successful reduction of GO.12,22 The intensity ratio of D to G (I(D/G)), correlating with the disordered and ordered crystal structures of carbon, is inverse to the average size of sp2 domains.23 The I(D/G) of G-130 is decreased from 1.10 to 1.01, which is due to the increase in the average size of the graphitic sp2 domains upon reduction.
C), localized alternant hydrocarbon,20,21 hydroxyl or epoxide group (C–OH/C–O–C) and carboxyl group (CO/O–CO), respectively. As listed in Table S1,† the relative atomic percentages of C–OH/C–O–C and CO/O–CO groups decrease to 14.2% and 6.5% for G-130 sheets, in comparison to 38.1% and 7.1% for GO sheets. The thermal reduction was further confirmed by Raman spectra. As shown in Fig. 3d, the G-band of G-130 red-shifts to 1581 cm−1, which is close to the value of pristine graphite (1579 cm−1), indicating the successful reduction of GO.12,22 The intensity ratio of D to G (I(D/G)), correlating with the disordered and ordered crystal structures of carbon, is inverse to the average size of sp2 domains.23 The I(D/G) of G-130 is decreased from 1.10 to 1.01, which is due to the increase in the average size of the graphitic sp2 domains upon reduction.
Electrical conductivity of the compressed G-130 sample was measured via the linear four-probe method. Despite the defective structure of G-130 sheets and some residual functional sites after the exfoliation and reduction step, the DC conductivity of compacts at a bulk density of ∼0.3 g cm−3 is ∼1200 S m−1. This value compares favorably with the conductivity (1000–2300 S m−1) of compressed high-temperature exfoliated graphene sheets at a similar bulk density,11,15 which makes the as-prepared G-130 possible in producing conductive composites, lithium batteries and electrochemical energy storage devices. As an application demonstration, the polystyrene (PS)/G-130 composites were prepared by solution blending and their electrical conductivity are shown in Fig. 4a. Clearly, the incorporation of G-130 greatly improved the electrical conductivity of PS, resulting in a sharp transition from an electrical insulator to a semiconductor with a low percolation threshold of ∼0.6 vol%, which is estimated by using the power law (Fig. S4†).16 A high electrical conductivity of ∼1 S m−1 was achieved with only 2.3 vol% of G-130. These results indicate that the as-prepared G-130 has great potential in improving electrical conductivity of polymers.
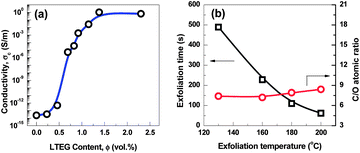 | ||
| Fig. 4 (a) Electrical conductivity of polystyrene/G-130 composites as a function of filler volume fraction. (b) The exfoliation time and C/O atomic ratio of graphene exfoliated at different temperatures. | ||
Furthermore, by using this approach, G-160, G-180, and G-200, thermally treated at 160, 180, and 200 °C under ambient atmosphere, respectively, were also prepared. Compared with G-130, G-160, G-180, and G-200 show no apparent difference from the morphological observations (Fig. S5†). As shown in Fig. 4b, the C/O ratios of the as-prepared graphene increase only from 7.4 to 8.4 by increasing the exfoliation temperature from 130 °C to 200 °C. However, the exfoliation times significantly reduced from ∼8 min (G-130) to ∼1 min (G-200), which might be due to a faster heating rate at higher temperature. Moreover, the BET surface areas of G-160, G-180, and G-200 were ∼620, ∼700, and ∼750 m2 g−1, respectively, indicating that higher exfoliation temperature could promote the efficiency of exfoliation. Research is now ongoing to further identify the HCl-assisted exfoliation mechanism and the impact of temperature on exfoliation behaviors.
In summary, we have demonstrated a facile thermal exfoliation and reduction method for scalable synthesis of high quality graphene from GO under ambient atmosphere at a low temperature of 130 °C in only 8 min. The obtained graphene has high BET specific surface area (∼500 m2 g−1) and excellent electrical conductivity (∼1200 S m−1). Similarly, we suppose that other volatile substances other than HCl would also be able to facilitate exfoliation of GO into graphene and more examples are underway to further prove this mechanism.
Acknowledgements
The authors are grateful to the National Natural Science Foundation of China (Grant 51003115), and Ningbo Natural Science Foundation (Grant no. 2011A6101118) for their financial support of this study.Notes and references
- A. K. Geim and K. S. Novoselov, Nat. Mater., 2007, 6, 183–191 CrossRef CAS.
- X. Li, X. Wang, L. Zhang, S. Lee and H. Dai, Science, 2008, 319, 1229–1232 CrossRef CAS.
- X. Wang, L. Zhi, N. Tsao, Ž. Tomović, J. Li and K. Müllen, Angew. Chem., Int. Ed., 2008, 47, 2990–2992 CrossRef CAS.
- E. Yoo, J. Kim, E. Hosono, H.-s. Zhou, T. Kudo and I. Honma, Nano Lett., 2008, 8, 2277–2282 CrossRef CAS.
- B. Shen, W. T. Zhai, C. Chen, D. D. Lu, J. Wang and W. G. Zheng, ACS Appl. Mater. Interfaces, 2011, 3, 3103–3109 CAS.
- B. Shen, W. T. Zhai, D. D. Lu, J. Wang and W. G. Zheng, RSC Adv., 2012, 2, 4713–4719 RSC.
- K. S. Kim, Y. Zhao, H. Jang, S. Y. Lee, J. M. Kim, K. S. Kim, J.-H. Ahn, P. Kim, J.-Y. Choi and B. H. Hong, Nature, 2009, 457, 706–710 CrossRef CAS.
- P. W. Sutter, J.-I. Flege and E. A. Sutter, Nat. Mater., 2008, 7, 406–411 CrossRef CAS.
- K. S. Novoselov, A. K. Geim, S. V. Morozov, D. Jiang, Y. Zhang, S. V. Dubonos, I. V. Grigorieva and A. A. Firsov, Science, 2004, 306, 666–669 CrossRef CAS.
- M. J. McAllister, J.-L. Li, D. H. Adamson, H. C. Schniepp, A. A. Abdala, J. Liu, M. Herrera-Alonso, D. L. Milius, R. Car, R. K. Prud'homme and I. A. Aksay, Chem. Mater., 2007, 19, 4396–4404 CrossRef CAS.
- H. C. Schniepp, J.-L. Li, M. J. McAllister, H. Sai, M. Herrera-Alonso, D. H. Adamson, R. K. Prud'homme, R. Car, D. A. Saville and I. A. Aksay, J. Phys. Chem. B, 2006, 110, 8535–8539 CrossRef CAS.
- H.-B. Zhang, J.-W. Wang, Q. Yan, W.-G. Zheng, C. Chen and Z.-Z. Yu, J. Mater. Chem., 2011, 21, 5392–5397 RSC.
- W. Lv, D.-M. Tang, Y.-B. He, C.-H. You, Z.-Q. Shi, X.-C. Chen, C.-M. Chen, P.-X. Hou, C. Liu and Q.-H. Yang, ACS Nano, 2009, 3, 3730–3736 CrossRef CAS.
- A. Kaniyoor, T. T. Baby and S. Ramaprabhu, J. Mater. Chem., 2010, 20, 8467–8469 RSC.
- H. Kim, A. A. Abdala and C. W. Macosko, Macromolecules, 2010, 43, 6515–6530 CrossRef CAS.
- H.-B. Zhang, W.-G. Zheng, Q. Yan, Y. Yang, J.-W. Wang, Z.-H. Lu, G.-Y. Ji and Z.-Z. Yu, Polymer, 2010, 51, 1191–1196 CrossRef CAS.
- J. C. Meyer, A. K. Geim, M. I. Katsnelson, K. S. Novoselov, T. J. Booth and S. Roth, Nature, 2007, 446, 60–63 CrossRef CAS.
- A. Peigney, C. Laurent, E. Flahaut, R. R. Bacsa and A. Rousset, Carbon, 2001, 39, 507–514 CrossRef CAS.
- H. Limin, C. Xiaodong, D. Gordana and P. O. B. Stephen, Nanotechnology, 2004, 15, 1450 CrossRef.
- J. I. Paredes, S. Villar-Rodil, P. Solís-Fernández, A. Martínez-Alonso and J. M. D. Tascón, Langmuir, 2009, 25, 5957–5968 CrossRef CAS.
- D.-Q. Yang, J.-F. Rochette and E. Sacher, Langmuir, 2005, 21, 8539–8545 CrossRef CAS.
- S. Stankovich, R. D. Piner, S. T. Nguyen and R. S. Ruoff, Carbon, 2006, 44, 3342–3347 CrossRef CAS.
- K. N. Kudin, B. Ozbas, H. C. Schniepp, R. K. Prud'homme, I. A. Aksay and R. Car, Nano Lett., 2007, 8, 36–41 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c2tc00044j |
| This journal is © The Royal Society of Chemistry 2013 |