A bifunctional approach for the preparation of graphene and ionic liquid-based hybrid gels†
Sampath
Srinivasan
a,
Weon Ho
Shin
a,
Jang Wook
Choi
a and
Ali
Coskun
*ab
aGraduate School of EEWS, NanoCentury KAIST Institute, 373-1 Guseong Dong, Yuseong Gu, Daejeon 305-701, Republic of Korea. E-mail: coskun@kaist.ac.kr; Fax: +82-42-350-1710; Tel: +82-42-350-1724
bDepartment of Chemistry, Korea Advanced Institute of Science and Technology (KAIST), 373-1 Guseong Dong, Yuseong Gu, Daejeon 305-701, Republic of Korea
First published on 27th September 2012
Abstract
The synthesis of high quality graphene in large quantities from graphene oxide (GO) and its dispersion in a chemical environment is essential for the real-life applications. Although GO can be converted to reduced graphene oxide (RGO) by using reducing agents, in the absence of suitable stabilizing agents, RGO undergoes an irreversible aggregation in solution. Herein, we propose and illustrate the concept of using bifunctional molecules which (1) can enable the solubilization of GO sheets in ionic liquids (ILs) and (2) can facilitate the highly efficient thermal reduction of GO to RGO on account of the high thermal stability of ILs. We have demonstrated this concept by incorporating an imidazolium cation onto pyrene which can interact with RGO via cation–π and π–π interactions to form highly stable, porous hybrid gel materials.
The allotropes of carbon, such as graphite, fullerenes, carbon nanotubes and graphene, contain the basic building block of sp2 hybridized carbon atoms.1–8 Graphene is a one-atom thick, two-dimensional (2D) sheet where sp2-carbon atoms are arranged in fused hexagons.9–17 The honeycomb structured graphene has attracted18–23 the attention of numerous scientific communities since its discovery in 2004. Graphene's unique 2D nanostructure exhibits properties, such as its self-assembling capability, high mechanical strength and good conductivity, which make it an ideal candidate for the development of functional hybrid materials.23–25 It remains, however, a challenge to produce single-layer graphene sheets of high quality in quantities.26–37 Graphene sheets have been produced by various methods including mechanical cleavage9 of natural graphite, epitaxial growth,38 exfoliation39 and chemical manipulation.26–30,40 In this context, production of a large amount of graphene at low cost is essential for mass production, leading to the utilization of graphene-based materials in real-life applications.41–43 From all the available methods, chemical manipulation can provide graphene at relatively low cost and in large quantities.18–28
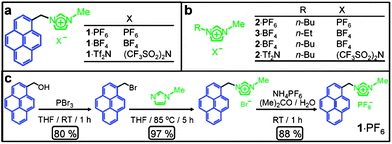 | ||
| Fig. 1 (a) Structural formulae of the bifunctional molecules. (b) Structural formulae of ionic liquids (ILs). (c) Synthetic scheme for the preparation of 1·PF6. | ||
Graphene oxide (GO) can be prepared from graphite by chemical manipulation.22–30,40 The chemically prepared GO can be reduced by different methods, involving either chemical or thermal reduction, to afford18,31–34 reduced graphene oxide (RGO). In the absence of suitable stabilizing agents or surfactants in the solution, the reduced graphene layers self-assemble irreversibly back to graphite, losing most of their unique properties.26–37 Reports related to the thermal reduction of GO in solution are rare because of the high volatility and low thermal stability of organic solvents.44 In order to address this problem, a high boiling solvent with high thermal stability needs to be used for the thermal reduction of GO to RGO. To date, ionic liquids (ILs) have received45 a significant amount of interest on account of their thermal stability, recyclability, non-flammability, wide electrochemical window and high conductivity.
Recently, Fukushima and coworkers46 discovered the preparation of bucky gels in ILs serendipitously. These bucky gels have found applications as actuators47 and sensors.48 Although there are several reports49–55 in the literature related to the formation of self-assembled three-dimensional (3D) network structures, which lead to the formation of hydrogels upon reduction of GO by chemical or hydrothermal methods, neither of these methods involve direct dispersion of GO and its thermal reduction in ILs. Herein, we demonstrate that a bifunctional molecule (Fig. 1a), 1·PF6, can (i) act as a solubilizing agent for GO in ILs, (ii) facilitate thermal reduction of GO to RGO and (iii) assist the self-assembly process of RGO with ILs to form highly stable hybrid gels. While 1·PF6 can interact with both GO and RGO through π–π and cation–π interactions, an imidazolium cation can effectively interact with ILs on account of the similarities in their molecular structures. Thus, we have utilized the low volatility and high thermal stability of ILs for the efficient thermal reduction of GO to RGO.
GO has been prepared from graphite by using modified literature procedures.26,40 GO can be dispersed in different solvents such as H2O, EtOH, DMF, etc.31,56,57 Dispersion of GO sheets in ILs, however, is hindered by their relatively strong π–π interactions.55,58,59 In order to disperse GO in ILs, we synthesized a pyrene derivative 1·PF6 functionalized with an imidazolium moiety. The salt 1·PF6, the structure of which is shown in Fig. 1a, was synthesized (Fig. 1c) in three steps in relatively high yields. 1-Bromopyrenemethanol was prepared60 in 80% yield by reacting 1-pyrenemethanol in THF with phosporous tribromide, followed by its functionalization with N-methylimidazole to yield the corresponding pyrene derivative in 97% yield. The bromide counterions were exchanged quantitatively for different counterions—namely, PF6−, BF4− and Tf2N−—by treating them with aqueous solutions of NH4PF6, KBF4 and LiTf2N, respectively.
While GO is dispersed (Fig. 2a) in H2O, it is insoluble (Fig. 2b) in imidazolium-based ILs,55 even after sonication for 2 h. The bifunctional compound 1+, however, acts as an anchoring cation on the surface of GO and the imidazolium cation interacts with the IL solvent molecules, thus rendering GO dispersible in ILs. The optical image of a solution of 1·PF6 in 2·PF6 is illustrated in Fig. 2c. On treating GO with 2·PF6 in the presence of 1·PF6, GO sheets disperse immediately in ILs forming (Fig. 2d) a brown solution upon sonication for 30 min. This experiment demonstrates clearly the ability of 1·PF6 to render GO layers dispersed in ILs. The brown color is the characteristic of amorphous and chemically functionalized graphene, reflecting the fact that the chemical functionalization converts sp2 hybridized carbon atoms to sp3 ones. Interestingly, the above brown solution forms (Fig. 2e) a black hybrid gel upon heating at 150 °C for 1 h. This hybrid gel, containing thermally reduced GO (RGO) with 1·PF6 in 2·PF6, is highly stable at room temperature for several months and can be easily dispersed (Fig. 2f) upon dilution in DMF. The resulting nanocomposite solution is stable at room temperature for several months without any precipitation. The black color of the nanocomposite solution indicates visually that the thermal treatment results in the formation of RGO, thus demonstrating the utility of the bifunctional approach for the thermal reduction of GO to RGO. The schematic representation of the proposed mechanism for the interaction of GO with 1·PF6 in ILs is depicted in Fig. 3. We believe that 1·PF6 can exfoliate the GO layers through π–π and cation–π interactions in ILs. Heating the composite dispersion at 150 °C for 1 h in ILs leads to the formation of a black hybrid gel. The thermal reduction of GO leads to the graphenization process which, in turn, increases its interaction with 1+. The isostructural relationship of the IL with the bifunctional molecule initiates the self-assembly process, which leads to the formation of a 3D gel network.
 | ||
| Fig. 2 (a) Picture of GO (0.1 mg mL−1) dispersed in H2O. (b) Picture of GO insoluble in 2·PF6. (c) Picture of the solution of 1·PF6 (25 mg) in 2·PF6 (1 g). (d) Picture of 1·PF6 (25 mg) and GO (10 mg) dispersed in 2·PF6 (1 g). (e) Picture of 1·PF6 with thermally reduced GO in 2·PF6 in a hybrid gel state. (f) Picture of the composite material dispersed (10 mg mL−1) in DMF. | ||
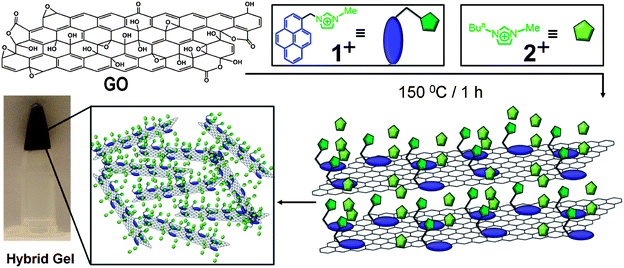 | ||
| Fig. 3 Schematic illustration of the proposed mechanism for the formation of the hybrid gel material from GO and the bifunctional compound 1+ in ILs through the thermal reduction process. | ||
In order to understand the effect of counterions on the gelation process, we have prepared (Fig. 1b) hybrid gels in ILs with various different counterions. We have synthesized bifunctional compounds with counterions, such as 1·BF4 and 1·Tf2N, in order to match the counterions of ILs. The 1·PF6 (25 mg, 78 mM, 2.5 wt%) and 2·PF6 (1 g) can gelate GO (10 mg) after thermal reduction. Similarly, 1·BF4 and 1·Tf2N have been used to prepare hybrid gels in 2·BF4, 3·BF4 and 2·Tf2N ILs. The amount of 1+ required to form stable gels at room temperature with other ILs was determined by varying the concentration of 1+ in ILs in the presence of GO. 1·BF4 was used to gelate the ILs, 2·BF4 and 3·BF4, while keeping the amount of GO at 10 mg (1 wt% with respect to IL). To gelate 2·BF4 and 3·BF4, 30 mg (95 mM, 3 wt%) and 28 mg (94 mM, 2.8 wt%) of 1·BF4 are required, respectively. 1·Tf2N can gelate 2·Tf2N at an amount of 55 mg (137 mM, 5.5 wt%). It is important to note that the gelling ability of 1·PF6 is better than those of 1·BF4 and 1·Tf2N in their corresponding ILs, based on their critical gelation concentrations. We believe that the size of the counterions could be responsible for the differences in the gelling ability of 1+ in different ILs. The difference in the gelling ability upon changing the counterions indicates that the π–π and cation–π interactions of ILs and 1+ with GO depend on the nature of counterions which, in turn, can alter the interactions and gel stability. The effect of the concentration of GO on the gelling ability of the hybrid gels was investigated. For GO with 15 mg (1.5 wt%), 20 mg (2 wt%), and 25 mg (2.5 wt%) in 2·PF6 (1 g), the amount of 1·PF6 required to form a stable gel is 20 mg (62 mM, 2 wt%), 18 mg (56 mM, 1.8 wt%) and 18 mg (56 mM, 1.8 wt%), respectively. When the weight percentage of GO in 2·PF6 is increased, the concentration of 1·PF6 required for gelation decreases. These observations indicate that the concentrations of both GO and 1·PF6 are important for ensuring the formation of stable hybrid gels.
In order to investigate the mechanical properties of hybrid gels, we have carried out rheology studies. The dynamic storage modulus (G′) and loss modulus (G′′) of RGO/1·PF6/2·PF6, RGO/1·BF4/2·BF4 and RGO/1·Tf2N/2·Tf2N hybrid gels at their respective critical gel concentrations are measured (Fig. 4a) as a function of angular frequency (ω) at a strain amplitude (γ) of 0.1. The hybrid gels showed a plateau region when the angular frequency was varied from 100 to 0.1 rad s−1. G′ value was higher than G′′ over the entire frequency range, thus exhibiting a substantial elastic response. The ratio of G′ and G′′ for RGO/1·PF6/2·PF6 is 12.7, for RGO/1·BF4/2·BF4 is 10 and for RGO/1·Tf2N/2·Tf2N is 14.8. The G′/G′′ values which are higher than 1 indicate that the hybrid gels retained their gel-like nature for a wide range of strain amplitudes, which commensurate with the reported gel property of RGO-based IL gels.55 The plot of variation in G′ and complex viscosity (η*) as a function of ω for hybrid gels shows a decrease in η* with increasing ω (see ESI†). Strain amplitude, γ, dependencies of G′ and G′′ of the RGO/1·PF6/2·PF6 hybrid gel at 25 °C with ω at a rate of 10 rad s−1 are shown in Fig. 4b. The value of G′/G′′ decreases with increasing γ indicating disruption of the gel structure. The G′ and G′′ curves cross each other and the G′/G′′ ratio becomes less than 1 at a strain amplitude of 46%. These results indicate that the hybrid gel retains its gel-like character up to 46% strain amplitude and above that the hybrid gel behaves like a viscous fluid. These results are comparable to the previously reported46,47,55,61 hybrid gels based on RGO and single walled carbon nanotubes in ILs. We believe that the stability of hybrid gel materials is arising from the cooperative interactions between bifunctional 1+ with RGO—cation–π and π–π interactions—and ILs. The molecular ordering of ILs on the surface of RGO was also supported by recent molecular dynamic simulations.62–64
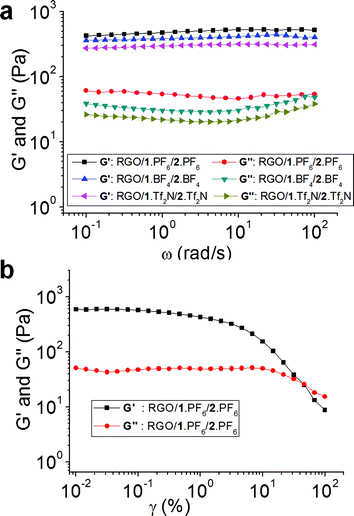 | ||
| Fig. 4 (a) Angular frequency (ω) dependencies of dynamic storage (G′) and loss (G′′) moduli of RGO/1·PF6/2·PF6, RGO/1·BF4/2·BF4 and RGO/1·Tf2N/2·Tf2N hybrid gels at their respective critical gel concentration at 25 °C at a strain amplitude (γ) of 0.1. (b) Strain amplitude, γ, dependencies of dynamic storage (G′) and loss (G′′) moduli of the RGO/1·PF6/2·PF6 hybrid gel at 25 °C with angular frequency (ω) at a rate of 10 rad s−1. | ||
It is known that the GO is more flexible because of the defects (functional groups) present in the graphene sheets. The thermal reduction of GO in ILs leads to the graphenization of GO layers, thus increasing the π-surface area. This structural change improves significantly π–π stacking interactions between the pyrene moiety of 1+ and RGO surface. While this strong π–π stacking interaction between RGO and 1+ allows the hybrid system to self-assemble strongly, the interactions between the imidazolium unit of 1+ and ILs lead to the entrapment of IL molecules between the RGO layers, resulting in the formation of a strong 3D gel network structure. The packing of ILs between the RGO layers and their interaction with 1+ are responsible for the formation of strong hybrid gels.
Raman spectroscopy is a very important technique for characterizing carbon-based materials such as graphite, carbon nanotubes, graphene, etc.65,66 There are three common peaks, one which appears at ∼1350 cm−1 is a D (defect) peak, the second one is called a G peak at ∼1580 cm−1, and the third one is a 2D peak at ∼2700 cm−1. The D peak appears because of the in-plane breathing mode of A1g symmetry caused by six-fold aromatic rings. The G peak originates from the E2g mode on account of the vibration of the sp2-hybridized carbon atoms within graphite layers.65 In graphite, owing to the presence of the sp2-hybridized carbon atoms, graphitic sheets are defect free, hence the D peak is very weak. The 2D peak arises from the second-order defect peak which is even present in the graphite with no defect, because defects are not needed to activate two phonons with the same momentum. The intensity ratio of the D and G peaks, ID/IG, is generally used66 to characterize the quantities of defects in graphene. The graphite reveals (Fig. 5a) a very weak D peak and a prominent G peak at 1574 cm−1. Nanophoton Raman studies of GO show the presence of an intense defect (D) band at 1337 cm−1, and upon thermal reduction, the RGO shows a relatively weaker D band at 1349 cm−1, indicating (Fig. 5a) that graphenization occurs during thermal reduction in ILs. The intensity ratio ID/IG for the GO and RGO is decreased from 1.28 to 0.48, proving that, during thermal reduction, the number of defects present in the GO is decreased‡ significantly in the RGO/1·PF6 nanocomposite. It is known67–69 that, while chemical reduction of GO67 using NaBH4 results in an intensity ratio (ID/IG) of slightly greater than 1, reduction involving the use of hydrazine hydrate68 and alcohols69 is associated with ratios of ca. 1.44 and 1, respectively. Hydrothermal reduction70 yields an intensity ratio of 0.90. It follows that the (ID/IG) ratio of 0.48 indicates clearly that GO is reduced significantly by thermal reduction in ILs. We believe that the simultaneous non-covalent interactions of 1+ with GO sheets and ILs enable direct dispersion and stabilization of GO sheets in ILs, thus facilitating highly efficient thermal reduction of GO to good quality RGO layers.
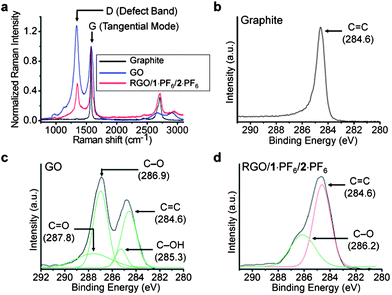 | ||
| Fig. 5 (a) Raman spectra of graphite (black), GO (blue), hybrid gel of RGO/1·PF6 in 2·PF6 (red). Raman spectra were measured by depositing the samples on a Si substrate and by exciting the samples with a laser source of 532 nm. Spectra were normalized at the G band peak at 1574 cm−1. The intensity ratio ID/IG for the GO is 1.28 and for RGO/1·PF6 in 2·PF6 is 0.48. High-resolution C1s XPS spectra of (b) graphite, (c) GO and (d) RGO/1·PF6 in 2·PF6. XPS samples were prepared by depositing onto a silicon substrate and samples were analyzed at a high vacuum (<10−8 Torr). All binding energies were calibrated using the C(1s) carbon peak (284.6 eV). | ||
Evidence for the interaction of RGO with 1·PF6 was also obtained (see ESI†) from micro-FT-IR analysis of the composite. In the IR spectrum of GO, the broad and weak band located at 1750 cm−1 is attributed to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibrations associated with COOH functional groups.71 The distinct features of the FT-IR spectrum of 1·PF6 are the C–H stretching band located at 3110 cm−1 and 3165 cm−1 and the CC stretching vibration at 1590 cm−1. The FT-IR spectrum of the RGO/1·PF6/2·PF6 composite exhibits peaks located at 3112 cm−1, 3168 cm−1 (C–H stretching vibration) and 1589 cm−1 (CC stretching vibration), indicating clearly the presence of 1·PF6 with RGO in the composite.
O stretching vibrations associated with COOH functional groups.71 The distinct features of the FT-IR spectrum of 1·PF6 are the C–H stretching band located at 3110 cm−1 and 3165 cm−1 and the CC stretching vibration at 1590 cm−1. The FT-IR spectrum of the RGO/1·PF6/2·PF6 composite exhibits peaks located at 3112 cm−1, 3168 cm−1 (C–H stretching vibration) and 1589 cm−1 (CC stretching vibration), indicating clearly the presence of 1·PF6 with RGO in the composite.
High-resolution core-level C1s X-ray photoelectron spectroscopy (XPS) analysis also revealed the decrease in the amount of sp3 carbon atoms in RGO–1·PF6–2·PF6, when compared to those in GO. The XPS spectrum of graphite shows (Fig. 5b) a peak at 284.6 eV, corresponding to the binding energy of a CC double bond. The deconvoluted spectra of GO reveal (Fig. 5c) peaks at 284.6, 285.3, 286.9 and 287.8 eV corresponding to the binding energies of CC, C–OH, C–O and CO bonds, respectively.66 When the thermally reduced GO was analyzed by XPS, a decrease in the peak intensities of the C–O functionalized carbon was noted. Moreover, the binding energy peaks of C–OH and CO become (Fig. 5d) weak to the extent that they almost disappear. The analysis of the XPS spectra of GO reveals that the CC is 32%, C–OH is 8%, C–O is 41%, and CO is 19%. Following its thermal treatment, however, RGO showed CC as 63% and C–O as 37%. The decrease in the percentage of oxygen-functionalized carbon indicates clearly that the graphenization of GO sheets occurs during thermal reduction in ILs.
The hybrid gel has been characterized using various microscopic techniques. Both the GO–H2O and RGO–1·PF6–2·PF6 hybrid gel dispersion in DMF were investigated by means of transmission electron microscopy (TEM). TEM samples were prepared by drop-casting a few drops of the solution onto copper grids, coated with a carbon film. The TEM images demonstrate the presence of single layer GO sheets in the chemically manipulated GO (see ESI†). The TEM image (Fig. 6a) of GO revealed its amorphous nature, which was confirmed by selected area electron diffraction (SAED). The absence of any prominent diffraction pattern in the SAED of the GO–H2O sample also indicates (Fig. 6c) the amorphous nature of GO. TEM studies reveal (Fig. 6b) the presence of single layer graphene sheets in the RGO obtained from the RGO/1·PF6/2·PF6 hybrid gel (see ESI†) which also shows (Fig. 6d) sharp electron diffraction peaks when analyzed by SAED.13,18
 | ||
| Fig. 6 Transmission electron microscopy (TEM) images of (a) GO and (b) RGO/1·PF6/2·PF6, scale bar, 300 nm. Selected area electron diffraction (SAED) images of (c) GO and (d) RGO/1·PF6/2·PF6, scale bar, 10 nm. (e) Line profile curve of SAED image (d), the intensity variation for 1−210 and 0−110 electron diffraction spots. TEM studies were conducted at 200 kV accelerating electron volt. TEM samples were prepared by drop-casting the sample solution on a carbon-coated copper grid. (f) AFM height image of RGO/1·PF6/2·PF6, scale bar, 0.5 μm. (g) Scanning electron microscopy (SEM) images of RGO/1·PF6/2·PF6, scale bar, 5 μm. | ||
Electron diffraction investigations revealed the reciprocal lattice spacing of the Miller–Bravais indices (hkil) at 0−110 (2.13 Å) and 1−210 (1.23 Å), which are characteristic of the graphene's electron diffraction pattern.13,72 The intensity profile (Fig. 6e) of the SAED reveals that the intensity of the 0−110 spots is nearly twice as intense as that of the 1−211 spots, an observation which is the fingerprint for the presence of single-layer graphenes. At the same time, folded and disordered graphene layers were also observed under TEM. RGO/1·PF6/2·PF6 hybrid gel dispersion in DMF was also investigated by means of atomic force microscopy (AFM). The AFM height image shown in Fig. 6f demonstrates the presence of single layer RGO sheets. The height profile of the RGO sheet shown in the bottom panel of Fig. 6f shows that the graphene sheet is about 0.9 nm thick and well dispersed in ILs. This observation indicates the presence of single layer graphene sheets which is comparable with the reported thickness of a single layer RGO sheet on a SiO2 substrate.31,55,56 The scanning electron microscopy (SEM) studies were carried out using a dried nanocomposite obtained from RGO/1·PF6/2·PF6. The SEM samples were prepared by diluting the hybrid gel in DMF and then drying by filtration under vacuum at room temperature. The composite film was peeled off from the filter paper and analyzed in the SEM. The SEM image (Fig. 6g) reveals the self-assembly of RGO sheets into a highly porous network. Similarly, SEM analyses of RGO/1·BF4/2·BF4, RGO/1·BF4/3·BF4 and RGO/1·TF2N/2·TF2N revealed the presence of porous networks.
In summary, we have demonstrated how a bifunctional approach has enabled the dispersion of GO sheets in ILs and, at the same time, allowed highly efficient thermal reduction of GO to RGO. Interaction of 1·PF6 with RGO sheets through π–π and cation–π interactions, and also with the imidazolium-based ILs, facilitates the formation of highly stable, porous gels. Thermal reduction in ILs decreases the amount of defects present in GO, while graphenization improves the interaction of the GO with bifunctional molecules and IL solvent molecules. This highly efficient thermal reduction can be employed in the preparation of RGO of high quality in quantities. Since the solvent used in this investigation is an ionic liquid, the graphene-based integrated molecular systems prepared by this method can potentially be used in energy storage applications.
Acknowledgements
We acknowledge the support from High Risk, High Return (HRHR) research support funds provided by the Korea Advanced Institute of Science and Technology (KAIST), Daejeon, South Korea.Notes and references
- A. K. Geim and K. S. Novoselov, Nat. Mater., 2007, 6, 183 CrossRef CAS.
- K. S. Novoselov, A. K. Geim, S. V. Morozov, D. Jiang, M. I. Katsnelson, I. V. Grigorieva, S. V. Dubonos and A. A. Firsov, Nature, 2005, 438, 197 CrossRef CAS.
- Y. Zhang, Y. W. Tan, H. L. Stormer and P. Kim, Nature, 2005, 438, 201 CrossRef CAS.
- D. R. Dreyer, R. S. Ruoff and C. W. Bielawski, Angew. Chem., Int. Ed., 2010, 49, 9336 CrossRef CAS.
- B. Standley, W. Bao, H. Zhang, J. Bruck, C. N. Lau and M. Bockrath, Nano Lett., 2008, 8, 3345 CrossRef CAS.
- C. Gomez-Navarro, R. T. Weitz, A. M. Bittner, M. Scolari, A. Mews, M. Burghard and K. Kern, Nano Lett., 2007, 7, 3499 CrossRef CAS.
- S. Stankovich, D. A. Dikin, G. H. B. Dommett, K. M. Kohlhaas, E. J. Zimney, E. A. Stach, R. D. Piner, S. T. Nguyen and R. S. Ruoff, Nature, 2006, 442, 282 CrossRef CAS.
- J. T. Robinson, F. K. Perkins, E. S. Snow, Z. Wei and P. E. Sheehan, Nano Lett., 2008, 8, 3137 CrossRef CAS.
- K. S. Novoselov, A. K. Geim, S. V. Morozov, D. Jiang, Y. Zhang, S. V. Dubonos, I. V. Grigorieva and A. A. Firsov, Science, 2004, 306, 666 CrossRef CAS.
- J. H. Chen, M. Ishigami, C. Jang, D. R. Hines, M. S. Fuhrer and E. D. Williams, Adv. Mater., 2007, 19, 3623 CrossRef CAS.
- Q. Liu, Z. Liu, X. Zhang, N. Zhang, L. Yang, S. Yin and Y. Chen, Appl. Phys. Lett., 2008, 92, 223303 CrossRef.
- X. Wang, L. J. Zhi, N. Tsao, Z. Tomovic, J. L. Li and K. Müllen, Angew. Chem., Int. Ed., 2008, 47, 2990 CrossRef CAS.
- J. C. Meyer, A. K. Geim, M. I. Katsnelson, K. S. Novoselov, T. J. Booth and S. Roth, Nature, 2007, 446, 60 CrossRef CAS.
- H. A. Becerril, J. Mao and Z. Liu, ACS Nano, 2008, 2, 463 CrossRef CAS.
- X. Wang, L. J. Zhi and K. Müllen, Nano Lett., 2008, 8, 323 CrossRef CAS.
- N. Mohanty and V. Berry, Nano Lett., 2008, 8, 4469 CrossRef CAS.
- J. Lu, I. Do, L. T. Drzal, R. M. Worden and I. Lee, ACS Nano, 2008, 2, 1825 CrossRef CAS.
- W. Gao, L. B. Alemany, L. Ci and P. M. Ajayan, Nat. Chem., 2009, 1, 403 CrossRef CAS.
- E. Yoo, J. Kim, E. Hosono, H. Zhou, T. Kudo and I. Honma, Nano Lett., 2008, 8, 2277 CrossRef CAS.
- Z. Liu, J. T. Robinson, X. Sun and H. Dai, J. Am. Chem. Soc., 2008, 130, 10876 CrossRef CAS.
- A. P. Yu, P. Ramesh, M. E. Itkis, E. Bekyarova and R. C. Haddon, J. Phys. Chem. C, 2007, 111, 7565 CAS.
- H. Bai, C. Li and G. Shi, Adv. Mater., 2011, 23, 1089 CrossRef CAS.
- Y. Zhu, S. Murali, W. Cai, X. Li, J. W. Suk, J. R. Potts and R. S. Ruoff, Adv. Mater., 2010, 22, 3906 CrossRef CAS.
- S. Gilje, S. Han, M. Wang, K. L. Wang and R. B. Kaner, Nano Lett., 2007, 7, 3394 CrossRef CAS.
- C. Sheng, Z. Junwu, W. Xiaodong, H. Qiaofeng and W. Xin, ACS Nano, 2010, 4, 2822 CrossRef.
- K. P. Loh, Q. Bao, P. K. Ang and J. Yang, J. Mater. Chem., 2010, 20, 2277 RSC.
- S. Stankovich, R. D. Piner, X. Chen, N. Wu, S. T. Nguyen and R. S. Ruoff, J. Mater. Chem., 2006, 16, 155 RSC.
- Y. Si and E. T. Samulski, Nano Lett., 2008, 8, 1679 CrossRef CAS.
- S. Wang, P. Chia, L. Chua, L. Zhao, R. Png, S. Sivaramakrishnan, M. Zhou, R. G. S. Goh, R. H. Friend, A. T. S. Wee and P. K. H. Ho, Adv. Mater., 2008, 20, 3440 CrossRef CAS.
- V. C. Tung, M. J. Allen, Y. Yang and R. B. Kaner, Nat. Nanotechnol., 2009, 4, 25 CrossRef CAS.
- S. Park, J. H. An, I. W. Jung, R. D. Piner, S. J. An, X. S. Li, A. Velamakanni and R. S. Ruoff, Nano Lett., 2009, 9, 1593 CrossRef CAS.
- X. Fan, W. Peng, Y. Li, X. Li, S. Wang, G. Zhang and F. Zhang, Adv. Mater., 2008, 20, 4490 CrossRef CAS.
- Y. Xu, H. Bai, G. Lu, C. Li and G. Shi, J. Am. Chem. Soc., 2008, 130, 5856 CrossRef CAS.
- J. Rodríguez-López, N. L. Ritzert, J. A. Mann, C. Tan, W. R. Dichtel and H. D. Abruña, J. Am. Chem. Soc., 2012, 134, 6224 CrossRef.
- Y. Y. Liang, D. Q. Wu, X. L. Feng and K. Müllen, Adv. Mater., 2009, 21, 1679 CrossRef CAS.
- S. J. Kang, B. Kim, K. S. Kim, Y. Zhao, Z. Chen, G. H. Lee, J. Hone, P. Kim and C. Nuckolls, Adv. Mater., 2011, 31, 3531 CrossRef.
- H. Wang, L. F. Cui, Y. Yang, H. S. Casalongue, J. T. Robinson, Y. Liang, Y. Cui and H. Dai, J. Am. Chem. Soc., 2010, 132, 13978 CrossRef CAS.
- C. Berger, Z. Song, X. Li, X. Wu, N. Brown, C. Naud, D. Mayou, T. Li, J. Hass, A. N. Marchenkov, E. H. Conrad, P. N. First and W. A. de Heer, Science, 2006, 312, 1191 CrossRef CAS.
- D. Y. Cai and M. Song, J. Mater. Chem., 2007, 17, 3678 RSC.
- W. S. Hummers Jr and R. E. Offeman, J. Am. Chem. Soc., 1958, 80, 1339 CrossRef.
- W. Xiangjian, L. Guankui, H. Lu and C. Yongsheng, Adv. Mater., 2011, 23, 5342 CrossRef.
- X. Li, G. Zhang, X. Bai, X. Sun, X. Wang, E. Wang and H. Dai, Nat. Nanotechnol., 2008, 3, 538 CrossRef CAS.
- S. Gilje, S. Han, M. Wang, K. Wang and R. B. Kaner, Nano Lett., 2007, 7, 3394 CrossRef CAS.
- Y. Zhu, M. D. Stoller, W. Cai, A. Velamakanni, R. D. Piner, D. Chen and R. S. Ruoff, ACS Nano, 2010, 4, 1227 CrossRef CAS.
- J. Dupont, R. F. De Souza and P. A. Z. Suarez, Chem. Rev., 2002, 102, 3667 CrossRef CAS.
- T. Fukushima, A. Kosaka, Y. Ishimura, T. Yamamoto, T. Takigawa, N. Ishii and T. Aida, Science, 2003, 300, 2072 CrossRef CAS.
- T. Fukushima and T. Aida, Chem.–Eur. J., 2007, 13, 5048 CrossRef CAS.
- J. Lee and T. Aida, Chem. Commun., 2011, 47, 6757 RSC.
- W. Chen and L. Yan, Nanoscale, 2011, 3, 3132 RSC.
- Y. Xu, K. Sheng, C. Li and G. Shi, ACS Nano, 2010, 4, 4324 CrossRef CAS.
- S. K. Xuan, X. Y. Xi, L. Chun and S. G. Quan, New Carbon Mater., 2011, 26, 9 CrossRef.
- Z. Sui, X. Zhang, Y. Lei and Y. Luo, Carbon, 2011, 49, 4314 CrossRef CAS.
- Y. Sun, Q. Wu and G. Shi, Phys. Chem. Chem. Phys., 2011, 13, 17249 RSC.
- L. Zhang and G. Shi, J. Phys. Chem. C, 2011, 115, 17206 CAS.
- B. Zhang, W. Ning, J. Zhang, X. Qiao, J. Zhang, J. He and C.-Y. Liu, J. Mater. Chem., 2010, 20, 5401 RSC.
- D. Li, M. B. Müller, S. Gilje, R. B. Kaner and G. G. Wallace, Nat. Nanotechnol., 2008, 3, 101 CrossRef CAS.
- S. Niyogi, E. Bekyarova, M. E. Itkis, J. L. McWilliams, M. A. Hamon and R. C. Haddon, J. Am. Chem. Soc., 2006, 128, 7720 CrossRef CAS.
- X. Zhou, T. Wu, K. Ding, B. Hu, M. Hou and B. Han, Chem. Commun., 2010, 46, 386 RSC.
- T. Y. Kim, H. W. Lee, J. E. Kim and K. S. Suh, ACS Nano, 2010, 4, 1612 CrossRef CAS.
- S. Y. Park, J. H. Yoon, C. S. Hong, R. Souane, J. S. Kim, S. E. Matthews and J. Vicens, J. Org. Chem., 2008, 73, 8212 CrossRef CAS.
- X. Yang, L. Qiu, C. Cheng, Y. Wu, Z.-F. Ma and D. Li, Angew. Chem., Int. Ed., 2011, 50, 7325 CrossRef CAS.
- M. L. Sha, F. C. Zhang, G. Z. Wu, H. P. Fang, C. L. Wang, S. M. Chen, Y. Zhang and J. Hu, J. Chem. Phys., 2008, 128, 134504 CrossRef.
- M. L. Sha, G. Z. Wu, H. P. Fang, G. L. Zhu and Y. S. Liu, J. Phys. Chem. C, 2008, 112, 18584 CAS.
- Y. S. Shim and H. J. Kim, ACS Nano, 2009, 3, 1693 CrossRef CAS.
- M. A. Pimenta, G. Dresselhaus, M. S. Dresselhaus, L. G. Cançado, A. Jorio and R. Saito, Phys. Chem. Chem. Phys., 2007, 9, 1276 RSC.
- D. Yanga, A. Velamakannia, G. Bozoklub, S. Parka, M. Stollera, R. D. Pinera, S. Stankovich, I. Junga, D. A. Fieldd, C. A. Ventrice Jr and R. S. Ruoff, Carbon, 2009, 47, 145 CrossRef.
- H.-J. Shin, K. K. Kim, A. Benayad, S.-M. Yoon, H. K. Park, I.-S. Jung, M. H. Jin, H. K. Jeong, J. M. Kim, J.-Y. Choi and Y. H. Lee, Adv. Funct. Mater., 2009, 19, 1987 CrossRef CAS.
- J. Yan, Z. Fan, T. Wei, W. Qian, M. Zhang and F. Wei, Carbon, 2010, 48, 3825 CrossRef CAS.
- C.-Y. Su, Y. Xu, W. Zhang, J. Zhao, A. Liu, X. Tang, C.-H. Tsai, Y. Huang and L.-J. Li, ACS Nano, 2010, 4, 5285 CrossRef CAS.
- Y. Zhou, Q. Bao, L. A. L. Tang, Y. Zhong and K. P. Loh, Chem. Mater., 2009, 21, 2950 CrossRef CAS.
- S. Park, D. A. Dikin, S. T. Nguyen and R. S. Ruoff, J. Phys. Chem. C, 2009, 113, 15801 CAS.
- H.-K. Jeong, Y. P. Lee, R. J. W. E. Lahaye, M.-H. Park, K. H. An, I. J. Kim, C.-W. Yang, C. Y. Park, R. S. Ruoff and Y. H. Lee, J. Am. Chem. Soc., 2010, 130, 1362 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available: Detailed description of the synthesis, characterization, and methods. See DOI: 10.1039/c2ta00192f |
| ‡ It is important to note that the prolonged reaction time did not improve the graphenization ratio. |
| This journal is © The Royal Society of Chemistry 2013 |