Improvement of the electrochemical performance of carbon-coated LiFePO4 modified with reduced graphene oxide
Bo
Wang
ab,
Dianlong
Wang
*a,
Qiuming
Wang
a,
Tiefeng
Liu
a,
Chenfeng
Guo
a and
Xiusong
Zhao
*b
aHarbin Institute of Technology, School of Chemical Engineering and Technology, Xidazhi Street, 150001 Harbin, China. E-mail: wangdianlongwbhit@163.com; Fax: +86 451 86413721; Tel: +86 451 86413751
bThe University of Queensland, Faculty of Engineering, Architecture and Information Technology, School of Chemical Engineering, St Lucia, Brisbane, QLD 4072, Australia. E-mail: george.zhao@uq.edu.au; Fax: +61 7 33654199; Tel: +61 7 33469997
First published on 30th October 2012
Abstract
In this work, carbon-coated LiFePO4 was further modified with reduced graphene oxide (RGO) using an ultrasonic-assisted rheological phase method coupled with carbothermal treatment. X-ray diffraction (XRD), field-emission scanning electron microscopy (FESEM), transmission electron microscopy (TEM), high-resolution transmission electron microscopy (HRTEM), and electrochemical methods were used to characterize the material's properties. The results showed that the composite material consisting of carbon-coated LiFePO4 and RGO sheets possesses a unique and effective three-dimensional “sheet-web” structure. In the structure, the LiFePO4 particle size can be maintained at nanosize to form abundant voids between the nanoparticles while the RGO sheets are significantly beneficial for Li+ diffusion. As a result, the electrochemical properties of the composite material have been greatly improved. A sample with 5 wt% RGO exhibited high specific capacity and superior rate performance with the discharge capacities of 160.4 mA h g−1 at 0.2 C and 115.0 mA h g−1 at 20 C. The sample also showed an excellent cycling stability with only about 10% capacity decay at 10 C after 1000 cycles.
Introduction
LiFePO4 is a promising cathode material for high-power lithium-ion batteries used in electric vehicles (EV) and hybrid-electric vehicles (HEV) owing to its low cost, safety, superior stability and relatively high theoretical specific capacity of 170 mA h g−1.1–3 However, one of the major challenges for the commercialization of LiFePO4 is poor high-rate performance (≥5 C), attributed to its intrinsically low electrical conductivity (10−9 to 10−10 s cm−2) and lithium-ion diffusivity (10−14 cm2 s−1).4–6 Considerable efforts have been devoted to solving this problem such as decorating the surface with a high electrical conductivity agent,7,8 mixing with an electronically conductive material,9,10 doping the host framework with alien ions,11,12 reducing the particle size13–16 and optimizing the morphology.17,18 Among them, carbon-coating is considered as one of the most effective and conventional ways to improve the electrochemical properties. On one hand, the coated carbon layer can effectively retard the growth of LiFePO4 crystallites,19 which will favor Li+ insertion/extraction due to the shortened distance for Li+ diffusion and increased contact area with the electrolyte.20 On the other hand, the carbon coating layer can dramatically improve the surface electrical conductivity of LiFePO4 particles and decrease the polarization resistance of the electrode, resulting in a significantly improved rate performance. Nonetheless, current carbon-coating technologies still cannot satisfy the requirements for high-rate performance in practical applications. Too much carbon in the LiFePO4-based cathode will decrease the energy density. Therefore, finding an effective approach to coating carbon with a proper carbon source is still challenging.Graphene,21 a one-atom thick two-dimensional (2D) carbon structure, is an emerging material with many potential applications. Graphene-based composites have been explored as supercapacitor and lithium-ion battery electrodes.22–24 LiFePO4–graphene composite materials have also been reported.20,25–33 In this work, LiFePO4 particles were coated with carbon, followed by reduced graphene oxide (RGO) to prepare a novel composite material, denoted as LFP/(C + RGO). An ultrasonic-assisted rheological phase method in combination with a carbothermal reduction process was employed to prepare the material. The rheological phase technology34 was used to synthesize the precursor, which can effectively decrease the calcination temperature and time for the carbothermal reduction process due to the uniform distribution of the solid powders and liquid substances in the rheological body. Simultaneously, the carbon from sucrose pyrolysis was exploited as the reducing agent, and the residual carbon can also form carbon layers and a conductive web which could improve the electrochemical performance of the product effectively. The LFP/(C + RGO) composite showed excellent electrochemical properties, especially at high rates, as compared with a carbon-coated LiFePO4 sample (designated as LFP/C), which was prepared using the sample method of LFP/(C + RGO) except for without the presence of RGO.
Experimental
Preparation of RGO
Graphene oxide was prepared according to the modified Hummers method from natural graphite powders.35 Then the prepared graphene oxide was reduced by thermal treatment as described previously.36 6.5 g graphite powder was placed in 150 mL cold (0 °C) concentrated H2SO4, and then 19.5 g KMnO4 was added gradually with stirring and cooling, and the temperature of the solution was maintained below 20 °C. After stirring for 40 min, 460 mL distilled water was added slowly. The temperature was maintained at 35 ± 3 °C for 30 min. Finally, 1.4 L distilled water and 100 mL of 30% H2O2 solution were added after the reaction. The solution was held at room temperature for 24 h and then the mixture was filtered and washed with 5% HCl aqueous solution until sulfate could not be detected with BaCl2. The graphene oxide obtained was dried at 50 °C for 72 h. Reduced graphene oxide (RGO) was obtained after thermal treatment of the graphene oxide at 800 °C for 30 s.Preparation of LFP/(C + RGO) and LFP/C
The LFP/(C + RGO) composite was prepared through an ultrasonic-assisted rheological phase method coupled with carbothermal treatment as illustrated in Scheme 1. An appropriate amount of Fe(NO3)3·9H2O (AR), NH4H2PO4 (AR), and LiNO3 (AR) was dissolved in 25 mL of distilled water with a Fe![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :P:Li molar ratio of 1:1:1.05. Then, an appropriate amount of sucrose (the weight ratio of sucrose to LiFePO4 was 6:10) was added into the above solution as a carbon source. After that, the resulting solution and appropriate amount of RGO (the weight ratio of RGO to LiFePO4 was 5:95) were added to 25 mL ethanol and dispersed by ultrasonication for 2 h. Then the mixed solution was heated at 80 °C in air with continuous stirring to obtain a homogeneous rheological body. The rheological body was dried at 100 °C under vacuum for 10 h. Then the obtained precursor was heat-treated under a reductive (5 vol% hydrogen in argon) atmosphere at 260 °C for 2 h, followed by sintering at 600 °C for 10 h under the aforementioned reductive atmosphere. For an electrochemical performance comparison purpose, LFP/C composite was synthesized by the same way without the addition of RGO.
:P:Li molar ratio of 1:1:1.05. Then, an appropriate amount of sucrose (the weight ratio of sucrose to LiFePO4 was 6:10) was added into the above solution as a carbon source. After that, the resulting solution and appropriate amount of RGO (the weight ratio of RGO to LiFePO4 was 5:95) were added to 25 mL ethanol and dispersed by ultrasonication for 2 h. Then the mixed solution was heated at 80 °C in air with continuous stirring to obtain a homogeneous rheological body. The rheological body was dried at 100 °C under vacuum for 10 h. Then the obtained precursor was heat-treated under a reductive (5 vol% hydrogen in argon) atmosphere at 260 °C for 2 h, followed by sintering at 600 °C for 10 h under the aforementioned reductive atmosphere. For an electrochemical performance comparison purpose, LFP/C composite was synthesized by the same way without the addition of RGO.
 | ||
| Scheme 1 The procedures used to prepare LFP/(C + RGO). | ||
Characterizations
X-ray diffraction (XRD) patterns were collected on a D/max-γB X-ray diffractometer (Rigaku, Japan) using Cu Kα radiation (λ = 1.54178 Å). The diffraction angle was scanned from 10° to 90° at the scanning speed of 0.02° s−1. The morphology and microstructure of the composites were characterized by field emission scanning electron microscopy (Hitachi, S-4800), transmission electron microscopy (Hitachi, S-7650) and high resolution transmission electron microscopy (JEM-2100). Carbon element was analyzed using vario EL cube (Elementar, Germany). The nitrogen adsorption and desorption isotherms at 77.3 K were obtained on a Micromeritics ASAP 2020 surface area analyzer. The electrical conductivities of the composites were measured at room temperature using a four-probe conductivity test metre (SB120; San Feng).Electrochemical measurements
The electrochemical performance of the as-prepared LiFePO4-based composites and pure RGO cathode materials were evaluated using a CR2025 coin-type cell. The working electrode was fabricated by dispersing the active materials (80 wt%), acetylene black (10 wt%), and a polyvinylidene fluoride (PVDF) binder (10 wt%) in N-methylpyrrolidone (NMP) solvent to form a homogeneous slurry. The slurry was plastered on an Al foil and then dried at 100 °C for 10 h in a vacuum oven. The coin-type cell was assembled in a glove box filled with pure Ar. A Li foil and a polypropylene micro-porous film (Celgard 2400) were used as the counter electrode and separator, respectively. The electrolyte used was EC/DMC/DEC-based (1:1:1 by weight) containing 1 M LiPF6. For fabrication of the electrode LFP/(C + RGO), we treated it as the active material. The average loading of active mass in both LiFePO4-based electrodes was about 2 mg. The specific capacity of the LFP/(C + RGO) composite electrode was calculated based on the total mass of LFP/(C + RGO), including the mass of RGO.30–32
The cells were charged and discharged over a voltage range of 2.5–4.2 V (vs. Li/Li+) at different rates independent of the test procedure used with a Battery Testing System (Neware, China). Note that “C/n” means that the charge current is set up to achieve the nominal capacity in “n” hours. The same rule applies to the discharge steps where “D/n” corresponds to a discharge in “n” hours (e.g. 10C corresponds to a charge in 6 min; 10D corresponds to a discharge in 6 min). Electrochemical impedance spectroscopy (EIS) and cyclic voltammetry (CV) were measured on an electrochemical workstation (PARSTAT 2273, Princeton Applied Research, U.S.A.). CV was carried out at a scanning rate of 0.1 mV s−1 between 2.5 and 4.2 V. EIS measurements were performed over a frequency range of 100 kHz to 10 mHz at both fully discharged state and fully charged state with an applied amplitude of 5 mV.
Results and discussion
Characterizations
Fig. 1a shows the XRD patterns of natural graphite and the as-prepared RGO. A very broad peak at around 24° is seen for the RGO sample. This is in sharp contrast with the XRD pattern of the natural graphite sample, which displays a strong reflection peak at about 26°, revealing that the RGO sample is highly amorphous in nature, in accordance with previous reports.36,37 The XRD patterns of LFP/(C + RGO) and LFP/C are shown in Fig. 1b. For both the composites, all Bragg peaks are indexed to a well-crystallized orthorhombic structure LiFePO4 phase with the space group of Pnmb. No evidence of diffraction peaks for RGO appears in the diffraction pattern of LFP/(C + RGO), indicating the presence of RGO does not influence the structure of LiFePO4, which is consistent with previous studies.21–25 Meanwhile, no crystalline carbon was detected, implying the residual carbon from sucrose pyrolysis is in an amorphous phase. The vario EL cube (Elementar, Germany) was used to measure the carbon content in both products. It was confirmed that the residual carbon of LFP/C and LFP/(C + RGO) were 3.96 wt% and 8.81 wt%, respectively, suggesting RGO in LFP/(C + RGO) was about 5 wt%. The crystal lattice parameters are a = 5.9947 Å, b = 10.3315 Å, c = 4.6855 Å for LFP/C, and a = 5.9888 Å, b = 10.3440 Å, c = 4.6900 Å for LFP/(C + RGO). Both sets of parameters are very close to the values of LiFePO4 (ICDD PDF no. 40-1499, a = 6.019 Å, b = 10.347 Å, c = 4.704 Å). | ||
| Fig. 1 XRD patterns of (a) natural graphite and RGO; (b) LFP/(C + RGO), LFP/C and LFP (ICDD PDF no. 40-1499). | ||
The morphology and microstructure of the as-prepared samples were characterized by FESEM, TEM and HRTEM. As shown in the FESEM images (Fig. 2), the RGO sample (Fig. 2a and b) shows a nanoporous structure resulting from thermal exfoliation. The LFP/C composite (Fig. 2c) consisting of quasi-spherical particles has an equable size distribution ranging from 100 to 200 nm. Simultaneously, a random aggregation of primary nanoparticles can also be observed. As for LFP/(C + RGO) (Fig. 2d), the particle morphology is similar to LFP/C. However, because of the confined effect of the RGO sheets the particle size was kept smaller (<100 nm), which is considered to shorten the distance for Li+ diffusion.20,28,29

The electrical conductivities of the LFP/C and LFP/(C + RGO) are measured at room temperature using a four-probe conductivity test metre with results of 6.67 × 10−6 S cm−1 and 1.38 × 10−3 S cm−1, respectively.
Some representative transmission electron micrographs and high resolution transmission electron micrographs of RGO, LFP/C and LFP/(C + RGO) are shown in Fig. 3 and 4, respectively. The typical morphology of agglomerated RGO is observed in Fig. 3a and b. From Fig. 3c and d and 4a, the primary LFP/C particle coated by carbon layer (about 3 nm thickness) displays a quasi-spherical shape with diameters of 100–200 nm in agreement with the SEM images. Meanwhile, the existence of carbon layers can not only dramatically increase the inter-granular electrical conductivity, but also efficiently hinder particle growth. In the LFP/(C + RGO) sample (Fig. 3e and f and 4b), both the carbon layers (about 3 nm thickness) and RGO can be observed. The carbon coated LiFePO4 particles are homogeneously adhered to the surface of RGO, wrapped by RGO film or embedded in the RGO sheets, which could further reduce the particle size to diameters less than 100 nm. The reduced particle size would be favorable for improving the rate of Li+ insertion/extraction, which could be due to four reasons: firstly, the short distance for Li+ diffusion within the crystals; secondly, the large specific surface area of the small particles ensuring a large contact area with the electrolyte and hence a higher flux across the interface; thirdly, the range of composition over which solid solutions exist being often more extensive for nanocrystals; and fourthly, the strain associated with Li+ intercalation being better accommodated by smaller crystals due to the larger pore volumes surrounding them.20


Simultaneously, the residual carbon can form a conductive web bridging carbon layers and RGO together. Thus, a more effective 3D “sheet-web” mode mixed (electron and ion) conductive network25–27,29,38 will be built around the active particles, which can also decrease the effect of the particle-agglomeration phenomenon effectively. The average particle size of the prepared LFP/(C + RGO) composite, as deduced from the XRD data (Fig. 1b) using Scherrer's equation is about 60 nm, which is consistent with the SEM and TEM observations. In addition, the interspace between particles and RGO can further facilitate the penetration of the electrolyte to the surface of active particles.
According to BET analysis, the specific surface area of LFP/C, LFP/(C + RGO) and RGO are 12.6 m2 g−1, 36.8 m2 g−1 and 328.3 m2 g−1, respectively, and the nitrogen adsorption/desorption isotherms of them are shown in Fig. 5.

The schematics of the possible structures of LFP/(C + RGO) are shown in Scheme 2. In view of the one-dimensional Li+ ion mobility in the framework,39 coating with a carbon layer, which ensures LiFePO4 particles get electrons from all directions, could further alleviate the polarization phenomenon of the electrode.19 Therefore, such a highly efficient and stable mixed (electron and ion) conducting network will provide superior electronic contact between the LiFePO4 particles and facilitate the diffusion of Li+, which will be more effective in enhancing the electrochemical performance of LFP/(C + RGO) in comparison with LFP/C.
 | ||
| Scheme 2 Schematics of the possible structures of LFP/(C + RGO): (a) LiFePO4 particles wrapped by RGO sheets; and (b) LiFePO4 particles embedded in RGO sheets. | ||
Electrochemical testing
Typical charge–discharge profiles of LFP/C and LFP/(C + RGO) at a current rate of 0.2C–0.2D are shown in Fig. 6a. As noted, LFP/(C + RGO) exhibited an improved specific capacity of 160 mA h g−1.30–32 whereas LFP/C displayed a capacitance of around 140 mA h g−1. As seen from the embedded figure, the voltage plateau for LFP/(C + RGO) was significantly flatter and longer than that of LFP/C with a decreased polarization between the charge and discharge plateaus from 50 mV to 25 mV, indicating that the kinetics of electron transport in electrode LFP/(C + RGO) was indeed improved by the addition of RGO. Moreover, as shown by the dot-line-box part, the typical capacitive charge–discharge profile with a capacity of about 15 mA h g−1 could be clearly observed at the beginning of both the charge and discharge processes of LFP/(C + RGO). The RGO electrode measured under the same experimental conditions showed a typical capacitive capacity of about 60 mA h g−1 (127 F g−1). However, considering that there was only 5 wt% of RGO in LFP/(C + RGO) electrode, this would contribute only about 3 mA h g−1 (6.4 F g−1) to the overall electrode capacity. This is much lower than the difference between the specific capacity of LFP/(C + RGO) and LFP/C. Therefore, we believed that LFP/(C + RGO) should not be simply divided into two independent parts, RGO and LFP/C. Instead there existed a “synergic effect” between the two components, as has been observed in previous papers.33,40,41 | ||
| Fig. 6 Charge–discharge profiles of (a) LFP/C (□) and LFP/(C + RGO) (△) (the inset shows part of the flat region magnified); and (b) pure RGO. | ||
The comparison of rate performance between LFP/C and LFP/(C + RGO) step by step from 0.2C–0.2D to 20C–20D is shown in Fig. 7. Obviously, the LFP/(C + RGO) composite exhibited a better electrochemical performance, especially at high rates, with discharge capacities of about 160, 150, 136, 127 and 116 mA h g−1 at 0.2 C, 1 C, 5 C, 10 C and 20 C, respectively. Whereas, the LFP/C composite exhibited 146, 120, 98, 85 and 70 mA h g−1 with an increased gap from 15 to 46 mA h g−1 compared to LFP/(C + RGO). The rate capability of LFP/(C + RGO) indicates that being co-modified with RGO and carbon layer dramatically improves the surface electrical conductivity of LiFePO4 particles and shortens the distance for Li+ diffusion, which decrease the polarization resistance of the electrode and results in a preeminent rate performance.
 | ||
| Fig. 7 Rate performances (herein referred to as discharge capacity) of LFP/C (□) and LFP/(C + RGO) (△). | ||
The time–(current and charge capacity) and time–(current and charge efficiency) curves of both the as-synthesized samples at 20C–1D are shown in Fig. 8a and b, respectively. It can be seen that it only took around 80 seconds to reach the designated upper limited voltage (4.2 V vs. Li/Li+) for the LFP/C electrode during the galvanostatic charge process (followed by a constant-voltage process) and the charge capacity of this process is 63 mA h g−1, which is about 52% of the total charge capacity (ca. 120 mA h g−1). As for the LFP/(C + RGO) electrode, the time for the galvanostatic charge process was approximately 150 seconds and the charge capacity during this process was around 130 mA h g−1, which is nearly 87% of the total charge capacity (ca. 150 mA h g−1). Additionally, after charging for 200 seconds, the charge efficiency of electrode LFP/(C + RGO) was approximate 95%, which is also better than that of electrode LFP/C (90%). The results indicate an excellent fast-charging performance of LFP/(C + RGO) which is promising as a cathode material for EVs and HEVs.
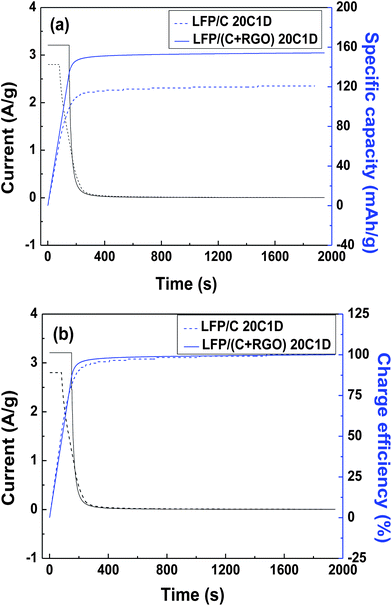 | ||
| Fig. 8 (a) Time–(current and charge capacity) and (b) time–(current and charge efficiency) curves. | ||
The high-rate cycling performances at 10C–10D of both the as-prepared electrode materials are shown in Fig. 9. It is found that LFP/C can deliver a discharge capacity of nearly 70 mA h g−1 after 1000 cycles which is about 85% of the initial capacity. In contrast, LFP/(C + RGO) showed a better cycling stability with a final discharge capacity of around 115 mA h g−1, which is nearly 90% of the initial discharge capacity.
 | ||
| Fig. 9 High-rate cycling performances (herein referred to as discharge capacity) of LFP/C (□) and LFP/(C + RGO) (△). | ||
In order to investigate the effect of RGO on the electrochemical properties of LFP in more detail, cyclic voltammetry and electrochemical impedance measurements were performed for both samples.
Cyclic voltammetry plots are shown in Fig. 10. The CV plot of LFP/(C + RGO) showed a more symmetrical and poignant shape of the anodic/cathodic peaks, suggesting a better electrochemical performance.30 Furthermore, the peak separation exhibited by LFP/(C + RGO) was 0.147 V, whereas that of the LFP/C was much bigger (0.217 V). Meanwhile, a large increase can also be observed between the corresponding peak currents of LFP/C and LFP/(C + RGO). The sharper peaks, smaller peak potential separation and larger peak currents all indicate the higher electrochemical reactivity and lower ohmic resistance of the LFP/(C + RGO) composite.
 | ||
| Fig. 10 Cyclic voltammetry profiles of LFP/C and LFP/(C + RGO). | ||
The Nyquist plots at fully discharged and charged states of both samples are presented in Fig. 11a and b, respectively. The EIS profiles consist of a partially overlapped semicircle in the high frequency region followed by a sloping line in the low frequency region. The intercept of the Z′ axis in the high frequency region corresponds to the ohmic resistance (Ro), which represents the sum of resistance of the electrolyte and electrodes. The semicircle in the high and middle frequency regions is due to the charge transfer resistance (Rct) and the sloping line in the lower frequency represents the Warburg impedance, which is associated with lithium-ion diffusion in the bulk of the electrode.20,25,31,32
 | ||
| Fig. 11 EIS profiles of LFP/C and LFP/(C + RGO) at (a) fully discharged state and (b) fully charged state. | ||
The semicircles of electrode LFP/(C + RGO) are found to have much smaller diameters than those of electrode LFP/C from both the Nyquist plots, indicating a lower resistance to charge transfer as a result of the improved electrical conductivity of the LFP/(C + RGO) composite, which is consistent with the results of the four-probe conductivity measurement. The electrical conductivities of LFP/C and LFP/(C + RGO) are 6.67 × 10−6 S cm−1 and 1.38 × 10−3 S cm−1, respectively. In addition, the impedance slopes of LFP/(C + RGO) in the low frequency range are also much higher than that of LFP/C, reflecting the enhanced mobility of Li ions in electrode LFP/(C + RGO).
These above results all indicate that the addition of RGO plays an important and positive role in improving the electrochemical performance of LFP/(C + RGO) composite which can be attributed to three main factors. Firstly, and most importantly, because of the confined effect of the RGO sheets the particle size can be reduced to nanosize, which would be favorable for improving the rate of Li+ insertion/extraction and the reasons are as follows: (i) the short distance for Li+ diffusion within the crystals; (ii) the large specific surface area of the small particles ensuring a large contact area with the electrolyte and hence a higher flux across the interface; (iii) the range of compositions over which solid solutions exist being often more extensive for nanocrystals; and (iv) the strain associated with Li+ intercalation being better accommodated by smaller crystals due to the larger pore volumes surrounding them. Secondly, the residual carbon can form a conductive web bridging carbon layers and RGO together. Thus, an effective 3D “sheet-web” mode mixed (electron and ion) conductive network will be built around the active particles, which can also decrease the effect of the particle-agglomeration phenomenon effectively. Meanwhile, the interspace between particles and RGO can further facilitate the penetration of the electrolyte to the surface of active particles. In addition, due to the high specific surface area of RGO, the interface reaction current density during the charge–discharge process decreases effectively. Thirdly, as we all know, RGO is an excellent capacitor material due to its distinctive properties which can instantly store and release a great quantity of charge. For the LFP/(C + RGO) composite, when in high-rate charge or discharge situations, RGO realizes a capacity response immediately which can ensure a preeminent rate performance. In addition, the RGO-unwrapped active particles can be coated by the carbon layers and linked by the carbon web which is also in favor of the improvement of electronic conductivity for the whole material.
Conclusions
In this paper, the LFP/(C + RGO) composite has been prepared successfully via an ultrasonic-assisted rheological phase method coupled with carbothermal treatment in a manner that the active particles are wrapped by RGO film or embedded in RGO sheets. By a comparative study on the electrochemical performance of LFP/C and LFP/(C + RGO), we have demonstrated that the specific capacity, rate capability, fast-charge performance and cycling stability are significantly improved due to the positive influence of RGO, such as the reduced active particle size, the construction of the 3D “sheet-web” mode mixed (electron and ion) conductive network and the capacitive property of RGO. In addition, this preparation strategy is of potential interest to other low conductive cathode materials and to develop high-power lithium-ion batteries for electric vehicles.Acknowledgements
We appreciate the support of National Natural Science Foundation of China (no. 50974045), the Ph.D. Programs Foundation of Ministry of Education of China (no. 20092302110052), the Natural Science Foundation of Heilongjiang Province, China (no. B200918) and China Scholarship Council (no. 201206120186). Australian Research Council (ARC) is also acknowledged for financial support under the ARC Future Fellow Program (FT100100897).Notes and references
- A. K. Padhi, K. S. Nanjundaswamy and J. B. Goodenough, J. Electrochem. Soc., 1997, 144, 1188 CrossRef CAS.
- M. Armand and J. M. Tarascon, Nature, 2008, 451, 652 CrossRef CAS.
- Y. Wang and G. Z. Cao, Adv. Mater., 2008, 20, 2251 CrossRef CAS.
- S. Y. Chung and Y. M. Chiang, Electrochem. Solid-State Lett., 2003, 6, A278 CrossRef CAS.
- A. S. Andersson, B. Kalska, L. Haggstrom and J. O. Thomas, Solid State Ionics, 2000, 130, 41 CrossRef CAS.
- P. P. Prosini, M. Lisi, D. Zane and M. Pasquali, Solid State Ionics, 2002, 148, 45 CrossRef CAS.
- Y. G. Wang, Y. R. Wang, E. J. Hosono, K. X. Wang and H. S. Zhou, Angew. Chem., Int. Ed., 2008, 47, 7461 CrossRef CAS.
- Y. S. Hu, Y. G. Guo, R. Dominko, M. Gaberscek, J. Jamnik and J. Maier, Adv. Mater., 2007, 19, 1963 CrossRef CAS.
- S. Kuroda, N. Tobori, M. Sakuraba and Y. Sato, J. Power Sources, 2003, 119, 924 CrossRef.
- E. M. Jin, B. Jin, K. H. Park, H. B. Gu, G. C. Park and K. W. Kim, J. Nanosci. Nanotechnol., 2008, 8, 5057 CrossRef CAS.
- S. Y. Chung, J. T. Bloking and Y. M. Chiang, Nat. Mater., 2002, 1, 123 CrossRef CAS.
- X. G. Yin, K. L. Huang, S. Q. Liu, H. Y. Wang and H. Wang, J. Power Sources, 2010, 195, 4308 CrossRef CAS.
- B. L. Ellis, W. R. M. Makahnouk, Y. Makimura, K. Toghill and L. F. Nazar, Nat. Mater., 2007, 6, 749 CrossRef CAS.
- C. Delmas, M. Maccario, L. Croguennec, F. Le Cras and F. Weill, Nat. Mater., 2008, 7, 665 CrossRef CAS.
- M. Gaberscek, R. Dominko and J. Jamnik, Electrochem. Commun., 2007, 9, 2778 CrossRef CAS.
- C. Delacourt, P. Poizot, S. Levasseur and C. Masquelier, Electrochem. Solid-State Lett., 2006, 9, A352 CrossRef CAS.
- J. Liu, T. E. Conry, X. Y. Song, M. M. Doeff and T. J. Richardson, Energy Environ. Sci., 2011, 4, 885 CAS.
- C. W. Sun, S. Rajasekhara, J. B. Goodenough and F. Zhou, J. Am. Chem. Soc., 2011, 133, 2132 CrossRef CAS.
- Y. G. Wang, Y. R. Wang, E. J. Hosono, K. X. Wang and H. S. Zhou, Angew. Chem., Int. Ed., 2008, 47, 7461 CrossRef CAS.
- S. W. Oh, Z. D. Huang, B. Zhang, Y. Yu, Y. B. He and J. K. Kim, J. Mater. Chem., 2012, 22, 17215 RSC.
- A. K. Geim and K. S. Novoselov, Nat. Mater., 2007, 6, 183 CrossRef CAS.
- M. D. Stoller, S. J. Park, Y. W. Zhu, J. H. An and R. S. Ruoff, Nano Lett., 2008, 8, 3498 CrossRef CAS.
- L. L. Zhang, R. Zhou and X. S. Zhao, J. Mater. Chem., 2010, 20, 5983 RSC.
- Y. Wang, Z. Q. Shi, Y. Huang, Y. F. Ma, C. Y. Wang, M. M. Chen and Y. S. Chen, J. Phys. Chem. C, 2009, 113, 13103 CAS.
- C. Su, X. D. Bu, L. H. Xu, J. L. Liu and C. Zhang, Electrochim. Acta, 2012, 64, 190 CrossRef CAS.
- L. Wang, H. B. Wang, Z. H. Liu, C. Xiao, S. M. Dong, P. X. Han, Z. Y. Zhang, X. Y. Zhang, C. F. Bi and G. L. Cui, Solid State Ionics, 2010, 181, 1685 CrossRef CAS.
- X. F. Zhou, F. Wang, Y. M. Zhu and Z. P. Liu, J. Mater. Chem., 2011, 21, 3353 RSC.
- Y. Ding, Y. Jiang, F. Xu, J. Yin, H. Ren, Q. Zhuo, Z. Long and P. Zhang, Electrochem. Commun., 2010, 12, 10 CrossRef CAS.
- Y. Wang, Z. S. Feng, J. J. Chen and C. Zhang, Mater. Lett., 2012, 71, 54 CrossRef CAS.
- J. L. Yang, J. J. Wang, D. N. Wang, X. F. Li, D. S. Geng, G. X. Liang, M. Gauthier, R. Y. Li and X. L. Sun, J. Power Sources, 2012, 208, 340 CrossRef CAS.
- Y. Shi, S. L. Chou, J. Z. Wang, D. Wexler, H. J. Li, H. K. Liu and Y. Wu, J. Mater. Chem., 2012, 22, 16465 RSC.
- Y. Zhang, W. C. Wang, P. H. Li, Y. B. Fu and X. H. Ma, J. Power Sources, 2012, 210, 47 CrossRef CAS.
- O. Toprakci, H. A. K. Toprakci, L. W. Ji, Z. Lin and R. P. Gu, J. Renewable Sustainable Energy, 2012, 4, 013121-1 CrossRef.
- J. T. Sun, W. Xie, L. J. Yuan, K. L. Zhang and Q. Y. Wang, Mater. Sci. Eng., B, 1999, 64, 157 CrossRef.
- Z. S. Wu, W. C. Ren, L. B. Gao, B. L. Liu, C. B. Jiang and H. M. Cheng, Carbon, 2009, 47, 493 CrossRef CAS.
- L. Wang and D. L. Wang, Electrochim. Acta, 2011, 56, 5010 CrossRef CAS.
- K. C. Jiang, S. Xin, J. S. Lee, J. Kim, X. L. Xiao and Y. G. Guo, Phys. Chem. Chem. Phys., 2012, 14, 2934 RSC.
- X. L. Wu, L. Y. Jiang, F. F. Cao, Y. G. Guo and L. J. Wan, Adv. Mater., 2009, 21, 2710 CrossRef CAS.
- D. Morgan, A. Van der Ven and G. Ceder, Electrochem. Solid-State Lett., 2004, 7, A30 CrossRef CAS.
- N. Böckenfeld, R.-S. Kühnel, S. Passerini, M. Winter and A. Balducci, J. Power Sources, 2011, 196, 4136 CrossRef.
- X. B. Hu, Z. J. Lin, L. Liu, Y. J. Huai and Z. H. Deng, J. Serb. Chem. Soc., 2010, 75, 1259 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2013 |