Interfacial shear rheology of DPPC under physiologically relevant conditions
Eline
Hermans
and
Jan
Vermant
*
Department of Chemical Engineering, KU Leuven, University of Leuven, Willem de Croylaan 46, Heverlee, Belgium. E-mail: jan.vermant@cit.kuleuven.be; Fax: +32 16322991; Tel: +32 16322355
First published on 24th October 2013
Abstract
Lipids, and phosphatidylcholines in particular, are major components in cell membranes and in human lung surfactant. Their ability to encapsulate or form stable layers suggests a significant role of the interfacial rheological properties. In the present work we focus on the surface rheological properties of dipalmitoylphosphatidylcholine (DPPC). Literature results are confusing and even contradictory; viscosity values have been reported differ by several orders of magnitude. Moreover, even both purely viscous and gel-like behaviours have been described. Assessing the literature critically, a limited experimental window has been explored correctly, which however does not yet include conditions relevant for the physiological state of DPPC in vivo. A complete temperature and surface pressure analysis of the interfacial shear rheology of DPPC is performed, showing that the monolayer behaves as a viscoelastic liquid with a domain structure. At low frequencies and for a thermally structured monolayer, the interaction of the molecules within the domains can be probed. The low frequency limit of the complex viscosity is measured over a wide range of temperatures and surface pressures. The effects of temperature and surface pressure on the low frequency viscosity can be analysed in terms of the effects of free molecular area. However, at higher frequencies or following a preshear at high shear rates, elasticity becomes important; most probably elasticity due to defects at the edge of the domains in the layer is probed. Preshearing refines the structure and induces more defects. As a result, disagreeing interfacial rheology results in various publications might be due to different pre-treatments of the interface. The obtained dataset and scaling laws enable us to describe the surface viscosity, and its dependence under physiological conditions of DPPC. The implications on functioning of lung surfactants and lung surfactant replacements will be discussed.
1. Introduction
Lipids, and in particular phosphatidylcholines, are major components of cell membranes and lung surfactants in mammals. Their ability to act as building blocks and encapsulants, or the possibility to form stable layers in the lungs, is important for several biological phenomena. The role of the interfacial rheological properties has been put forward previously in several studies on DPPC (1,2-dipalmitoyl-sn-glycero-3-phosphocholine) and in a range of applications.DPPC is the main component of lung surfactant, which is a complex mixture of lipids and proteins lining the surface of the alveoli in the lungs and it is essential for breathing. The interfacial viscosity was measured to be rather high and it was suggested that this is important for keeping the lung surfactant in place in the alveoli.1 It is believed that the surfactant prevents the alveoli from collapsing by lowering the surface tension.2–5 However, during breathing, the area of the alveoli continuously changes while the area in the terminal bronchioles remains constant. This area change in the alveoli may lead to a change in surface concentration of the surfactant and as a consequence varying surface tension, while the surface tension in the bronchioles remains constant. This dynamic difference in surface tension could lead to a Marangoni flow of the surfactant and the underlying liquid out of the alveoli during exhaling and back towards the alveoli during inhaling. To date, no Marangoni flow has been observed in situ in a mammal lung. It has been suggested that a high interfacial viscosity would limit this surfactant induced flow and keep fluids in place.1
On the other hand, prematurely born babies often completely lack the surfactant and experience possibly fatal breathing problems denoted as Neonatal Respiratory Distress Syndrome (NRDS).5 During instillation of the replacement lung surfactant in the lungs of prematurely born babies, spreading is shown to occur within a minute by experiments with neonatal piglets.6 Although the spreading of the bolus is mostly dominated by respiration, the delivery to the final bronchioles and alveoli is believed to be controlled by a Marangoni flow, now directed to spread the surfactant in the lung.7 For this to occur, a low interfacial viscosity would be required to allow easy spreading of the replacement surfactant in the baby's lung. This is consistent with more recent measurements of DPPC rheology,8 which suggest low values of the complex viscosity although measurements were only reported for low to moderate surface pressures up to 30 mN m−1 and room temperature conditions.
DPPC rheology has also been suggested to play an important role in raft dynamics, and it has been reported to behave as a viscoelastic gel9 under exactly the same conditions for which other research groups have reported an essentially viscous behaviour.10 From the current state of the literature it is difficult to deduce what the behaviour of DPPC and similar phospolipids will be and how the rheological properties relate to the compositional variables.
In the past, the interfacial rheology of DPPC has been characterized only in a limited experimental window of relatively low surface pressures (Π) and at temperatures (T) close to room temperature8,11,12 which however does not yet include conditions relevant to the physiological state of DPPC in the human body (high surface pressures and temperatures). A few attempts were made to characterize DPPC under physiological conditions in the lung at a temperature of 37 °C.1,9,10 However, most of these data were obtained below the actual sensitivity limits of the experimental devices and the surface pressures did not mimic the high surface pressures that are believed to be present inside the lung.
In the present work, the experimental parameter window (Π, T) was enlarged by increasing sensitivity during interfacial rheology experiments combined with the controlled realization of high temperatures and surface pressures. The geometries for interfacial shear rheology, which allowed the expansion of the experimental window, are a magnetic needle and an adapted double wall ring (DWR) accessory. In combination with a ribbon trough and environmental control of temperature and humidity, the interfacial shear rheology of DPPC is determined at temperatures ranging from 20 to 45 °C and surface pressures from 20 up to 60 mN m−1, including the physiological condition of DPPC in the lung.
2. Methods
2.1. Monolayer preparation
DPPC was purchased from Avanti Polar Lipids (Alabaster, USA) and dissolved in chloroform (VWR, Prolabo grade) at a concentration of 1 mM. The solution was spread droplet-wise on a pure water subphase (MilliQ) after preheating it up to 45 °C. Subsequently, the system was left for 30 min for the chloroform to evaporate. The high temperature accelerates adsorption of DPPC and evaporation of chloroform. But most importantly, this ensures the homogeneous spreading of the monolayer over the entire area of the trough, since this temperature exceeds the melting temperature of DPPC, approximately 41.3 °C.13,14 Above this temperature the monolayer is in a liquid expanded (LE) state. Afterwards the system was cooled to 20 °C from which the temperature was raised to the actual measurement condition. This procedure ensures reproducible monolayers at the interface13 with a high surface pressure after spreading and a LC structure with presumably large domains due to the slow cooling during the preparation procedure. We hereafter refer to this monolayer as “thermally structured”. The temperature is set by circulating water coming from a Julabo thermostat under the bottom of the trough. The monolayer was characterized at five different temperatures, namely 20, 25, 30, 37 and 45 °C. Surface pressures varied from 20 mN m−1 up to 60 mN m−1. Above 60 mN m−1, monolayer collapse was too fast.To investigate the effect of size of the head groups on the interfacial rheology of DPPC a buffer subphase (150 mM NaCl, 2 mM CaCl2 and 0.2 mM NaHCO3) with an ionic strength of 0.152 M was used. To quantify the effect of the tails, the interfacial rheology of 1-hexadecanol (Aldrich) is determined. Hexadecanol consists of an alkyl chain identical to DPPC. Subsequently a 1 mg ml−1 solution of hexadecanol in chloroform (VWR, Prolabo grade) was spread on a MilliQ interface. In both cases the procedure to prepare the interface is similar as described above.
2.2. Surface pressure area isotherms
Surface pressure area isotherms were measured in a Langmuir trough with ribbon barriers (KSV NIMA). The use of a ribbon ensures a better stability at high surface pressures compared to conventional barriers, which are prone to leakage. The compression speed of the ribbon was set to 5 mm min−1 while surface tension was measured with a platinum Wilhelmy plate (KSV NIMA).Although the amount of surfactant spread gives an indication of how much of the interfacial area will be covered, this does not allow for an accurate determination of the molecular area as the amount really present at the interface depends on the details of the experimental protocol like spreading, compression rate, apparatus and possible experimental artifacts.15 The molecular area in the liquid condensed (LC) regions can be quantified by grazing incidence X-ray diffraction (GIXD),16 but this was not feasible in the present case for every combination of temperature and surface pressure. As a result, an equation of state was fitted to literature data of molecular area determined with GIXD at a certain surface pressure and temperature.17–19 Subsequently, this equation can be used to predict molecular area of DPPC in a fully LC state when measuring surface pressure and temperature.
As equation of state, the van der Waals equation for interfaces (eqn (1))20 was fitted. It describes the relationship between molecular area (A), surface pressure (Π) and temperature (T) for an isotropic system. Although the monolayer consists of anisotropic domains in the liquid condensed state, as will be shown below, the combination of different domains results in a macroscopically isotropic layer and the van der Waals equation is used as an effective, averaged description. A0 represents the excluded molecular area (an area around the center of the molecule which cannot contain a second molecule), B is a measure of the attractive interactions between the molecules and kB is Boltzmann's constant. Once the molecular area at a single point in the isotherm is known, the entire isotherm can be scaled based on the trough area.
 | (1) |
The van der Waals (VdW) equation of state only includes short range VdW interactions. However, between the DPPC molecules more interacting forces are present like hydrophobic forces between the tails and electrostatic forces between the polar head groups. All these forces reach further than van der Waals forces. As a result, the fraction of the interactions coming from van der Waals forces will be most dominant when molecules are closer together. Consequently, the van der Waals equation is only used to determine the molecular area at a single point in the LC domain of the surface pressure area isotherm at every temperature, the one with the highest stable surface pressure, 45 mN m−1. This procedure is further evaluated in Appendix A.
2.3. Fluorescence microscopy
To visualize the microstructure of the monolayer, a 1 mol% fluorescent marker was added to the DPPC spreading mixture in chloroform. This fluorescent marker, 1-palmitoyl-2-(12-[(7-nitro-2-1,3-benzoxadiazol-4-yl)amino]dodecanoyl)-sn-glycero-3-phosphocholine (PC-NBD) (Avanti Polar Lipids, Alabaster, USA), has a molecular structure similar to that of DPPC but has one shorter alkyl chain to which a fluorescent group (NBD) is attached. Due to the fluorescent molecule's slightly different structure, it segregates to the LE phase during compression21 or to the domain boundaries in the case of a fully LC structure. The preparation procedure for the fluorescent interfaces was identical to that of the non-fluorescent interfaces, described above. The desired surface pressure is obtained and maintained by compressing the monolayer. Visualization of the monolayer microstructure in the trough was performed with a fixed stage upright microscope (Olympus BX51WI) with mercury light (100 V), an internal filter U-MNIBA3 with a narrow bandpass region of 470–495 nm and a 50× objective (0.45 NA, Olympus, Japan). Images were captured with a C8800 Hamamatsu camera.2.4. Interfacial shear rheometry
The interfacial shear rheology of the DPPC monolayer has been characterized by small amplitude oscillatory experiments. Unless specified otherwise, no preshearing was performed after the formation and compression of the bilayer. Two different devices were used, both allowing interfacial shear rheometry at controlled temperature and surface pressure. The sensitivity of an interfacial rheology measurement is quantified in a macroscopic Boussinesq (Bo) number, which represents the ratio of viscous forces exerted by the interface divided by viscous forces coming from the subphase or the liquid underneath the interface. | (2) |
 | ||
| Fig. 1 Langmuir trough with an adapted double wall ring accessory attached to a stress controlled rheometer. The two openings in the cup allow a uniform compression inside and outside the cup. Additionally, the openings in the ring ensure a uniform interface on both sides of the geometry. | ||
3. Results
3.1. Surface pressure area isotherms
The best fit result of the GIXD literature data with the van der Waals equation yields a value of A0 equal to 39.6 Å2 and a B of 52.76 × 10−40 J. In Fig. 2, the scaled interfacial isotherms of DPPC at 20, 25, 30 and 37 °C are shown. The transition of a liquid expanded (LE) to a liquid condensed monolayer (LC) can be identified by the presence of a plateau in the curve. This plateau region clearly is temperature dependent and shifts to higher surface pressures with increasing temperature. The interfacial rheology at the four temperatures displayed in Fig. 2 was only characterized in the LC region, at a surface pressure above the plateau once the slope of the curve has stabilized to ensure a full LC layer. The surface pressure area isotherm at 45 °C is not shown in Fig. 2. Due to the melting transition of DPPC at 41.3 °C monolayers above this temperature will be in the LE state at every surface pressure. As a result the isotherm cannot be scaled by the use of GIXD data which only predicts A for an LC interface. | ||
| Fig. 2 Surface pressure area isotherms of DPPC at different temperatures. The molecular area is scaled by molecular areas obtained by best fits to GIXD data from the literature. | ||
Care should be taken in analysing the data at high surface pressures. Although it is possible to compress the monolayer up to values of 50 mN m−1 and higher, the maximum stable surface pressure that can be reached by spreading is only 45 mN m−1.26 When the bulk concentration exceeds the critical micellar concentration (0.46 nM for DPPC), high surface pressures lead to molecules detaching from the interface.27 Higher pressures can only be maintained in a Langmuir trough under continuous compression and the necessary compression rate increases with temperature.
3.2. Interfacial shear rheology of DPPC resembles a 2D liquid crystal
Fig. 3 shows the modulus of the complex interfacial viscosity (|η*s|) as a function of frequency (ω) for strain amplitudes within the linear region ranging from 3% for low surface pressures down to 0.5% at the highest pressures. The interfacial rheology of DPPC showed no significant frequency dependence in the observed range for low surface pressures. | ||
| Fig. 3 Frequency dependence of the magnitude of the complex interfacial viscosity (|η*s|) and phase angle of DPPC at 25 °C and five different surface pressures (strain amplitude from 3% down to 0.5%). | ||
However, at high surface pressures the effect of frequency becomes more pronounced with a decreasing complex viscosity and increasing elasticity. Rheological measurements following a well-defined preshear provide indirect evidence of a shear sensitive microstructure. This indication of the presence of a microstructure at the interface led to the examination of the effect of a preshear on the interfacial rheology at high surface pressures.
Fig. 4 shows the magnitude of the complex interfacial viscosity (|η*s|) and phase angle (δ) as a function of frequency (ω) for a DPPC monolayer at 25 °C and 40 mN m−1. By applying a preshear to the surface, the dominantly viscous behaviour at low frequencies remains unchanged while the elasticity fundamentally increases at high frequencies. These results suggest the presence of a domain structure at the interface which becomes smaller at higher shear rates.
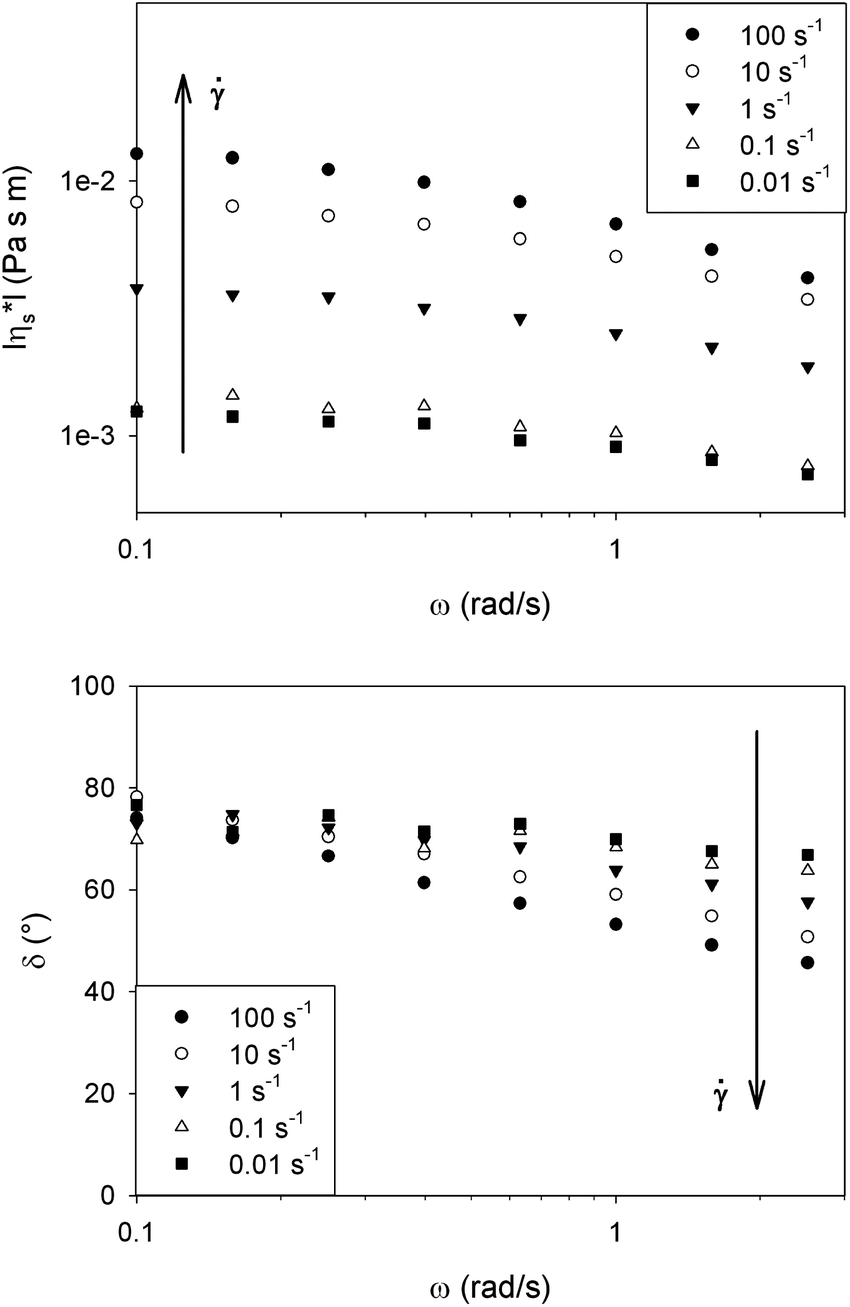 | ||
| Fig. 4 Effect of preshear on the complex interfacial viscosity (|η*s|) and phase angle (δ). At low frequencies the viscous dissipation of the molecules in the domains is probed, while at higher frequencies the elastic contribution of the domain defects starts dominating. Increasing preshear is believed to refine the structure leading to a more elastic behavior at high frequencies. | ||
Since the DPPC monolayer is in a fully liquid condensed state at the high surface pressures observed here, it can be considered as 2D liquid crystal polydomains with different orientations. Due to the use of the specific temperature protocol in preparing the monolayer, a coarse polydomain structure is expected. Shear subjects the domains to a tumbling motion which consequently leads to the breaking of domains during continuous shearing accompanied by a texture refinement.28
At low frequencies the dominantly viscous behaviour is caused by a combination of the in-domain structure plus the viscous friction between domains, while at high frequencies interactions between the domains dominate the interfacial rheology. At the edge of the domains, molecules with different orientations face each other and can be considered as defects which apparently cause elasticity at high frequencies, possibly similar to the effects of Frank or distortional elasticity in liquid crystal rheology.28 By increasing the preshear, the decrease in domain sizes increases the total amount of defects and as a result the elastic behaviour at high frequencies is increased. This morphological feature indicates a shear induced microstructure, which is also confirmed by the non-applicability of the Cox–Merz analogy, as shown in Fig. 5. In these experiments the sample was just presheared and consequently the shear rate was lowered, until some steady state was obtained. The corresponding shear viscosity differs from the magnitude of the complex viscosity of the quiescent structure obtained after the preshear, even in the low frequency limit. This difference is also observed for bulk suspensions, emulsions or textured systems; it demonstrates that the mobility of the system at equilibrium does not directly govern the dynamics under flow, due to changes in the microstructure caused by the flow.
 | ||
| Fig. 5 Comparison of a rate sweep and frequency sweep on a DPPC interface at 25 °C and a surface pressure of 40 mN m−1 shows that the Cox–Merz analogy is not valid and indicates the presence of a shear rate dependent structure. | ||
3.3. DPPC monolayer microstructure
The DPPC monolayer structure was visualised by the addition of a fluorescently labeled lipid to show the contrast between ordered and disordered phases. In theory this could not lead to a visualisation of a fully ordered, LC layer. In practice, the fluorescent molecules segregate to the domain boundaries at intermediate surface pressures above the transition surface pressure, but are probably squeezed out at higher surface pressures rendering no significant morphological features. In Fig. 6 the microstructures of a DPPC monolayer at 25 °C and a moderate surface pressure of approximately 30 mN m−1 before and after preshearing are shown. It is clear that the microstructure is refined after preshearing, confirming the origin of the increased elasticity as described above. | ||
| Fig. 6 Effect of preshear on the microstructure of a DPPC monolayer at a moderate surface pressure of approximately 30 mN m−1 and a temperature of 25 °C. | ||
3.4. Low frequency interfacial shear rheology of DPPC monolayers
In the following, the low frequency behaviour of a DPPC monolayer is examined more closely for interfaces prepared as described above without any preshearing after setting temperature and surface pressure. The observed dominantly viscous rheology is due to the creation of large domains during the annealing of the interface. This creates an opportunity to probe the rheology of the DPPC molecules within the domains with minimal interference of the domain boundaries. In this way an averaged in-domain viscosity of the DPPC can be probed.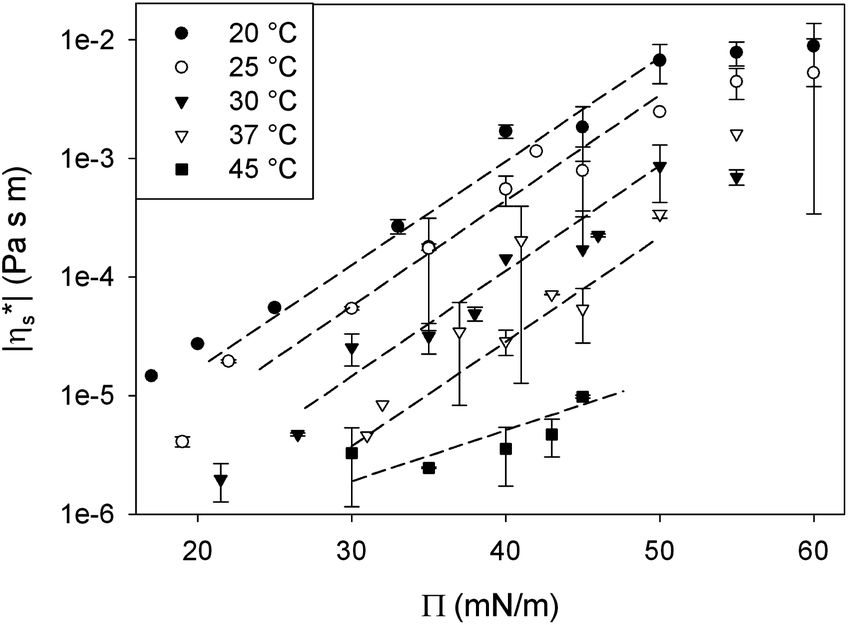 | ||
| Fig. 7 Effect of surface pressure (Π) on the low frequency limit of the complex interfacial shear viscosity (|η*s|) of a “thermally structured” DPPC monolayer at constant temperature. To guide the eye, the dashed lines highlight the exponential relationship. The error bars represent the standard deviation of a set of measurements of at least 3. | ||
For all temperatures, an exponential relationship between |η*s| and surface pressure is observed, similar to the relationships observed for simple bulk liquids under pressure. In a 3D liquid, an increase in pressure (P) results in a decrease in free molecular volume which reduces the mobility of the molecules, causing the bulk viscosity (η) to rise. In analogy an increase in surface pressure, by compressing the monolayer, decreases the molecular area and as such, the mobility of the surfactant molecules in the monolayer. However, at high surface pressures above the maximum spreading pressure of DPPC, the increase in viscosity levels off. This can be explained by the limits to the stability of the monolayer at high pressures. At surface pressures above the maximum spreading pressure (45 mN m−1), molecules detach from the interface and are forced into the subphase.27 Consequently these high surface pressures can only be maintained by continuous compression. This extra out of plane mobility of the molecules may explain the weaker dependency at high surface pressures.
For simple bulk liquids the pressure dependence of viscosity is described by a Barus or pressure coefficient β29 as denoted in eqn (3). Similarly, also for monolayers, an interfacial surface pressure coefficient βs can be defined in eqn (4).
 | (3) |
 | (4) |
Applying eqn (4) to the data in Fig. 7 results in the interfacial pressure coefficients βs for DPPC monolayers as a function of temperature as shown in Fig. 8. For the temperatures below the melting temperature of DPPC, only the data at surface pressures which correspond to a stable and fully LC interfaces are used. As can be seen in Fig. 8, a constant pressure coefficient of approximately 180 Pa−1 m−1 is observed at these temperatures. The pressure coefficient obtained at a temperature of 45 °C, above the melting temperature, is significantly lower. This monolayer does not have a LC, but LE configuration. The intermolecular interactions are less strong and thermal energy becomes dominant causing a high molecular area. At high free molecular areas effects of surface pressure and as such molecular area change on the interfacial viscosity are smaller.
 | ||
| Fig. 8 Interfacial pressure coefficient of DPPC monolayers as a function of temperature. At the highest temperature the monolayer is in the LE state, and the other T values correspond to a LC phase. The error bars indicate the 99% confidence interval obtained via a least square fit. | ||
By multiplying the pressure coefficient with the approximate thickness of the monolayer (10−9–10−10 m), the parameter can be compared to a corresponding bulk property β. Pressure coefficients of e.g. polyethylene (10 × 10−9 Pa−1)30 and even air (1.3 × 10−8 Pa−1) are of the same order of magnitude. This indicates that the pressure dependence of the interfacial viscosity of the LC monolayer is very similar to that of 3D liquids, related to a change in free area of the molecules in the monolayer.
 | (5) |
 | ||
| Fig. 9 Effect of temperature on the interfacial shear viscosity of DPPC at constant surface pressure. The error bars represent the standard deviation of a set of measurements of at least 3. | ||
R is the universal gas constant and T is temperature. E, the viscous activation energy, is the energy necessary for a molecule to jump from one equilibrium position in the liquid to another. The activation energies determined by fitting eqn (5) for each surface pressure are shown in Fig. 10. Most striking are the remarkably high values of the viscous activation energy. While the viscous activation energy varies around 15–30 kJ mol−1 for 3D liquids, the values for the DPPC interfaces are in the order of 150 kJ mol−1. The reduction of degrees of freedom of molecules on a 2D interface instead of a 3D liquid is expected to increase E, but not with a factor 10. This indicates the presence of other interactions which are not present in a simple liquid, namely the presence of electrostatic interactions between the polar head groups on the one hand, or hydrophobic interactions between the alkyl chains on the other hand.
 | ||
| Fig. 10 Viscous activation energy for DPPC monolayers in the LC phase at different surface pressures. The error bars indicate the 99% confidence interval obtained via a least square fit. | ||
To evaluate the interaction between the head groups, the interfacial viscosity of DPPC is compared for two different subphases in Fig. 11, namely pure water (MilliQ) and a buffer solution with an ionic strength of 0.152 M. By increasing the ionic strength of the subphase, the electrostatic interactions of the head groups are shielded. However, the viscous activation energy remained unchanged. From this it can be concluded that electrostatic interactions between the head groups are not responsible for the high activation energy.
 | ||
| Fig. 11 Effect of the ionic strength of the subphase medium on the interfacial viscosity of DPPC at a surface pressure of 40 mN m−1. | ||
The absence of effects of electrolyte concentrations suggests that the strong hydrophobic interaction between the tails is responsible for the high values. To evaluate this further, hexadecanol, which consists of an alkyl chain identical to DPPC and a small head group from which the effect on viscosity or activation energy can be neglected, was studied. Fig. 11 shows the effect of temperature on the interfacial shear viscosity of hexadecanol at 40 mN m−1. Although the interfacial viscosity of hexadecanol is much higher than that of DPPC due to its smaller head group and hence molecular area, the activation energy is 145 kJ mol−1, similar to that of DPPC. Although the data in Fig. 11 correspond to a surface pressure of 40 mN m−1, the same trends were observed at other surface pressures. As a result, it can be concluded that strong hydrophobic interactions between the chains are most probably responsible for the high activation energy in the monolayers.
Israelachvili and Pashley32 predict a hydrophobic interaction potential between hydrophobic molecules described in eqn (6) with r the molecular radius, D the intermolecular distance and D0 the intermolecular distance at contact. When applying this equation to estimate the hydrophobic interaction between two methyl groups (ΔGmeth) of two different tails, D and D0 can respectively be estimated from A and A0. The molecular radius is approximated with 0.2 nm based on twice the C–H bond length. This results in an approximation of (ΔGmeth) by 1.9 kJ mol−1. Within the domains, the DPPC molecules are stacked in an orthorhombic lattice. Assuming a hexagonal close packed structure, both tails with 16 methyl groups each face 3 neighbouring tails. According to eqn (7), this results in an approximate total hydrophobic interaction of 180 kJ mol−1, which matches the observed order of magnitude.
 | (6) |
| ΔGDPPC = ΔGmeth·16·2·3 ≈ 180 kJ mol−1 | (7) |
 | (8) |
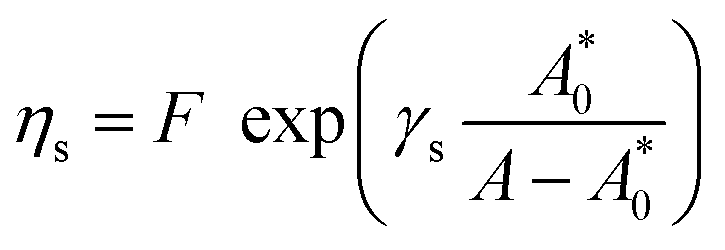 | (9) |
From the relationship of surface pressure and molecular area, shown in the scaled surface pressure isotherms (Fig. 2), a molecular area is determined for the data points shown in Fig. 7. As a result, Fig. 12 shows the modulus of the complex viscosity as a function of molecular area together with a fit of Doolittle's equation with the fitting parameter γs equal to 13.18 (±1.47). From Fig. 12, it can be concluded that the low frequency limit of the interfacial viscosity scales with molecular area, independent of the temperature and surface pressure combination that is present to obtain this molecular area.
 | ||
| Fig. 12 Effect of free molecular area on the low frequency limit of the modulus of the complex interfacial shear viscosity of DPPC. | ||
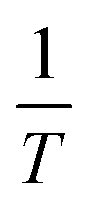 . From this result eqn (10) and (11).
. From this result eqn (10) and (11). | (10) |
 | (11) |
In eqn (11) the partial derivatives of ηs can be calculated from eqn (4), (5) and (9) resulting in eqn (12).
 | (12) |
Substituting the differential dA as a function of dΠ and d 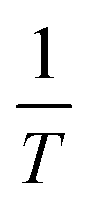
 | (13) |
 are independent variables and rendering the groups dimensionless by multiplying with (A − A0), the physical parameters βs, E and γs should relate as stated in eqn (14).
are independent variables and rendering the groups dimensionless by multiplying with (A − A0), the physical parameters βs, E and γs should relate as stated in eqn (14). | (14) |
In Fig. 13 the three terms of eqn (14) are evaluated and denoted as respectively A = Π = T. They are shown as their absolute values, since all are negative, together with the errors corresponding to the 99% confidence interval. The graph clearly shows that the terms are equal. The error on the third term (T) is quite large, due to the large error on  (see Appendix B for the numerical values). In conclusion, these equations can form a powerful tool to predict the interfacial rheology of untextured interfaces once its behavior in the function of one of the physical parameters is known.
(see Appendix B for the numerical values). In conclusion, these equations can form a powerful tool to predict the interfacial rheology of untextured interfaces once its behavior in the function of one of the physical parameters is known.
 | ||
| Fig. 13 The three dimensionless terms corresponding to eqn (14) denoted as A = Π = T which show the relationship among the physical parameters βs, E and γs. The error bars represent the 99% confidence interval. | ||
4. Discussion
So far the interfacial rheology of DPPC had only been characterized under ambient conditions and moderate surface pressures.8,11,12 In the present work a range of temperatures and surface pressures was used which enables one to estimate the role of the interfacial rheology of a DPPC rich layer inside the human lung (or in cell membranes). It is known that most phospholipid mixtures in living organisms are in a liquid disordered state, indicating that their melting temperatures (or those of their mixtures) are below body temperature. Besides DPPC with a melting temperature of 41.3 °C other lipids are present in the lung surfactant which have lower melting temperatures and hence decrease the melting temperature of the mixture.34 As a result, the melting temperature of the lipid mixture is believed to be below body temperature and consequently the monolayer will be in the LE state.34 To obtain a first estimate of the properties under in vivo conditions, it seems more appropriate to consider the rheological properties of DPPC in the LE state, since it corresponds most likely to the physical state of the surfactant in the lung.Although the main mode of deformation in the alveoli is a dilatational motion during breathing, there is a significant amount of shear flow. Due to the fact that the alveoli are not perfectly spherical, the flow field will be a combination of shear and dilatation. Additionally, in the small pores connecting the alveoli to each other and the airway, an important shearing motion is invoked in the surface layer during flow.
Lung surfactant is believed to function at high surface pressures and vary during breathing between 50 and 65 mN m−1.4,35 As the layer is in a liquid expanded state, experiments show a limited pressure dependence, resulting in a very low modulus of the surface shear viscosity of approximately 10−5 Pa s m and dominantly viscous behaviour. Whether this is sufficient to keep the lung surfactant in place by limiting the Marangoni flow depends on the driving force present in the lung. This driving force is dominated by the change in surface tension during breathing and the profile of the surface tension from the alveoli up to the bronchioles. Although surface pressure has been characterized at certain locations in the lung, e.g. 50 mN m−1 in the bronchioles connected to the alveoli36 decreasing down to 40 ± 3 mN m−1 in the trachea,37 the actual gradients are unknown to date. As a result, it cannot be stated for certain whether or not this viscosity is sufficient to limit Marangoni flow. The presented low interfacial rheology results also agree with the more recent results of Kim et al.38 On the other hand is a low interfacial viscosity consistent with the easy spreading of the surfactant during instillation in a baby's lung, which is claimed to be partially driven by Marangoni flow.7 Due to the absence of lung surfactant in a baby's lungs, the driving forces for Marangoni flow are much bigger than those during breathing.
Finally, the increased elasticity of a DPPC monolayer after preshear might indicate an important consequence for lung surfactant functioning. Since the lung functions at rather high frequencies (±0.2 Hz), the shearing motion at the alveoli openings might have a significant effect on the lung surfactant microstructure, by breaking up the domains in the liquid crystal structure and as a result, induction of elasticity to the layer. This additional elasticity should be beneficial in limiting Marangoni flow during breathing.
In addition to the lung surfactant, in living animals phospholipids are also an important component of the cell membranes.34 Although this is a bilayer, and the studied system in this work is a monolayer, the result can give some insight into the bilayer's fluidity. This fluidity is of importance when studying lipid rafts, dynamic assemblies of sphingolipids and cholesterol in the membranes. It is believed that these rafts can easily break up and recombine on the surface.39 For this purpose, the flow resistance experienced by the surrounding lipid bilayer during displacements on the surface should preferably be small. The molecular area of phospholipids in the cell membrane at 37 °C resembles that of DPPC in a monolayer at a surface pressure of 30 mN m−1.40 The observed interfacial viscosity of approximately 10−6 Pa s m is low. However, in contrast to lung surfactants, cell membranes contain a significant amount of cholesterol which has been shown to affect interfacial rheology38 and make the layer even more fluid.
5. Conclusion
In this work, a systematic analysis of the temperature and surface pressure dependence of the interfacial shear rheology of DPPC was carried out. Maintaining sensitivity over these large ranges was possible by the selection of suitable geometries for interfacial shear rheology. DPPC monolayers prepared with well controlled thermal history appear to be behaving like 2D liquid crystals in their liquid condensed state. At low frequencies and without preshearing, the monolayer shows a dominantly viscous behaviour similar to simple 3D liquids. Since the domains are rather large due to the protocol for preparation, the rheology is dominated by interactions between the molecules within the different domains. The effects of temperature and surface pressure on the interfacial viscosity then originate from the effect of molecular area. For DPPC, the surface pressure coefficient corresponds to the pressure dependence of known materials in the bulk. However the temperature dependence, quantified by the viscous activation energy, appears to be much higher than that of bulk liquids, probably because of strong hydrophobic interactions between the alkyl chains in the molecule.However, at higher frequencies of a monolayer subjected to a preshear the interaction between the domains is probed. At the edges of the domains, molecules with different orientation face each other and cause defects which induce distortional elasticity in the layer, similar to Frank elasticity. This effect becomes more pronounced after preshearing, which refines the structure and induces more defects. Consequently, diverging interfacial rheology results in various publications might be due to different pre-treatments of the DPPC interface.
Finally, the present dataset enables us to evaluate the interfacial rheology of DPPC under conditions representative of those in the human body. The interfacial viscosity appears to be very low, and confirms the ability of easy spreading of the lung surfactant in a baby's lung driven by Marangoni flow. However, since the driving force for Marangoni flow during instillation in a baby's lung is believed to be much more important than during normal breathing, it cannot be stated for certain whether the interfacial viscosity is too low to keep the lung surfactant in place. Further research is necessary to identify the surface tension profile in the lung to determine whether the initial hypothesis of a high interfacial viscosity as a resistance to Maragoni flow has to be reconsidered.
Appendix
A GIXD data and van der Waals fit
The molecular area in the liquid condensed (LC) regions can be quantified by grazing incidence X-ray diffraction (GIXD),16 but this was not feasible in the present case for every combination of temperature and surface pressure. As a result, an equation of state was fitted to relate the available molecular area data from the literature (Fig. 14)17–19 to the present surface pressure and temperature. Subsequently, this equation can be used to predict molecular area of DPPC in a fully LC state. | ||
| Fig. 14 GIXD data from the literature used to determine the van der Waals parameters.17–19 | ||
As equation of state, the van der Waals equation for interfaces (eqn (15))20 was fitted. It describes the relationship between molecular area (A), surface pressure (Π) and temperature (T). Once the molecular area at a single point in the isotherm is known, the entire isotherm can be scaled based on the trough area.
 | (15) |
The best fit result of the GIXD literature data with the van der Waals equation yields a value of A0 equal to 39.6 Å2 and a B of 52.76 × 10−40 J. In Fig. 2, the scaled interfacial isotherms of DPPC at 20, 25, 30 and 37 °C are shown. The transition of a liquid expanded (LE) to a liquid condensed monolayer (LC) can be identified by the presence of a plateau. This plateau region clearly is temperature dependent and shifts to higher surface pressures with increasing temperature. The interfacial rheology at the four temperatures displayed in Fig. 2 was only characterized in the LC region, at a surface pressure above the plateau once the slope of the curve had stabilized to ensure a full LC layer. The surface pressure area isotherm at 45 °C is not shown in Fig. 2. Due to the melting transition of DPPC at 41.3 °C monolayers above this temperature will be in the LE state at every surface pressure. As a result the isotherm cannot be scaled by the use of GIXD data which only predict A for an LC interface.
In Fig. 15 a comparison is made between the surface pressure area isotherms scaled with the van der Waals equation at a single surface pressure (45 mN m−1) and the isotherm predicted by the van der Waals equation at that temperature. The van der Waals equation of state only includes van der Waals interactions, which are short range interactions. However, between the DPPC molecules more interacting forces are present like hydrophobic forces between the tails and electrostatic forces between the polar head groups. All these forces reach further than van der Waals forces. As a result, the surface pressure in the real isotherm will turn up at lower molecular areas than that predicted by van der Waals. Since the fraction of the interactions coming from van der Waals forces will be most dominant when molecules are closer together, the van der Waals equation is only used to determine the molecular area at a single point in the LC domain of the surface pressure area isotherm at every temperature, the one with the highest stable surface pressure, 45 mN m−1.
 | ||
| Fig. 15 The experimentally determined surface pressure –area isotherms with a molecular area scaled at a single surface pressure of 45 mN m−1 (1 point vdW) compared to the surface pressure area isotherms in which for all data points the molecular area is determined via the van der Waals equation (all points vdW) for three different temperatures 20, 25 and 30 °C. | ||
B Relationship among βs, E and γs
The partial derivatives and
and  are derived from the surface pressure area isotherms for different data points together with the size of the 99% confidence intervals (Table 1 and Table 2).
are derived from the surface pressure area isotherms for different data points together with the size of the 99% confidence intervals (Table 1 and Table 2).
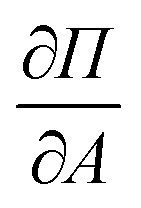 for all surface pressure area isotherms together with error which corresponds to the size of the 99% confidence interval
for all surface pressure area isotherms together with error which corresponds to the size of the 99% confidence interval
| T °C |
|
Error
|
|---|---|---|
| 20 | −2.56 | 0.02 |
| 25 | −1.65 | 0.02 |
| 30 | −1.28 | 0.01 |


 for all data points at constant surface pressure together with the error which corresponds to the size of the 99% confidence interval
for all data points at constant surface pressure together with the error which corresponds to the size of the 99% confidence interval
|
Π
|
|
Error
|
|
Error
|
|
Error
|
|---|---|---|---|---|---|---|
| 30 | −3.44 × 1015 | −1.08 × 1014 | −2.18 × 1015 | −6.83 × 1013 | −1.23 × 1015 | −3.85 × 1013 |
| 31 | −3.72 × 1015 | −2.30 × 1014 | −2.40 × 1015 | −1.48 × 1014 | −1.38 × 1015 | −8.53 × 1013 |
| 32 | −3.93 × 1015 | −2.27 × 1015 | −2.63 × 1015 | −1.52 × 1015 | −1.47 × 1015 | −8.52 × 1014 |
| 33 | −4.22 × 1015 | −3.50 × 1015 | −2.89 × 1015 | −2.40 × 1015 | −1.62 × 1015 | −1.34 × 1015 |
| 34 | −4.55 × 1015 | −5.20 × 1015 | −3.20 × 1015 | −3.66 × 1015 | −1.78 × 1015 | −2.04 × 1015 |
| 35 | −4.95 × 1015 | −3.47 × 1015 | −3.67 × 1015 | −2.57 × 1015 | −2.04 × 1015 | −1.43 × 1015 |
| 36 | −5.44 × 1016 | −1.05 × 1016 | −4.10 × 1016 | −7.91 × 1015 | −2.31 × 1016 | −4.45 × 1015 |
| 37 | −5.79 × 1015 | −1.35 × 1016 | −4.46 × 1015 | −1.04 × 1016 | −2.46 × 1015 | −5.74 × 1015 |
| 38 | −6.18 × 1015 | −2.15 × 1016 | −5.08 × 1015 | −1.76 × 1016 | −2.66 × 1015 | −9.23 × 1015 |
| 39 | −6.73 × 1015 | −3.05 × 1016 | −5.87 × 1015 | −2.66 × 1016 | −3.03 × 1015 | −1.37 × 1016 |
| 40 | −6.98 × 1015 | −3.79 × 1016 | −6.34 × 1015 | −3.44 × 1016 | −3.05 × 1015 | −1.66 × 1016 |
| 41 | −9.01 × 1015 | −4.44 × 1016 | −8.08 × 1015 | −3.98 × 1016 | −4.33 × 1015 | −2.13 × 1016 |
| 42 | −1.13 × 1016 | −3.00 × 1016 | −9.05 × 1015 | −2.40 × 1016 | −5.18 × 1015 | −1.37 × 1016 |
| 43 | −1.36 × 1016 | −2.31 × 1016 | −1.01 × 1016 | −1.72 × 1016 | −5.76 × 1015 | −9.78 × 1015 |
| 44 | −1.64 × 1016 | −1.25 × 1016 | −1.05 × 1016 | −8.03 × 1015 | −5.26 × 1015 | −4.01 × 1015 |
| 45 | −1.73 × 1016 | −1.25 × 1017 | −1.70 × 1016 | −1.22 × 1017 | −3.42 × 1015 | −2.46 × 1016 |






Acknowledgements
The authors like to thank Prof.em. H. Devlieger and Dr Anja Vananroye for fruitful discussions and a continued interest in this work. Ir. Dirk Abbeloos is acknowledged for the support with the mathematical analysis. FWO Vlaanderen (Ph.D. Fellowship for E.H.) and the Hercules foundation (A-KUL 05/024) are gratefully acknowledged for their financial support.References
- C. Alonso, A. Waring and J. A. Zasadzinski, Biophys. J., 2005, 89, 266–273 CrossRef CAS PubMed.
- R. E. Pattle, Nature, 1955, 175, 1125–1126 CrossRef CAS.
- R. E. Pattle, Physiol. Rev., 1965, 45, 48–79 CAS.
- E. S. Brown, R. P. Johnson and J. A. Clements, J. Appl. Physiol., 1959, 14, 717–720 CAS.
- J. A. Clements, R. F. Hustead, R. P. Johnson and I. Gribetz, J. Appl. Phys., 1961, 16, 444–450 CAS.
- J. M. Davis, G. A. Russ, L. Metlay, B. Dickerson and B. S. Greenspan, Pediatr. Res., 1992, 31, 445–450 CrossRef CAS PubMed.
- D. Halpern, O. E. Jensen and J. B. Grotberg, J. Appl. Physiol., 1998, 85, 333–352 CAS.
- K. Kim, S. Q. Choi, J. A. Zasadzinski and T. M. Squires, Soft Matter, 2011, 7, 7782–7789 RSC.
- G. Espinosa, I. López-Montero, F. Monroy and D. Langevin, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 6008–6013 CrossRef CAS PubMed.
- J. Ding, H. E. Warriner, J. A. Zasadzinski and D. K. Schwartz, Langmuir, 2002, 18, 2800–2806 CrossRef CAS.
- S. Y. Nishimura, G. M. Magana, H. A. Ketelson and G. G. Fuller, Langmuir, 2008, 24, 11728–11733 CrossRef CAS PubMed.
- D. L. Leiske, C. Monteux, M. Senchyna, H. A. Ketelson and G. G. Fuller, Soft Matter, 2011, 7, 7747–7753 RSC.
- N. Albon and J. M. Sturtevant, Proc. Natl. Acad. Sci. U. S. A., 1978, 75, 2258–2260 CrossRef CAS.
- B. G. Tenchov, H. Yao and I. Hatta, Biophys. J., 1989, 56, 757–768 CrossRef CAS.
- S. L. Duncan and R. G. Larson, Biophys. J., 2008, 94, 2965 CrossRef CAS PubMed.
- S. G. Wolf, L. Leiserowitz, M. Lahav, M. Deutsch, K. Kjaer and J. Als-Nielsen, Nature, 1987, 328, 63–66 CrossRef CAS.
- M. Christoforou, E. Leontidis and G. Brezesinski, J. Phys. Chem. B, 2012, 116, 14602–14612 CrossRef CAS PubMed.
- A. Aroti, E. Leontidis, E. Maltseva and G. Brezesinski, J. Phys. Chem. B, 2004, 108, 15238–15245 CrossRef CAS.
- M. Flasiński, M. Broniatowski, J. Majewski and P. Dynarowicz-atka, J. Colloid Interface Sci., 2010, 348, 511–521 CrossRef PubMed.
- A. W. Adamson, Physical chemistry of surfaces, Interscience Publishers, 1960 Search PubMed.
- M. Flörsheimer and H. Möhwald, Chem. Phys. Lipids, 1989, 49, 231–241 CrossRef.
- T. Verwijlen, P. Moldenaers, H. A. Stone and J. Vermant, Langmuir, 2011, 27, 9345–9358 CrossRef CAS PubMed.
- C. F. Brooks, G. G. Fuller, C. W. Frank and C. R. Robertson, Langmuir, 1999, 15, 2450–2459 CrossRef CAS.
- S. Reynaert, C. F. Brooks, P. Moldenaers, J. Vermant and G. G. Fuller, J. Rheol., 2008, 52, 261 CrossRef CAS.
- S. Vandebril, A. Franck, G. G. Fuller, P. Moldenaers and J. Vermant, Rheol. Acta, 2009, 49, 131–144 CrossRef PubMed.
- H. M. Mansour and G. Zografi, Langmuir, 2007, 23, 3809–3819 CrossRef CAS PubMed.
- S. T. Milner, J. F. Joanny and P. Pincus, Europhys. Lett., 1989, 9, 495–500 CrossRef CAS.
- G. Marrucci, Pure Appl. Chem., 1985, 57, 1545–1552 CrossRef CAS.
- C. Barus, Proc. Natl. Acad. Sci. U. S. A., 1891, 27, 13–18 CrossRef.
- A. Goubert, J. Vermant, P. Moldenaers, A. Göttfert and B. Ernst, Appl. Rheol., 2001, 11, 26–37 CAS.
- H. Eyring and W. J. J. Moore, J. Chem. Phys., 1938, 6, 391–394 CrossRef.
- J. Israelachvili and R. Pashley, Nature, 1982, 300, 341–342 CrossRef CAS.
- A. K. Doolittle and D. B. Doolittle, J. Appl. Phys., 1957, 28, 901 CrossRef CAS.
- H. Lodish, A. Berk, P. Matsudaira, C. A. Kaiser, M. Krieger, M. P. Scott, S. L. Zipursky and J. Darnell, Molecular cell biology, W. H. Freeman, New York, 5th edn, 2004 Search PubMed.
- S. Schurch, J. Goerke and J. A. Clements, Proc. Natl. Acad. Sci. U. S. A., 1976, 73, 4698–4702 CrossRef CAS.
- M. Geiser, S. Schürch and P. Gehr, J. Appl. Physiol., 2003, 94, 1793–1801 Search PubMed.
- V. Im Hof, P. GEHR, V. Gerber, M. M. Lee and S. Schurch, Respir. Physiol., 1997, 109, 81–93 CrossRef CAS.
- K. Kim, S. Q. Choi, Z. A. Zell, T. M. Squires and J. A. Zasadzinski, Proc. Natl. Acad. Sci. U. S. A., DOI:10.1073/pnas.1303304110.
- K. Simons and E. Ikonen, Nature, 1997, 387, 569 CrossRef CAS PubMed.
- D. Marsh, Chem. Phys. Lipids, 2006, 144, 146–159 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2014 |