TiO2 nanotube arrays grafted with Fe2O3 hollow nanorods as integrated electrodes for lithium-ion batteries
Le
Yu
,
Zhiyu
Wang
,
Lei
Zhang
,
Hao Bin
Wu
and
Xiong Wen (David)
Lou
*
School of Chemical and Biomedical Engineering, Nanyang Technological University, 70 Nanyang Drive, Singapore 637457. E-mail: xwlou@ntu.edu.sg; Web: http://www.ntu.edu.sg/home/xwlou/
First published on 18th October 2012
Abstract
Novel nano-heterostructures composed of Fe2O3 hollow nanorods on both the outer and inner surface of the aligned TiO2 nanotube arrays (TNAs) have been fabricated by growing FeOOH nanospindles onto the TNAs, and then followed by thermal transformation in air. Owing to their structural advantages, such a hierarchical architecture exhibits superior electrochemical performance with a remarkable areal capacity of over 600 μA h cm−2 at a current density of 100 μA cm−2 for 50 cycles (∼680 μA h cm−2 at 100 μA cm−2). This hierarchal structure might be easily integrated as a binder-free electrode for microscale lithium-ion batteries considering the improved performance and the simple synthesis.
1. Introduction
Rechargeable lithium-ion batteries (LIBs) have long been considered as attractive power sources for a variety of applications ranging from electronic devices to electric vehicles and stationary energy storage systems because of their high energy density, long lifespan and environmental benignity.1–8 The continuously surging demand for high-performance LIBs has stimulated extensive interest in seeking novel electrode materials that enable faster delivery of more energy.9–12 Amongst the available materials, iron oxides have been considered very appealing substitutes for the conventional graphite (theoretical capacity is 372 mA h g−1) owing to their much higher specific capacity of 900–1000 mA h g−1, nontoxicity, high corrosion resistance and enhanced safety.13–15 The lithium storage within iron oxides mainly relies on the reversible conversion reactions, where they are reduced to metallic nanocrystals dispersed in the Li2O matrix upon lithiation and then are reversibly restored to the initial state after successive delithiation.13,16,17 The fully utilized oxidation states and multi-electron redox reactions involved in the charge–discharge process render iron oxides higher specific capacities than those of the metal oxides which store lithium by addition-type reaction (e.g., TiO2).18 The highly reversible formation of Li2O matrix not only reduces the irreversible capacity loss, but also enhances the structural stability upon electrochemical cycling, resulting in the superior performance over the metal or metal alloy electrodes.19,20 Therefore, iron oxides are very promising for lithium storage due to the balanced lithium storage capacities and calendar life.The practical performance of bulk iron oxides, however, is far below the expectation as fast capacity fading even at low current rates is often observed.21,22 This problem is known to be mainly caused by their drastic volume variations (>200%) during the repetitive lithium intake/extraction process, which leads to severe disintegration and pulverization of the electrode, and hence a loss of electrical contacts between adjacent particles.8,23,24 The intrinsically sluggish electronic/ionic conductivity is another factor that hampers the lithium storage within iron oxides. One effective approach to prolong the cycling lifespan of iron oxides is to engineer delicate nanostructures. In nanostructured electrodes, lithium diffusion can be facilitated because of the short lithium diffusion path, large surface area and enhanced reactivity.1,3,25,26 Meanwhile, the strain associated with lithium insertion is also better accommodated, enabling significantly improved electrochemical performance.23,27–29 So far, many iron oxide nanostructures ranging from zero-dimensional nanoparticles to complex hybrid architectures have been synthesized and evaluated as negative electrodes for lithium-ion batteries with encouraging results.30–35 Nevertheless, the synthesis of truly durable iron oxide based electrodes still remains as a significant challenge.
The construction of three-dimensional (3D) nanoframeworks also holds the key for making high-performance electrodes because of their substantial advantages over conventional planar electrodes.36–39 For example, the nanosized Fe3O4 supported on 3D Cu nanopillars can achieve a six-fold improvement in power density over the planar electrode.27 Moreover, the 3D frameworks not only ensure much higher lithium ion flux across the electrode due to the dramatically increased electrode–electrolyte interface, but also serve as the pathway for efficient electron transport.10,40 Therefore, the diffusion kinetics for lithium storage is significantly enhanced. Furthermore, the free space within porous 3D electrodes can provide sufficient tolerance for the volume variation of the entire electrode during lithium storage.27,41 As a result of improved kinetics and electrode stability, excellent performance for reversible lithium storage is anticipated with well-designed 3D electrodes.
Herein, we report the design and synthesis of a 3D electrode by assembling Fe2O3 hollow nanorods onto highly oriented TiO2 nanotube arrays (TNAs). The TNAs are chosen as the 3D framework for their particular advantages: (a) they can be readily synthesized in large scale by common methods such as anodization;42 (b) TiO2 can also contribute to the lithium storage capacity with negligible lattice change;41,43 (c) the tubular nanostructure may offer much larger surface area and easier access for the lithium ions than the solid rods or pillars; (d) TiO2 is highly stable in solution to support the growth of iron oxide nanostructures. By combining with Fe2O3 hollow nanostructures, the formed TNAs@Fe2O3 hierarchical structure can be directly used as the integrated electrode in LIBs without any ancillary additives such as polymer binder and conductive agents. The TNAs@Fe2O3 electrode manifests excellent electrochemical performance, as characterized by high areal capacities of around 680 μA h cm−2 at a high current density of 100 μA cm−2 and nearly 100% capacity retention in 50 cycles.
2. Experimental section
Synthesis of TNAs
TiO2 nanotube arrays were synthesized by two-step electrochemical anodization of Ti foil at room temperature.44 Prior to anodization, a Ti foil with a thickness of 0.3 mm was successively cleaned with ethanol and deionized (DI) water in an ultrasonic bath for 30 min. Then it was subjected to potentiostatic anodization for 3 h in a two-electrode electrochemical cell with a Pt foil as the counter electrode. A constant voltage of 60 V was employed for the anodization and the electrolyte used is 0.12 M NH4F in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :9 (w/w) mixture of DI water and ethylene glycol (EG). Afterward, the Ti foil was taken out and ultrasonically cleaned in DI water for a few seconds for the next round of potentiostatic anodization under identical conditions. After reaction, the anodized Ti foil was ultrasonically rinsed with ethanol and DI water several times and annealed at 400 °C for 2 h with a ramp rate of 1 °C min−1 to obtain crystalline TNAs on the Ti foil.
:9 (w/w) mixture of DI water and ethylene glycol (EG). Afterward, the Ti foil was taken out and ultrasonically cleaned in DI water for a few seconds for the next round of potentiostatic anodization under identical conditions. After reaction, the anodized Ti foil was ultrasonically rinsed with ethanol and DI water several times and annealed at 400 °C for 2 h with a ramp rate of 1 °C min−1 to obtain crystalline TNAs on the Ti foil.
Growth of FeOOH nanospindles on TNAs (denoted as TNAs@FeOOH)
The growth of FeOOH nanospindles on TNAs was conducted by immersing the as-obtained TNAs in 40 mL of aqueous FeCl3 solution (80 mM) at 80 °C for 12 h. The final products were washed with DI water and ethanol several times before being fully dried in air at 60 °C.Synthesis of TNAs@Fe2O3 hierarchical structure
For the formation of hollow Fe2O3 nanorods on the TNAs, the TNAs@FeOOH hierarchical structure was annealed in air at 400 °C for 2 h with a slow ramp rate of 1 °C min−1. As a reference, pure Fe2O3 powder from the reaction solution was also collected and annealed in air at 400 °C for 2 h.Materials characterization
The morphology and structure of the samples were examined using a field-emission scanning electron microscope (FESEM, JEOL, JSM-7600F) and a transmission electron microscope (TEM, JEOL, JEM-2010). The elemental compositions of the samples were analyzed with energy-dispersive X-ray (EDX) spectroscopy attached to a FESEM. XRD patterns were obtained on a Bruker D2 Phaser X-ray Diffractometer (Cu Kα, λ = 1.5406 Å). The TNAs@Fe2O3 and TNAs samples were scraped from Ti foil for the XRD test.The electrochemical measurements were conducted using two-electrode Swagelok cells with pure lithium foil as the counter and reference electrode at room temperature. The Ti foil supported TNAs or TNAs@Fe2O3 were directly used as the working electrodes without ancillary binders or conductive agents. The electrolyte was composed of 1.0 M LiPF6 in a 50:50 (w/w) mixture of ethylene carbonate and diethyl carbonate. Cell assembly was carried out in an Ar-filled glovebox with concentrations of moisture and oxygen below 1.0 ppm. The galvanostatic charge–discharge tests were performed using a Neware battery tester at a constant current rate of 100 μA cm−2 with a cut-off voltage window of 0.05–3.0 V.
3. Results and discussion
The synthesis process of Fe2O3 hollow nanorods on TNAs is straightforward, as schematically illustrated in Fig. 1. Well-oriented TNAs are firstly synthesized as the nanostructured 3D substrate by electrochemical anodization of Ti foil at room temperature. Then β-FeOOH nanospindles are spontaneously grown and assembled on both the outer and inner surface of TNAs by forced hydrolysis of FeCl3 without the need for any structure-directing agent. Due to the good material compatibility, surface pre-treatment is not required for the growth of FeOOH on TiO2, which greatly simplifies the synthesis process. Secondly, β-FeOOH nanospindles grafted on TNAs can be easily transformed to α-Fe2O3 hollow nanorods as a result of thermal dehydroxylation and simultaneous lattice shrinkage at 400 °C, leading to the formation of nano-heterostructures composed of α-Fe2O3 hollow nanorods on TNAs.45 | ||
| Fig. 1 Schematic illustration of the formation of Fe2O3 hollow nanorods on TiO2 nanotube arrays (TNAs): (I) growth of FeOOH nanospindles on TNAs by hydrolysis of Fe3+ ions; (II) thermal transformation of FeOOH nanospindles to Fe2O3 hollow nanorods on TNAs (TNAs@Fe2O3). | ||
The crystallographic structure of TNAs@Fe2O3 is determined by X-ray powder diffraction (XRD) analysis, as shown in Fig. 2a. The XRD pattern of TNAs confirms the crystal phase of the TiO2 nanotubes to be anatase TiO2 (JCPDS card no. 21-1272) and the existence of titanium (JCPDS card no. 44-1294). Compared to the TNAs, the Fe2O3 phase gives rather weak signals. Nonetheless, it is obvious that one major diffraction peak indexed to the (110) plane of hexagonal α-Fe2O3 (hematite, JCPDS card no. 33-0664) can be observed at around 35° in the TNAs@Fe2O3 hybrid structure. The presence of the Fe element is confirmed by energy dispersive X-ray (EDX) spectroscopy, indicating a relatively small atomic fraction of around 8%, as shown in Fig. 2b.
 | ||
| Fig. 2 (a) XRD patterns of TNAs@Fe2O3, TNAs and Fe2O3; (b) EDX spectrum of TNAs@Fe2O3. The asterisks in (a) indicate the main diffraction peak of Fe2O3. | ||
The morphological characteristics of TNAs and TNAs@Fe2O3 are characterized using a field-emission scanning electron microscope (FESEM) and a transmission electron microscope (TEM). A panoramic FESEM image reveals that the sample consists of entirely the dense array of uniform nanotubes without impurity particles or aggregates at the surface (Fig. 3a). These TiO2 nanotubes have an average length of as long as 10 μm with a uniform diameter of around 160 nm, corresponding to a high aspect ratio of around 100 (Fig. 3b). After the reaction in FeCl3 solution, the surface of TNAs is covered with large quantities of FeOOH nanospindles as a result of the hydrolysis of Fe3+ and simultaneously heterogeneous growth of FeOOH on TiO2 nanotubes, as shown in Fig. 3c–e. Benefitting from the precisely controlled reaction, the FeOOH nanospindles have a length of less than 60 nm, enabling their growth inside the TiO2 nanotubes. In this process, the initial concentration of FeCl3 in the solution plays a critical role in the formation of FeOOH nanospindles on TNAs. A very low concentration (e.g., 4 mM or 20 mM) results in few FeOOH nanorods on TNAs due to insufficient growth; whereas the excessive amount of FeCl3 (e.g., 160 mM) induces severe homogeneous growth of FeOOH particles (Fig. 3f–h).
 | ||
| Fig. 3 (a and b) FESEM images of TNAs, in which the TEM image of a TiO2 nanotube is shown as the inset in (a); (c and d) top view of the TNAs@FeOOH structure; (e) the cross-section of the TNAs@FeOOH structure; (f–h) the TNAs@FeOOH samples obtained by the hydrolysis of FeCl3 with various concentrations of 4 mM, 20 mM and 160 mM, respectively. | ||
TEM analysis is carried out to provide further insight into the detailed structures of the as-prepared samples. It is obvious from the cross-sectional view and top view of TEM observation that highly dispersed FeOOH nanospindles have assembled onto both the outer and inner surface of TNAs without the formation of large particles or aggregates, as shown in Fig. 4a–c. Being in good agreement with the FESEM examination, the FeOOH nanospindles are relatively short with a small diameter of several nanometers. The composition of each component of the TNAs@FeOOH hybrid structure can be varied without damaging the overall structure. By careful annealing in air at 400 °C, TNAs@Fe2O3 can be derived through phase transformation of FeOOH to polycrystalline α-Fe2O3 on robust TNAs, as shown in Fig. 4d–e. After annealing, the hollow interior is generated in the resultant Fe2O3 structures as a result of the volume contraction associated with the transformation from low density FeOOH (3 g cm−3) to hematite with a denser density of 5.3 g cm−3 (Fig. 4e). Despite the huge mass loss, this conversion undergoes a topotactic transformation process due to the structural similarity of FeOOH nanospindles and Fe2O3 nanorods. With high porosity, the Fe2O3 hollow nanorods might be beneficial in offering sufficient interface to facilitate the electrolyte absorption and electrochemical reactions on TNAs.22,45 Further high resolution TEM (HRTEM) examination of the Fe2O3 hollow nanorods is also provided in Fig. 4f. Consistent with XRD analysis, there is a distinct set of visible lattice fringes in TNAs@Fe2O3 with an inter-planar distance of 0.25 nm, which corresponds to the (110) plane of α-Fe2O3. Both the XRD and HRTEM analyses show the existence of crystalline α-Fe2O3 in the TNAs@Fe2O3 heterostructure.
 | ||
| Fig. 4 (a) TEM images of the TNAs@FeOOH structure; (b) several free-standing FeOOH nanospindles on the surface of TiO2 nanotube; (c) a top-view TEM image showing the formation of FeOOH on the inner and outer surface of TNAs; (d) FESEM; (e and f) TEM images of the TNAs@Fe2O3 architecture; (f) HRTEM image of a Fe2O3 nanorod on the TNAs@Fe2O3 hybrid structure. | ||
It is well known that the electrochemical performance is not only dependent on the intrinsic crystalline texture and surface properties, but also greatly related to the morphology and assembled structure of active materials. In TNAs@Fe2O3, the electrochemically active TiO2 nanotube arrays can act as a stable 3D framework to support the growth of Fe2O3 hollow nanorods that deliver extra capacity for lithium storage. The unique tubular structure provides open channels for high lithium ion flux across the entire electrode and reduces the internal stress. On the other hand, the Fe2O3 nanorods are largely separated from each other because they are grown directly on the TNAs, thus making them fully accessible to lithium ions in the electrolyte. This is in distinct contrast to the electrode composed of nanoparticles, where severe agglomeration will occur and eliminate most of the active interfaces for lithium storage. Meanwhile, the extremely reduced dimensions of free-standing Fe2O3 nanorods, adequate inter-space between them, and the formation of hollow interiors also enhance the electrode stability by effectively mitigating the internal mechanical stress. Motivated by these structural features, we have evaluated the electrochemical lithium storage properties of TNAs@Fe2O3 for their potential use as an anode material in microscale LIBs. Fig. 5a shows the respective discharge–charge voltage profiles of TNAs@Fe2O3, taken at a current density of 100 μA cm−2 within a cut-off voltage window of 0.05–3.0 V at room temperature. Due to the presence of iron oxides, the TNAs@Fe2O3 sample exhibits high initial discharge and charge capacities of 770 and 550 μA h cm−2, respectively; whereas the bare TNAs can only deliver nearly half of the values (Fig. 5b). The irreversible capacity loss of 28% may be mainly attributed to irreversible processes such as the inevitable formation of inorganic solid electrolyte interface (SEI) film and electrolyte decomposition, which are common to most anode materials. From the second cycle onwards, the capacity of TNAs@Fe2O3 hierarchical structures increases gradually from 570 μA h cm−2 to 680 μA h cm−2 within 50 cycles, as shown in Fig. 5c. This result is superior to most reported TiO2-based 3D electrodes.43,46–48 The specific capacity of TNAs@Fe2O3 hierarchical structures is also estimated as shown by the vertical axis on the right in Fig. 5c, where it increases gradually to about 400 mA h g−1 after 50 cycles. The capacity rise with cycling is not uncommon for various nanostructured transition metal oxide electrodes.22,49,50 In theory, the reversible formation of organic polymeric/gel-like layer by electrolyte decomposition at low potential has been suggested as one of the possible reasons for this phenomenon due to its capability of coating around the active materials to ensure the mechanical cohesion and delivering excess capacity at low potential through the so-called “pseudo-capacitance-type behavior”.51 For cobalt and iron oxides, such an effect has resulted in the stable and gradual capacity increase during the electrochemical cycling.22,49 Moreover, the activation of the active materials may also contribute to the capacity rise in metal oxide electrodes. With the aim of demonstrating the advantages of TNAs@Fe2O3 architectures on lithium storage, the cycling performance of bare TNAs is also investigated under the identical conditions. They deliver an initial discharge capacity of 400 μA h cm−2, which then stabilizes at around 300 μA h cm−2 after 50 cycles, clearly indicating the beneficial effect of the TNAs@Fe2O3.
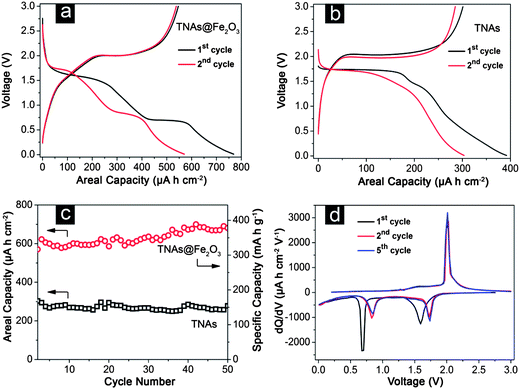 | ||
| Fig. 5 Galvanostatic discharge–charge profiles of (a) TNAs@Fe2O3 and (b) TNAs for the first and second cycles; (c) comparative cycling performances of TNAs@Fe2O3 with bare TNAs; (d) the differential capacity versus voltage plots of TNAs@Fe2O3. All measurements were conducted in the voltage range of 0.05–3.0 V at a current density of 100 μA cm−2. | ||
To further investigate the mechanism of lithium storage in TNAs@Fe2O3, the differential capacity versus voltage curves of various cycles are carefully studied (Fig. 5d). At the 1st cycle, two peaks at 1.6 V and 0.7 V are observed, corresponding to the insertion of lithium into the Fe2O3–TiO2 and complete reduction of Fe2O3 to metallic Fe and SEI film formation, respectively.52,53,30,39 The peak at 2.0 V is ascribed to the delithiation of TiO2 (from orthorhombic LixTiO2 to tetragonal anatase) and the restoration of iron oxides.11,46,54 The subsequent curves show good reproducibility with two cathodic peaks at 0.9 and 1.75 V, and an anodic peak at 2.0 V. The slight shift of the two cathodic peaks to higher potentials after the first cycle is probably related to some possible activation process for the Li+ insertion in the first cycle, which has also been observed in previous reports.11,14,21,22,31,54 Apparently, the lithium storage mechanism in TNAs@Fe2O3 is similar to that in other iron oxide and TiO2 electrodes.47,49
4. Conclusions
In summary, a three-dimensional (3D) metal oxide electrode is fabricated by assembling FeOOH nanospindles onto both the inner and outer surface of well-aligned TiO2 nanotube arrays, followed by thermal conversion to Fe2O3 hollow nanorods. The good contact between Fe2O3 nanostructures and TiO2 nanotubes enables their application as promising integrated electrodes for microscale lithium-ion batteries without adding other ancillary materials such as carbon black or polymer binders. As a result of the desirable electrode nano-architecture, these hybrid Fe2O3–TiO2 electrodes can deliver high capacities of over 600 μA h cm−2 at a current density of 100 μA cm−2 with excellent capacity retention over 50 cycles. This result is superior to most of the reported TiO2-based 3D electrodes. Clearly, the combination of the 3D nanostructure and electrochemical active metal oxides could be an effective way to develop a new class of high-performance anodes for high-energy lithium-ion batteries.References
- A. S. Arico, P. Bruce, B. Scrosati, J. M. Tarascon and W. Van Schalkwijk, Nat. Mater., 2005, 4, 366 CrossRef CAS.
- M. Armand and J. M. Tarascon, Nature, 2008, 451, 652 CrossRef CAS.
- P. G. Bruce, B. Scrosati and J. M. Tarascon, Angew. Chem., Int. Ed., 2008, 47, 2930 CrossRef CAS.
- J. B. Goodenough and Y. Kim, Chem. Mater., 2010, 22, 587 CrossRef CAS.
- C. Liu, F. Li, L. P. Ma and H. M. Cheng, Adv. Mater., 2010, 22, E28 CrossRef CAS.
- F. Y. Cheng, J. Liang, Z. L. Tao and J. Chen, Adv. Mater., 2011, 23, 1695 CrossRef CAS.
- B. Scrosati, J. Hassoun and Y. K. Sun, Energy Environ. Sci., 2011, 4, 3287 CAS.
- Z. Y. Wang, L. Zhou and X. W. Lou, Adv. Mater., 2012, 24, 1903 CrossRef CAS.
- B. Kang and G. Ceder, Nature, 2009, 458, 190 CrossRef CAS.
- H. G. Zhang, X. D. Yu and P. V. Braun, Nat. Nanotechnol., 2011, 6, 277 CrossRef CAS.
- J. S. Chen, Y. L. Tan, C. M. Li, Y. L. Cheah, D. Y. Luan, S. Madhavi, F. Y. C. Boey, L. A. Archer and X. W. Lou, J. Am. Chem. Soc., 2010, 132, 6124 CrossRef CAS.
- L. W. Ji, Z. Lin, M. Alcoutlabi and X. W. Zhang, Energy Environ. Sci., 2011, 4, 2682 CAS.
- J. Chen, L. N. Xu, W. Y. Li and X. L. Gou, Adv. Mater., 2005, 17, 582 CrossRef CAS.
- M. V. Reddy, T. Yu, C. H. Sow, Z. X. Shen, C. T. Lim, G. V. S. Rao and B. V. R. Chowdari, Adv. Funct. Mater., 2007, 17, 2792 CrossRef CAS.
- W. M. Zhang, X. L. Wu, J. S. Hu, Y. G. Guo and L. J. Wan, Adv. Funct. Mater., 2008, 18, 3941 CrossRef CAS.
- D. Larcher, C. Masquelier, D. Bonnin, Y. Chabre, V. Masson, J. B. Leriche and J. M. Tarascon, J. Electrochem. Soc., 2003, 150, A133 CrossRef CAS.
- D. Larcher, D. Bonnin, R. Cortes, I. Rivals, L. Personnaz and J. M. Tarascon, J. Electrochem. Soc., 2003, 150, A1643 CrossRef CAS.
- H. B. Wu, J. S. Chen, H. H. Hng and X. W. Lou, Nanoscale, 2012, 4, 2526 RSC.
- M. G. Kim and J. Cho, Adv. Funct. Mater., 2009, 19, 1497 CrossRef CAS.
- C. Kim, M. Noh, M. Choi, J. Cho and B. Park, Chem. Mater., 2005, 17, 3297 CrossRef CAS.
- B. Wang, J. S. Chen, H. B. Wu, Z. Y. Wang and X. W. Lou, J. Am. Chem. Soc., 2011, 133, 17146 CrossRef CAS.
- Z. Y. Wang, D. Y. Luan, S. Madhavi, Y. Hu and X. W. Lou, Energy Environ. Sci., 2012, 5, 5252 CAS.
- X. W. Lou, L. A. Archer and Z. C. Yang, Adv. Mater., 2008, 20, 3987 CrossRef CAS.
- Z. Y. Wang, D. Y. Luan, S. Madhavi, C. M. Li and X. W. Lou, Chem. Commun., 2011, 47, 8061 RSC.
- Y. Y. Liu, D. W. Liu, Q. F. Zhang and G. Z. Cao, J. Mater. Chem., 2011, 21, 9969 RSC.
- G. N. Zhu, Y. G. Wang and Y. Y. Xia, Energy Environ. Sci., 2012, 5, 6652 CAS.
- L. Taberna, S. Mitra, P. Poizot, P. Simon and J. M. Tarascon, Nat. Mater., 2006, 5, 567 CrossRef.
- W. Li, Y. X. Yin, S. Xin, W. G. Song and Y. G. Guo, Energy Environ. Sci., 2012, 5, 8007 CAS.
- D. Deng, M. G. Kim, J. Y. Lee and J. Cho, Energy Environ. Sci., 2009, 2, 818 CAS.
- Z. M. Cui, L. Y. Hang, W. G. Song and Y. G. Guo, Chem. Mater., 2009, 21, 1162 CrossRef CAS.
- X. L. Wu, Y. G. Guo, L. J. Wan and C. W. Hu, J. Phys. Chem. C, 2008, 112, 16824 CAS.
- H. Liu, G. X. Wang, J. Park, J. Wang, H. Liu and C. Zhang, Electrochim. Acta, 2009, 54, 1733 CrossRef CAS.
- J. S. Chen, T. Zhu, X. H. Yang, H. G. Yang and X. W. Lou, J. Am. Chem. Soc., 2010, 132, 13162 CrossRef CAS.
- G. M. Zhou, D. W. Wang, F. Li, L. L. Zhang, N. Li, Z. S. Wu, L. Wen, G. Q. Lu and H. M. Cheng, Chem. Mater., 2010, 22, 5306 CrossRef CAS.
- J. P. Liu, Y. Y. Li, H. J. Fan, Z. H. Zhu, J. Jiang, R. M. Ding, Y. Y. Hu and X. T. Huang, Chem. Mater., 2010, 22, 212 CrossRef CAS.
- J. W. Long, B. Dunn, D. R. Rolison and H. S. White, Chem. Rev., 2004, 104, 4463 CrossRef CAS.
- M. M. Shaijumon, E. Perre, B. Daffos, P. L. Taberna, J. M. Tarascon and P. Simon, Adv. Mater., 2010, 22, 4978 CrossRef CAS.
- T. S. Arthur, D. J. Bates, N. Cirigliano, D. C. Johnson, P. Malati, J. M. Mosby, E. Perre, M. T. Rawls, A. L. Prieto and B. Dunn, MRS Bull., 2011, 36, 523 CrossRef CAS.
- J. A. Jiang, Y. Y. Li, J. P. Liu and X. T. Huang, Nanoscale, 2011, 3, 45 RSC.
- W. Wang, M. Tian, A. Abdulagatov, S. M. George, Y. C. Lee and R. G. Yang, Nano Lett., 2012, 12, 655 CrossRef CAS.
- G. D. Du, Z. P. Guo, P. Zhang, Y. Li, M. B. Chen, D. Wexler and H. K. Liu, J. Mater. Chem., 2010, 20, 5689 RSC.
- G. K. Mor, O. K. Varghese, M. Paulose, K. Shankar and C. A. Grimes, Sol. Energy Mater. Sol. Cells, 2006, 90, 2011 CrossRef CAS.
- G. F. Ortiz, I. Hanzu, P. Lavela, P. Knauth, J. L. Tirado and T. Djenizian, Chem. Mater., 2010, 22, 1926 CrossRef CAS.
- M. D. Ye, X. K. Xin, C. J. Lin and Z. Q. Lin, Nano Lett., 2011, 11, 3214 CrossRef CAS.
- Y. Piao, J. Kim, H. Bin Na, D. Kim, J. S. Baek, M. K. Ko, J. H. Lee, M. Shokouhimehr and T. Hyeon, Nat. Mater., 2008, 7, 242 CrossRef CAS.
- G. F. Ortiz, I. Hanzu, T. Djenizian, P. Lavela, J. L. Tirado and P. Knauth, Chem. Mater., 2009, 21, 63 CrossRef CAS.
- G. F. Ortiz, I. Hanzu, P. Lavela, J. L. Tirado, P. Knauth and T. Djenizian, J. Mater. Chem., 2010, 20, 4041 RSC.
- L. G. Xue, Z. Wei, R. S. Li, J. L. Liu, T. Huang and A. S. Yu, J. Mater. Chem., 2011, 21, 3216 RSC.
- P. Poizot, S. Laruelle, S. Grugeon, L. Dupont and J. M. Tarascon, Nature, 2000, 407, 496 CrossRef CAS.
- Y. F. Shi, B. K. Guo, S. A. Corr, Q. H. Shi, Y. S. Hu, K. R. Heier, L. Q. Chen, R. Seshadri and G. D. Stucky, Nano Lett., 2009, 9, 4215 CrossRef CAS.
- S. Laruelle, S. Grugeon, P. Poizot, M. Dolle, L. Dupont and J. M. Tarascon, J. Electrochem. Soc., 2002, 149, A627 CrossRef CAS.
- K. Y. Xie, Z. G. Lu, H. T. Huang, W. Lu, Y. Q. Lai, J. Li, L. M. Zhou and Y. X. Liu, J. Mater. Chem., 2012, 22, 5560 RSC.
- H. B. Wu, J. S. Chen, X. W. Lou and H. H. Hng, Nanoscale, 2011, 3, 4082 RSC.
- Z. Y. Wang and X. W. Lou, Adv. Mater., 2012, 24, 4124 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2013 |