 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTuning the porous texture and specific surface area of nanoporous carbons for supercapacitor electrodes by adjusting the hydrothermal synthesis temperature
Huajun Liu*, Yu Zhang, Qingqing Ke, Kuan Hung Ho, Yating Hu and John Wang*
Department of Materials Science and Engineering, National University of Singapore, Singapore. E-mail: mseliuh@nus.edu.sg; msewangj@nus.edu.sg
First published on 30th August 2013
Abstract
Nanoporous carbons with ordered mesopores were synthesized by the hydrothermal treatment of environmentally friendly carbohydrate precursor and Pluronic F127 as the soft template. The pore texture, pore size distribution and specific surface area of the nanoporous carbons were found to be very sensitive to the hydrothermal synthesis temperature. Long-range ordered mesoporous carbon with a specific surface area of above 1100 m2 g−1 was achieved at the hydrothermal synthesis temperature of 130 °C, and showed a large electrochemical capacitance of ∼290 F g−1 at scan rate of 1 mV s−1 in 6 M KOH aqueous electrolyte together with electrochemical stability for up to 6000 cycles of galvanostatic charge–discharge at 10 A g−1.
1. Introduction
Electrochemical capacitors or supercapacitors have attracted much attention recently as energy storage systems, filling the gap between batteries and conventional capacitors.1 The advantage of electrochemical capacitors, as compared to batteries, is the high power density, leading to extremely fast charge and discharge.2 Currently, three types of electrochemical capacitors are extensively studied based on the charge storage mechanism: electrochemical double layer capacitors (EDLCs), pseudo-capacitors and hybrid capacitors.1 Among the three types, EDLCs are especially promising as they involve only electrostatic charge storage by reversible ion adsorption onto electrode materials with negligible charge transfer chemical reactions, leading to a much longer cycle life.3 However, the low energy density limits the wide application of EDLCs.To achieve higher energy density, the capacitance of EDLCs has to be increased, as the energy density (E) is directly related to the capacitance (C) by E = ½CV2, where V is the operating voltage. The double layer capacitance is generally described by a parallel plate capacitor C = εrε0A/d, where εr is the dielectric constant of the electrolyte, ε0 is the vacuum permittivity, A is the surface area and d is the charge separation distance.4 From this relationship, a larger electrochemical capacitance is expected for materials with higher specific surface areas. Indeed, the linear relationship between C and A has been found in previous work.5,6 But in some other work, there is no correlation observed.7,8 One possible reason for this divergence is that the specific surface area measured by gas adsorption may not be equal to the accessible surface area by ions in the electrolytes, which is dependent on the pore shape, pore size distribution, and types of pores (open or closed).9 Besides the specific surface area, the pore size is another important factor for EDLCs. A recent experimental study shows an anomalous increase of capacitance for pore sizes below 1 nm, challenging the conventional wisdom that micropores are not accessible to large hydrated ions.10 A theoretical study suggests that the parallel plate capacitor model is over simplified for microporous and mesoporous electrochemical capacitors, and does not consider the effect of pore curvature.11 These recent advances in the understanding of the EDLC mechanism indicate that an increase of microporosity and proper matching of the pore size with the electrolyte ions are among the key considerations in achieving a higher capacitance.
Carbon is one of the most promising candidates as an electrode material for electrochemical capacitors, due to its high and tunable surface area, good electrical conductivity, and relatively low cost.12 The synthesis of nanoporous carbon materials for application in electrochemical capacitors has been extensively studied. To develop ordered nanoporous carbons, two approaches are generally employed, a hard template method and a soft template method.13,14 The tedious steps of forming hard templates and subsequent removal by a hazardous chemical method limit the wide applications of hard templates. Therefore, recent research has been focused on the development of soft template methods through the self-assembly of surfactants and copolymers.13 Among the soft template methods, phenol–formaldehyde, resols or resin are often employed as carbon precursors, which are toxic and not environmentally friendly.15
In this work, a direct hydrothermal method by a soft templating approach is employed to synthesize carbon with ordered pores by using D-fructose as the precursor.16 The hydrothermal synthesis temperature is tuned to control the pore size distribution and specific surface area, in order to optimize the nanoporous carbon for the application of supercapacitor electrodes. As the hydrothermal synthesis temperature increases from 130 °C to 200 °C, the nanoporous carbon transforms from a hierarchical microporous and mesoporous structure with long-range ordered mesopores to a disordered mesoporous structure with reduced microporosity and surface area. A micelle model is proposed to explain the change of microporosity and surface area based on the temperature dependent properties of PPO (polypropylene oxide) and PEO (polyethylene oxide) chains. Electrochemical characterization shows a large capacitance of ∼290 F g−1 at scan rate of 1 mV s−1 in 6 M KOH aqueous electrolyte for the ordered mesoporous carbon with a surface area above 1100 m2 g−1. In addition, a high electrochemical stability is shown by repeating galvanostatic charge–discharging at 10 A g−1 for up to 6000 cycles. The dependency of the capacitance on the specific surface area, pore size distribution and pore volume of nanoporous carbons is discussed for further improvement of the electrochemical performance.
2. Experimental details
2.1 Synthesis of nanoporous carbon
Appropriate amounts of the carbon precursor, D-fructose (1.2 g), and triblock-copolymer F127 (0.25 g) were first dissolved in 10 ml H2O by stirring overnight. The solution was then sealed in an autoclave for thermal treatment for 24 h. The hydrothermal synthesis temperature used was 80 °C, 100 °C, 130 °C, 150 °C, 180 °C or 200 °C. The solutions treated at 80 and 100 °C showed a clear solution without any precipitate after 24 h. This hydrothermal synthesis temperature is not high enough for cross-linking between precursors to stabilize the pore framework, which required the polymerization of hydroxymethyl furfural, which occurred at a hydrothermal synthesis temperature of around 120–140 °C.17,18 Therefore, only the remaining four samples at higher temperatures were carbonized and characterized. The carbonization of the solid precipitate after hydrothermal treatment was done at 550 °C for 2 h under an Ar atmosphere.2.2 Structural characterization
The morphology and microstructure of the carbon samples were studied by scanning electron microscopy (SEM) (XL 30 FEG-SEM Philips) and transmission electron microscopy (TEM) (JEM2010F). N2 adsorption at 77 K (Micromeritics ASAP 2020) was employed to study the pore texture and specific surface area of the as-synthesized carbon samples. To characterize small micropores, high resolution measurements in a low relative pressure (P/P0) range from 0 to 0.01, using CO2 adsorption at 298 K and N2 adsorption at 77 K, were made. The pore size distribution was calculated based on the Barrett–Joyner–Halenda (BJH) model for mesopores, and the Dubinin–Astakhov model for micropores. The surface function groups were investigated by Fourier-transform infrared spectroscopy (FTIR) (Perkin-Elmer FT-IR 2000) and X-ray photoelectron spectroscopy (XPS) (AXIS Ultra).2.3 Electrochemical characterization
The electrode was prepared by mixing carbon black (10 wt%), polytetrafluoroethylene (PTFE) (10 wt%) and as-synthesized carbon powder (80 wt%) in ethanol to form a slurry. The slurry was dropped onto a Ni foam current collector, which was then dried at 80 °C for 12 h. Both the three-electrode configuration and two-electrode symmetric configuration were employed for electrochemical characterization. In the three-electrode configuration, Pt was used as the counter electrode and Ag/AgCl as the reference electrode. The two-electrode symmetric configuration was tested by using Swagelok type cells to assemble two Ni foam electrodes loaded with carbon slurry, which were isolated by a porous separator. The electrolyte for both test configurations was 6 M KOH aqueous solution.3. Results and discussion
SEM images (Fig. 1) show a spherical morphology for the carbon samples synthesized at hydrothermal synthesis temperatures of 130, 150, 180 and 200 °C. The average size of these spheres decreases and becomes more uniform as the hydrothermal synthesis temperature increases from 130 to 200 °C. Fig. 2 shows the high resolution TEM images. Long-range ordered mesoporous structures are observed for samples at low hydrothermal synthesis temperatures of 130 and 150 °C. The mesopore size is ∼3 nm for both samples. For the samples using high hydrothermal synthesis temperatures of 180 and 200 °C, the pore structure becomes disordered. The strong interaction between the precursors and the templating block-copolymer is critical to obtain the ordered pore structure and prevent macrophase separation.15 The interaction mechanism here is probably hydrogen-bonding between the hydroxyl group of fructose and the PEO chains.19,20 At high hydrothermal synthesis temperatures of 150 and 180 °C, the polymerization of the precursors may be too fast, which weakens the hydrogen-bonding and leads to disorder of the mesopores. | ||
| Fig. 1 SEM images of the carbon samples using hydrothermal synthesis temperature of (a) 130 °C, (b) 150 °C, (c) 180 °C and (d) 200 °C. | ||
 | ||
| Fig. 2 TEM images of the carbon samples using hydrothermal synthesis temperatures of (a) 130 °C, (b) 150 °C, (c) 180 °C and (d) 200 °C. | ||
N2 adsorption–desorption isotherms in the relative pressure range P/P0 from 0.01 to 1.00 were measured to study the pore size distribution and specific surface area of the carbon samples. For the sample with a hydrothermal synthesis temperature of 130 °C, as shown in Fig. 3(a), the observed typical type I isotherm is characteristic of microporous solids.21 Pore size analysis using the BJH model shows an average mesopore size of ∼2.71 nm and a significant number of micropores. In order to characterize the micropore structure, CO2 adsorption at 298 K was measured in the relative pressure range from 0.00 to 0.01, as shown in Fig. 4(a). Based on the Dubinin–Astakhov model, the micropores have a peak pore size of ∼1.03 nm (Fig. 4(b)). For the sample with a hydrothermal synthesis temperature of 150 °C, the N2 adsorption also shows the type I micropore isotherm in Fig. 3(b) with an average mesopore size of ∼2.79 nm. CO2 adsorption was measured for this 150 °C sample, but the pore size distribution peak was larger than 1.1 nm, and could not be obtained for CO2 measured at the relative pressure range from 0.00 to 0.01, as shown in Fig. 4(c) and (d). N2 adsorption at low relative pressure from 0.00 to 0.01 (Fig. 4(e)) was measured to fully characterize the micropore size, which shows a peak pore size of ∼1.43 nm in Fig. 4(f). For the samples with hydrothermal synthesis temperatures of 180 °C and 200 °C, N2 adsorption shows type IV isotherms, with a characteristic hysteresis from the capillary condensation of the mesopores. The mesopore sizes for the samples with hydrothermal synthesis temperatures of 180 °C and 200 °C are 3.48 nm and 3.88 nm, respectively. The observed dependency of the BET (Brunauer–Emmett–Teller) surface area and mesopore size on the hydrothermal synthesis temperature is shown in Fig. 5(a). The sample with a hydrothermal synthesis temperature of 130 °C exhibits the largest BET specific surface area of ∼1100 m2 and a hierarchical microporous (∼1.03 nm) and mesoporous (∼2.71 nm) structure. To understand the pore size change observed, a schematic micelle model is proposed, as shown in Fig. 5(b). Micelle structures formed by block copolymers/surfactants are well documented. Cryogenic TEM has been used to measure the micelle size of F127 in solution at room temperature.22 The concentration of surfactant F127 employed in this experiment was ∼2.5 wt% or 1.98 mM, which is much higher than the critical micelle concentration (CMC) for F127 at 25 °C.23 Therefore, the micelles would be formed in the solution before the hydrothermal treatment for all the samples. The hydrophobic cores of the micelles, formed by PPO chains, are the source for mesopores after carbonization. The PEO chains (shown by curved lines in Fig. 5(b)) at the shell, trapped in the carbon precursor network, are the source for micropores after being removed by carbonization.24 Although it is almost impossible to measure the micelle size at various temperatures in situ, especially above 100 °C under high pressure conditions during the hydrothermal process, the temperature dependence of the hydrophilicity of the PEO and PPO chains has been reported.24–26 When the hydrothermal synthesis temperature is increased, the hydrophilicity of the PEO chains decreases,25–27 and they retract from the carbon network into the PPO cores. This leads to the increased micelle core size and thus a larger mesopore size at higher hydrothermal synthesis temperatures as observed in this work. At the same time, this retraction of the PEO chains reduces the number of micropores in the shell, which explains the reduced microporosity and surface area as the hydrothermal synthesis temperature increases.28
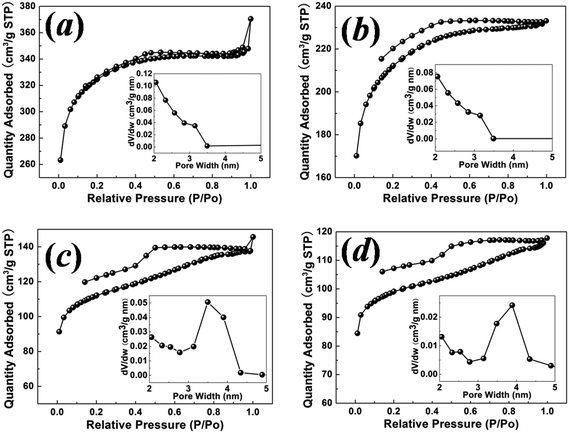 | ||
| Fig. 3 N2 (77 K) adsorption–desorption isotherms in the relative pressure range from 0.01 to 1.00 for the carbon samples with hydrothermal synthesis temperatures of (a) 130 °C, (b) 150 °C, (c) 180 °C and (d) 200 °C. Insets show the pore size distribution as calculated from the BJH model. | ||
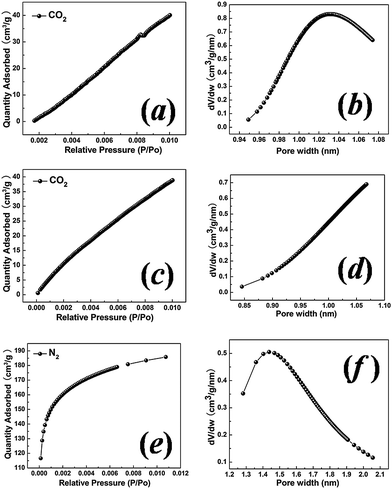 | ||
| Fig. 4 CO2 (298 K) adsorption isotherms and the Dubinin–Astakhov pore size distribution for the carbon samples with hydrothermal synthesis temperatures of 130 °C (a and b) and 150 °C (c and d). (e) High resolution low pressure N2 (77 K) adsorption isotherm for the sample with a hydrothermal synthesis temperature of 150 °C with the Dubinin–Astakhov pore size distribution (f). | ||
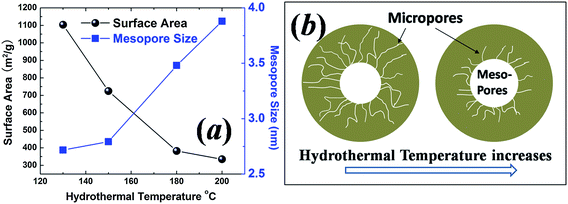 | ||
| Fig. 5 (a) Specific surface area and the average mesopore size as a function of hydrothermal synthesis temperature. (b) Schematic micelle model showing PEO chains retracting to the hydrophobic core as the hydrothermal synthesis temperature increases. | ||
XPS and FTIR spectroscopy were conducted to study the surface functional groups of the as-synthesized carbon samples, and are shown in Fig. 6. The high resolution C 1s spectrum of the 130 °C sample shows three resolved peaks corresponding to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O groups (288.3 eV), C–O groups (286.1 eV) and C–C groups (284.6 eV).29 The ratio of the peak area from the graphitic carbon C–C group to that of the oxygenated groups (C–O and CO) is around 3
O groups (288.3 eV), C–O groups (286.1 eV) and C–C groups (284.6 eV).29 The ratio of the peak area from the graphitic carbon C–C group to that of the oxygenated groups (C–O and CO) is around 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2. The percentage of oxygen atoms is calculated to be ∼19%, assuming the carbon to oxygen ratio is 3:2 in the C–O groups and 1:1 in the CO groups.30 The other three samples (spectra not shown) show similar spectra and ratios between the carbon and oxygenated functional groups. It is likely that the high oxygen content in these samples resulted from the same low carbonization temperature of 550 °C. FTIR spectra also suggest the presence of oxygen functional groups with peaks matching the C–O stretching mode at 1150–1050 cm−1, conjugated CC/olefinic species CC–O at 1650–1450 cm−1, and the CO mode from lactone groups at 1800–1520 cm−1.16
:2. The percentage of oxygen atoms is calculated to be ∼19%, assuming the carbon to oxygen ratio is 3:2 in the C–O groups and 1:1 in the CO groups.30 The other three samples (spectra not shown) show similar spectra and ratios between the carbon and oxygenated functional groups. It is likely that the high oxygen content in these samples resulted from the same low carbonization temperature of 550 °C. FTIR spectra also suggest the presence of oxygen functional groups with peaks matching the C–O stretching mode at 1150–1050 cm−1, conjugated CC/olefinic species CC–O at 1650–1450 cm−1, and the CO mode from lactone groups at 1800–1520 cm−1.16
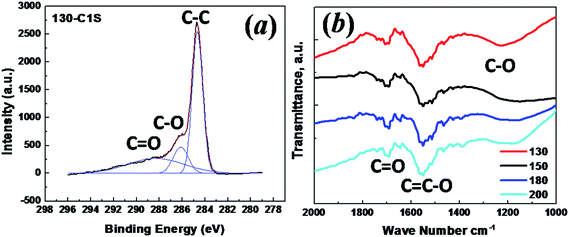 | ||
| Fig. 6 (a) XPS spectrum of the C 1s peak for the sample with a hydrothermal synthesis temperature of 130 °C; (b) FTIR curves for the carbon samples with hydrothermal synthesis temperatures of 130 °C, 150 °C, 180 °C and 200 °C. | ||
The cyclic voltammetry (CV) curves using the three-electrode configuration at three scan rates of 1, 5 and 10 mV s−1 for the sample with a hydrothermal synthesis temperatures of 130 °C are shown in Fig. 7(a). They are not rectangular in shape as observed in an ideal double layer electrochemical capacitor.31 Instead they show asymmetric triangular-like shapes, which may well be related to the high oxygen content detected in the carbon samples. The mass specific capacitance Cm is calculated from Cm = Qm/ΔV, where Qm is the specific charge integrated from 0 to −1 V in the CV scans, and ΔV is the potential range. The scan rate dependence of Cm is shown in Fig. 7(c) for all four samples with hydrothermal synthesis temperatures varying from 130 to 200 °C. The largest capacitance is shown for the sample with a hydrothermal synthesis temperature of 130 °C, with a value of ∼480 F g−1 at 1 mV s−1. The galvanostatic charge–discharge curves using the three-electrode configuration for the sample with a hydrothermal synthesis temperature of 130 °C at four current densities are shown in Fig. 7(b). The specific capacitance C from the galvanostatic charge–discharge is calculated from C = I/(dV/dt), where I is the constant current during charging and discharging, and dV/dt is derived from the slope of the discharge curve. In an agreement with the Cm results from the CV, the sample with a hydrothermal synthesis temperature of 130 °C shows the largest capacitance value of ∼350 F g−1 at 1 A g−1, as shown in Fig. 7(d). The capacitance decreases as the hydrothermal synthesis temperature increases, and is correlated with the pore size and surface area. The likely contribution to the capacitance from the oxygen surface function groups should not affect the different capacitances of these samples, as the oxygen content is almost the same for all four samples. To characterize the capacitance fading behavior, galvanostatic charge–discharge for the sample with a hydrothermal synthesis temperature of 130 °C was measured, and is shown in Fig. 7(e). 94% retention of the capacitance was observed after 6000 cycles at 10 A g−1, indicating good long term stability of the nanoporous carbon electrode.
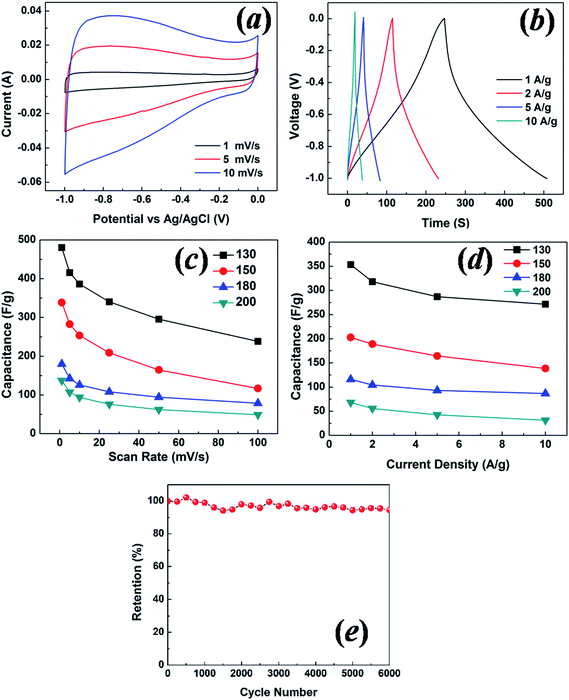 | ||
| Fig. 7 (a) Cyclic voltammetry test for the 130 °C sample at scan rates of 1, 5 and 10 mV s−1, (b) galvanostatic charge–discharge test for the 130 °C sample at current densities of 1, 2, 5 and 10 A g−1. Specific capacitance as a function of scan rate (c) and current density (d) for the samples with hydrothermal synthesis temperatures of 130 °C, 150 °C, 180 °C and 200 °C. (e) Cycle test for galvanostatic charge–discharge at 10 A g−1 for the sample with a hydrothermal synthesis temperature of 130 °C. The tests were conducted with the three-electrode configuration. | ||
To illustrate the dependence of capacitance on the surface area and pore texture of the carbon samples with different hydrothermal synthesis temperatures, the mass specific capacitance and normalized capacitance were plotted against the specific surface area, pore volume and average mesopore size. Fig. 8(a) and (b) show that the mass specific capacitance exhibits a very good linear dependence on the surface area, regardless of the different scan rates in the CV tests and different current densities in the charge–discharge tests. This agrees with the general expectation that a higher surface area gives rise to a larger capacitance. However, the intercepts of the linear fits are not zero, indicating that the interaction behavior of the electrolyte ions with small nanometer-sized pores cannot be completely accounted for by the simple parallel capacitor model.11Fig. 8(c) and (d) also show a linear dependence of the mass specific capacitance on the total pore volume, which is derived from the N2 adsorption–desorption measurements in Fig. 3. In previous reports, a linear relationship between the capacitance and the micropore volume was observed, indicating that the micropores are the main contributor to the capacitance.32,33 The correlation of capacitance with total pore volume in this work shows that both micropores and mesopores contribute to the charge storage, suggesting that electrolyte ions are able to diffuse through a well developed network between mesopores and micropores. To isolate the pore size effect, irrespective of the surface area, mass specific capacitance is normalized against surface area and plotted as a function of average mesopore size, as shown in Fig. 8(e) and (f). In the mesopore region, the normalized capacitance should increase as the average pore size increases, according to the electric double-cylinder capacitor model.11 However, a comparable or even slightly decreased normalized capacitance is observed here as the pore size increases. This is due to the large contribution from micropores for the 130 °C and 150 °C samples, which have smaller mesopore sizes. The size of a hydrated K+ ion is ∼0.33 nm and that of a hydrated OH− ion is ∼0.30 nm,34,35 and so they are small enough to diffuse into the smallest pore (∼1 nm) measured for the carbon samples in this work. The electrolyte ions in the small micropores therefore give rise to a significant additional capacitance for samples with low hydrothermal synthesis temperatures of 130 °C and 150 °C. This suggests that increased microporosity is beneficial for better electrochemical performance of supercapacitor electrodes.
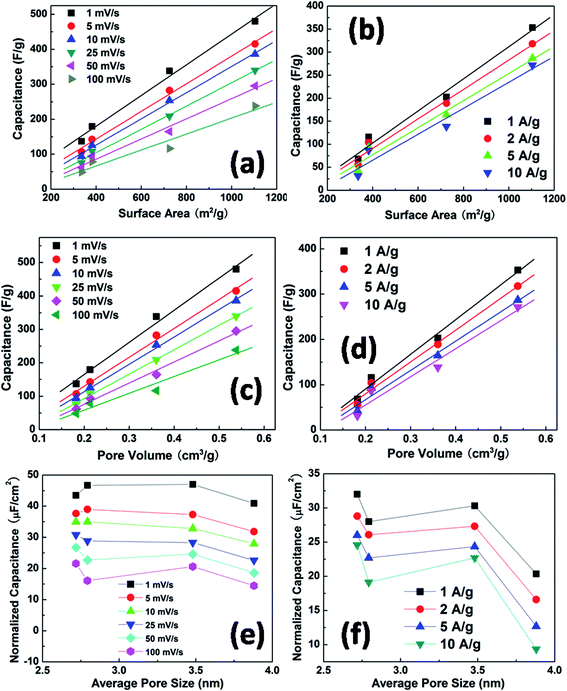 | ||
| Fig. 8 Capacitance derived from CV as a function of the specific surface area (a), and pore volume (c). Capacitance derived from galvanostatic charge–discharge measurements as a function of specific surface area (b) and pore volume (d). Normalized capacitance derived from CV (e) and charge–discharge measurements (f) as a function of mesopore size. | ||
The three-electrode test fixture tends to overestimate the actual capacitance.36 In order to evaluate the large capacitance shown by the three electrode configuration, two-electrode tests with symmetric carbon electrodes were conducted for the same batch of samples. Similar to the three-electrode configuration results shown in Fig. 7, the CV curves measured by the two-electrode configuration shown in Fig. 9(a) do not have an ideal rectangular shape. This indicates the possible contribution of pseudocapacitance from redox reactions of certain oxygen-containing surface function groups,37 as the amount of oxygen is quite large (∼19%) in our samples, as determined by XPS. The capacitance values reported for porous carbon-based materials are typically below 200 F g−1. The galvanostatic charge–discharge curves for the sample with a hydrothermal synthesis temperature of 130 °C at different current densities are shown in Fig. 9(b). For two-electrode supercapacitor cells, the electrode capacitance can be derived from cell capacitance Ccell using 1/Ccell = 1/C1 + 1/C2. As the two electrodes are symmetric, the electrode capacitance can thus be expressed as C = C1 = C2 = 2Ccell. The specific electrode capacitances for all four samples at different scan rates were calculated, as shown in Fig. 9(c). The calculated electrode capacitances are plotted as a function of current density in Fig. 9(d). The largest capacitance of ∼290 F g−1 at 1 mV s−1 is again shown by the sample with the hydrothermal synthesis temperature of 130 °C, although this is smaller than the value measured using the three-electrode configuration as expected. On the one hand, the three-electrode test may well overestimate the capacitance value.38 On the other hand, both the three-electrode and two-electrode configuration show a similar dependence of the capacitance on the hydrothermal synthesis temperature. The three-electrode configuration is reported to be highly sensitive towards redox reactions.36,38 The difference between the three-electrode capacitance and the two-electrode capacitance supports our expectation that the redox reactions of the oxygen-containing surface function groups, such as olefinic species CC–O and CO lactone groups present on the carbon surface, contribute to the overall capacitance. This is indeed demonstrated by the non-ideal rectangular shape of the CV curves.
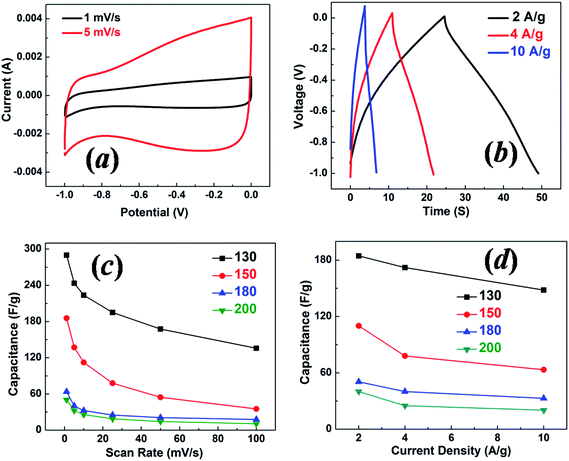 | ||
| Fig. 9 (a) Cyclic voltammograms for the 130 °C sample at scan rates of 1 and 5 mV s−1, (b) galvanostatic charge–discharge tests for the 130 °C sample at current densities of 2, 4 and 10 A g−1. Specific capacitance as a function of scan rate (c) and current density (d) for the samples with hydrothermal synthesis temperatures of 130 °C, 150 °C, 180 °C and 200 °C. Tests were conducted with the two-electrode symmetric configuration. | ||
4. Conclusion
A low temperature hydrothermal approach involving soft templating has been developed to synthesize nanoporous carbons with ordered mesopores for application as the electrode in electrochemical supercapacitors. The effect of hydrothermal synthesis temperature on the pore structure and electrochemical behavior of mesoporous carbon derived from carbohydrates has been studied. As the hydrothermal synthesis temperature increased from 130 °C to 200 °C, the nanoporous carbons lost the long-range ordering of the mesopores with a decrease of the microporosity and specific surface area, which is explained well by the proposed micelle model. The hierarchically porous carbon with a hydrothermal synthesis temperature of 130 °C shows the highest surface area of ∼1100 m2 g−1 and the best electrochemical capacitance in both the two-electrode and three-electrode test configurations. The capacitor performance of the nanoporous carbon samples with different hydrothermal synthesis temperatures suggests that the microporous structure with a higher surface area is advantageous for capacitance. A high electrochemical stability is demonstrated for the nanoporous carbon produced in the present work by repeated galvanostatic charge–discharge measurements at 10 A g−1 for 6000 cycles. This low temperature, soft template hydrothermal approach provides a promising way to develop highly ordered porous carbon electrodes for electrochemical capacitors with a high energy density and long cycle life.Acknowledgements
This work is supported by the Agency for Science, Technology and Research (A-Star, Singapore), grant no. 1121202013, conducted at the National University of Singapore.References
- P. Simon and Y. Gogotsi, Nat. Mater., 2008, 7, 845 CrossRef CAS PubMed.
- E. Frackowiak and F. Beguin, Carbon, 2001, 39, 937 CrossRef CAS.
- R. Kotz and M. Carlen, Electrochim. Acta, 2000, 45, 2483 CrossRef CAS.
- E. Frackowiak, Phys. Chem. Chem. Phys., 2007, 9, 1774 RSC.
- D. Lozano-Castello, D. Cazorla-Amoros, A. Linares-Solano, S. Shiraishi, H. Kurihara and A. Oya, Carbon, 2003, 41, 1765 CrossRef CAS.
- G. Gryglewicz, J. Machnikowski, E. Lorenc-Grabowska, G. Lota and E. Frackowiak, Electrochim. Acta, 2005, 50, 1197 CrossRef CAS.
- M. Endo, Y. J. Kim, T. Takeda, T. Maeda, T. Hayashi, K. Koshiba, H. Hara and M. S. Dresselhaus, J. Electrochem. Soc., 2001, 148, A1135 CrossRef CAS.
- D. Qu and H. Shi, J. Power Sources, 1998, 74, 99 CrossRef CAS.
- J. Gamby, P. L. Taberna, P. Simon, J. F. Fauvarque and M. Chesneau, J. Power Sources, 2001, 101, 109 CrossRef CAS.
- J. Chmiola, G. Yushin, Y. Gogotsi, C. Portet, P. Simon and P. L. Taberna, Science, 2006, 313, 1760 CrossRef CAS PubMed.
- J. Huang, B. G. Sumpter and V. Meunier, Chem.–Eur. J., 2008, 14, 6614 CrossRef CAS PubMed.
- A. G. Pandolfo and A. F. Hollenkamp, J. Power Sources, 2006, 157, 11 CrossRef CAS.
- C. Liang, Z. Li and S. Dai, Angew. Chem., Int. Ed., 2008, 47, 3696 CrossRef CAS PubMed.
- J. Lee, J. Kim and T. Hyeon, Adv. Mater., 2006, 18, 2073 CrossRef CAS.
- L. Chuenchom, R. Kraehnert and B. M. Smarsly, Soft Matter, 2012, 8, 10801 RSC.
- S. Kubo, R. J. White, N. Yoshizawa, M. Antonietti and M.-M. Titirici, Chem. Mater., 2011, 23, 4882 CrossRef CAS.
- C. Yao, Y. Shin, L.-Q. Wang, C. F. Windisch, W. D. Samuels, B. W. Arey, C. Wang, W. M. Risen and G. J. Exarhos, J. Phys. Chem. C, 2007, 111, 15141 CAS.
- M.-M. Titirici, R. J. White, C. Falco and M. Sevilla, Energy Environ. Sci., 2010, 5, 6796 Search PubMed.
- G. J. d. A. A. Soler-Illia, C. Sanchez, B. Lebeau and J. Patarin, Chem. Rev., 2002, 102, 4093 CrossRef PubMed.
- Y. Wan and Zhao, Chem. Rev., 2007, 107, 2821 CrossRef CAS PubMed.
- F. Beguin and E. Frackowiak, Carbons for electrochemical energy storage and conversion systems, CRC Press LLC, 2010 Search PubMed.
- K. Mortensen and Y. Talmon, Macromolecules, 1995, 28, 8829 CrossRef CAS.
- P. Alexandridis, J. F. Holzwarth and T. A. Hatton, Macromolecules, 1994, 27, 2414 CrossRef CAS.
- S. Ruthstein, V. Frydman, S. Kababya, M. Landau and D. Goldfarb, J. Phys. Chem. B, 2003, 107, 1739 CrossRef CAS.
- C. Guo, H. Z. Liu and J. Y. Chen, Colloid Polym. Sci., 1999, 277, 376 CAS.
- P. Bloss, W. D. Hergeth, E. Döring, K. Witkowski and S. Wartewig, Acta Polym., 1989, 40, 260 CrossRef CAS.
- G. Wanka, H. Hoffmann and W. Ulbricht, Macromolecules, 1994, 27, 4145 CrossRef CAS.
- A. Galarneau, H. Cambon, F. Di Renzo and F. Fajula, Langmuir, 2001, 17, 8328 CrossRef CAS.
- S. Biniak, G. Szymanski, J. Siedlewski and A. Swiatkowski, Carbon, 1997, 35, 1799 CrossRef CAS.
- B. P. Payne, M. C. Biesinger and N. S. McIntyre, J. Electron Spectrosc. Relat. Phenom., 2011, 184, 29 CrossRef CAS.
- Y. Zhu, S. Murali, M. D. Stoller, K. J. Ganesh, W. Cai, P. J. Ferreira, A. Pirkle, R. M. Wallace, K. A. Cychosz, M. Thommes, D. Su, E. A. Stach and R. S. Ruoff, Science, 2011, 332, 1537 CrossRef CAS PubMed.
- C. Vix-Guterl, E. Frackowiak, K. Jurewicz, M. Friebe, J. Parmentier and F. Beguin, Carbon, 2005, 43, 1293 CrossRef CAS.
- E. Frackowiak, G. Lota, J. Machnikowski, C. Vix-Guterl and F. Beguin, Electrochim. Acta, 2006, 51, 2209 CrossRef CAS.
- B. E. Conway and B. E. Conway, Ionic hydration in chemistry and biophysics, Elsevier, Amsterdam, 1981, vol. 741 Search PubMed.
- A. G. Volkov, S. Paula and D. W. Deamer, Bioelectrochem. Bioenerg., 1997, 42, 153 CrossRef CAS.
- M. D. Stoller and R. S. Ruoff, Energy Environ. Sci., 2010, 3, 1294 CAS.
- C.-T. Hsieh and H. Teng, Carbon, 2002, 40, 667 CrossRef CAS.
- V. Khomenko, E. Frackowiak and F. Beguin, Electrochim. Acta, 2005, 50, 2499 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2013 |