 Open Access Article
Open Access ArticleMicrophase separation induced by competitive hydrogen bonding interactions in semicrystalline triblock copolymer/homopolymer complexes
Nisa V.
Salim
a,
Nishar
Hameed
a,
Tracey L.
Hanley
b and
Qipeng
Guo
*a
aPolymers Research Group, Institute for Frontier Materials, Deakin University, Locked Bag 2000, Geelong, Victoria 3220, Australia. E-mail: qguo@deakin.edu.au
bInstitute of Materials Engineering, Australian Nuclear Science and Technology Organization, Locked Bag 2001, Kirrawee DC, NSW 2232, Australia
First published on 20th May 2013
Abstract
Microphase separation through competitive hydrogen bonding interactions in ABC/D triblock copolymer/homopolymer complexes is studied for the first time. This study investigated self-assembled nanostructures that are obtained in the bulk, by the complexation of a semicrystalline polystyrene-block-poly(4-vinylpyridine)-block-poly(ethylene oxide) (SVPEO) triblock copolymer with a poly(4-vinyl phenol) (PVPh) homopolymer in tetrahydrofuran (THF). In these complexes, microphase separation takes place due to the disparity in intermolecular interactions among PVPh/P4VP and PVPh/PEO pairs. At low PVPh concentrations, PEO interacts relatively weakly with PVPh, whereas in the complexes containing more than 30 wt% PVPh, the PEO block interacts considerably with PVPh, leading to the formation of composition-dependent nanostructures. SAXS and TEM results indicate that the cylindrical morphology of a neat SVPEO triblock copolymer changes into lamellae structures at 20 wt% of PVPh then to disordered lamellae with 40 wt% PVPh. Wormlike structures are obtained in the complex with 50 wt% PVPh, followed by disordered spherical microdomains with size in the order of 40–50 nm in the complexes with 60–80 wt% PVPh. Moreover, when the content of PVPh increases to 80 wt%, the complexes show a completely homogenous phase of PVPh/P4VP and PVPh/PEO with phase separated spherical PS domains. The fractional crystallization behavior in SVPEO and complexes at lower PVPh content was also examined. A structural model was proposed to explain the microphase separation and self-assembled morphologies of these complexes based on the experimental results obtained. The formation of nanostructures and changes in morphologies depend on the relative strength of hydrogen bonding interactions between each component block of the copolymer and the homopolymer.
1 Introduction
Block copolymers are an important class of “soft materials” and they can self-assemble to form various nanostructures.1 The repulsive and attractive interactions within and between the blocks as well as their covalent linkages are the driving force for producing self-assembled nanostructures. In diblock copolymers, the microphase behavior is governed by the thermodynamic balance between the segment–segment interaction parameter (χ) and the degree of polymerization (N). When the values of χN exceed a critical value, block copolymers microphase separate into periodically ordered structures, with length scales that vary from a few nanometers to several hundred nanometers. Blending of a block copolymer with a homopolymer is a convenient technique that offers a rich variety of self-organized nanostructures with diverse applications.2–6 There are many theories regarding the microphase separation in block copolymer/homopolymer systems. One of them is random phase approximation (RPA)7 where χ and ξ are the two interaction parameters used for characterizing such systems; χ denotes repulsive interactions (χ > 0) and ξ represents attractive interactions (ξ < 0). RPA, with mean field calculations, explains the microphase separation that takes place in systems consisting of block copolymers and a homopolymer that can selectively interact with one of the blocks. Hellmann et al.8 have shown that in systems where microphase separation occurs (avoiding the homogeneous state or macrophase separation), there is always a repulsive interaction between the homopolymer and one block of the block copolymer (χ > 0) which induces the microphase separation.Unlike diblock copolymers, the microphase separation in ABC triblock copolymers results in a rich variety of nanostructures because of the three different components A, B and C. The disordered phase of a triblock copolymer melt is more stable than that of a diblock copolymer of the same length and composition. Triblock copolymer systems have revealed a wide range of well-ordered complex micro-domain morphologies.9,10 In ABC triblock copolymers, with one or more crystallizable blocks, a much more complex behavior can be expected because of the crystallization process, which either disturbs an already organized structure and microphase separation or induces a transition between two different morphologies.11 Generally, there are six parameters that determine the equilibrium structure of a given triblock copolymer. (1) For an ABC triblock copolymer, there are three different Flory–Huggins interaction parameters χAB, χBC and χAC. The relative immiscibility is expressed by the interfacial tension çij, or by the interaction parameter øij, between the directly connected A/B, B/C and the “nonlinked” blocks A/C.7,12 (2) The formation of microphase separated assemblies is also influenced by two independent composition parameters, the volume fractions of each block (fA, fB and fC), and the total length of the chain (N). According to the relative magnitude of χAC compared to χAB and χBC there may be three types of systems; F0, F1 or F2.13 Systems are denoted F0system or no frustration, where the χAC interaction is stronger than χAB or χBC interactions. When the value of χAC is smaller than that of χAB and χBC, such systems are denoted as F2system or type II frustrations, while systems where χAC is intermediate between the other two neighboring blocks (i.e., χAB < χAC < χBC) are denoted F1system or type I frustrations. Systems with no frustration show core–shell structures like core–shell spheres, cylinders, gyroids, and lamellae, and alternating versions of the sphere, cylinder, and gyroid phases, with no A/C interface, for example, poly(butadiene-b-styrene-b-vinylpyridine),14 poly(isoprene-b-styrene-b-vinylpyridine),15 and poly(methyl methacrylate-b-styrene-b-butadiene).16 Systems with F1 display phases include core–shell gyroids, core–shell cylinders, perforated-lamellae, etc. Systems of this class that have been studied include poly(styrene-b-isoprene-b-ethylene oxide),17 poly(styrene-b-butadiene-b-vinylpyridine)18 and poly(styrene-b-isoprene-b-vinylpyridine).19 Systems with type II frustration exhibit morphologies such as spheres on spheres, spheres on cylinders and cylinders in lamellae. This occurs by minimizing the area of A/B and B/C interfaces due to the segregation of B domains into spheres, cylinders, or rings, for the formation of larger areas of A/C interface. Examples include poly(styrene-b-ethylpropylene-b-methyl methacrylate),20 poly(styrene-b-butadiene-b-methyl methacrylate),21 and poly(styrene-b-butadiene-b-caprolactone).22
In recent years, more attention has been focused on blending block copolymers of different compositions or adding a homopolymer to a block copolymer involving secondary interactions, although there are a few reports which have dealt with the influence of association on nanophase separated structures.23 The phase behavior and properties of polymer blends are highly dependent on the extent and strength of intermolecular interactions. In many polymer blends, hydrogen bonding is an important secondary interaction, where the strength of this interaction depends on the relative affinities between hydrogen bond acceptors and donors.24 When a strong intermolecular interaction exists between different polymers and the solvent interacts with the polymers, a miscible polymer blend can be formed. Where the interaction is sufficiently strong, i.e. the polymer–polymer interaction prevails over the polymer–solvent interaction, the two polymers co-precipitate to form highly associated mixtures known as polymer complexes. Very recently, Guo et al.25–28 and Chang et al.29–31 have reported a versatile method to develop self-assembled nanostructured block copolymer blends25,26,29–31 and complexes27,28 through competitive hydrogen bonding interactions. The concept is based on the competition between different blocks of the block copolymer to form more than one kind of intermolecular interaction with the complementary polymer in the complex. This important advance directs a new strategy for the design of self-assembled nanostructures for diverse applications.
In the present study the system of self-assembly has been extended with the introduction of a fourth component, the homopolymer. This study investigates, for the first time, the microphase separation induced by competitive hydrogen bonding in self-assembled semicrystalline triblock copolymer/homopolymer complexes in THF. The self-assembly, hydrogen bonding interactions, phase behavior, crystallization and morphology of polystyrene-block-poly(4-vinylpyridine)-block-poly(ethylene oxide) (SVPEO)/poly(4-vinyl phenol) (PVPh) complexes have been studied. A series of morphologies including hexagonal cylinders, lamellae, disordered and spherical microdomains was documented as a function of the copolymer concentration. The phase behavior of the complexes is correlated with the results obtained from small-angle X-ray scattering (SAXS) and transmission electron microscopy (TEM). This work, for the first time, demonstrates how the competitive hydrogen bonding determines the self-assembly and causes morphological transitions in ABC/D triblock copolymer/homopolymer complexes.
2 Experimental section
2.1 Materials and preparation of blends
Poly(4-vinyl phenol) (PVPh) with an average molecular weight Mw of 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 and a polydispersity Mw/Mn of 1.70 was obtained from Aldrich Chemical Co., Inc. Polystyrene-block-poly(4-vinyl pyridine)-block-poly(ethylene oxide) (SVPEO), the triblock copolymer used in the present study, was purchased from Polymer Source Inc., with Mn(PS) = 60000, Mn(P4VP) = 32000, Mn(PEO) = 39500 and Mw/Mn = 1.2. All these polymers were used as received. The complexes of PVPh/SVPEO were prepared by solution mixing. Tetrahydrofuran (THF) solutions containing 1% (w/v) of the individual polymers were mixed and stirred well until the complexes were precipitated. The solvent was allowed to evaporate slowly at room temperature. The complexes were dried under vacuum for 72 h before the measurements in order to reach equilibrium.
000 and a polydispersity Mw/Mn of 1.70 was obtained from Aldrich Chemical Co., Inc. Polystyrene-block-poly(4-vinyl pyridine)-block-poly(ethylene oxide) (SVPEO), the triblock copolymer used in the present study, was purchased from Polymer Source Inc., with Mn(PS) = 60000, Mn(P4VP) = 32000, Mn(PEO) = 39500 and Mw/Mn = 1.2. All these polymers were used as received. The complexes of PVPh/SVPEO were prepared by solution mixing. Tetrahydrofuran (THF) solutions containing 1% (w/v) of the individual polymers were mixed and stirred well until the complexes were precipitated. The solvent was allowed to evaporate slowly at room temperature. The complexes were dried under vacuum for 72 h before the measurements in order to reach equilibrium.
2.2 Fourier transform infrared (FTIR) spectroscopy
The FTIR spectra of all the samples were recorded on a Bruker Vertex 70 FTIR spectrometer. The spectra of all the samples were determined by using the conventional KBr disk method. The THF sample solution was cast onto KBr pellets and dried under vacuum at 80 °C to completely remove the solvent and then allowed to cool to room temperature. The spectra were recorded with an average of 32 scans in the standard wavenumber range of 400–4000 cm−1 at a resolution of 4 cm−1.2.3 Differential scanning calorimetry (DSC)
The glass transition temperatures of the complexes were determined by a TA Q200 differential scanning calorimeter using 5–10 mg of the sample under a nitrogen atmosphere. A heating rate of 10 °C min−1 was employed. All the samples were first heated to 150 °C and kept at that temperature for 3 min, subsequently cooled to −70 °C at 10 °C min−1, held for 5 min, and heating continued from −70 to 200 °C. The midpoints of the second heating scan of the plot were taken as the glass transition temperatures (Tg).2.4 Small angle X-ray scattering (SAXS)
The SAXS measurements were carried out on a Bruker NanoStar 3 pin-hole small angle X-ray scattering instrument. Two dimensional scattering patterns were recorded using a Vantec detector. Annealed samples having 1 mm thickness were prepared for SAXS measurements and all the experiments were carried out at room temperature (25 °C) using Cu Kα radiation (λ = 1.54 Å, wavelength). The intensity profiles were interpreted as the plot of scattering intensity (I) versus the magnitude of scattering vector, q = (4/λ)sin(θ/2) (θ = scattering angle).2.5 Transmission electron microscopy (TEM)
TEM experiments were performed on a JEOL JEM-2100 transmission electron microscope at an acceleration voltage of 100 kV. The samples were cut into ultrathin sections of approximately 70 nm thickness at room temperature with a diamond knife using a Leica EM UC6 ultra microtome machine. The bulk samples were annealed at 180 °C for about 72 h before microtoming. The thin sections were stained by ruthenium tetroxide before TEM observation.3 Results and discussion
3.1 Hydrogen bonding interactions
FTIR spectroscopy is an excellent tool for providing information on the specific interactions between the components of polymer blends and complexes.32–34Fig. 1 shows the possible hydrogen bonding interactions in the PVPh/SVPEO complexes by detecting hydroxyl, pyridine and ether regions. PVPh has excellent potential as a proton donor for hydrogen bonding interactions with proton-acceptor polymers, because the hydroxyl group of the repeating unit is easily accessible in the 4-position of the aromatic ring.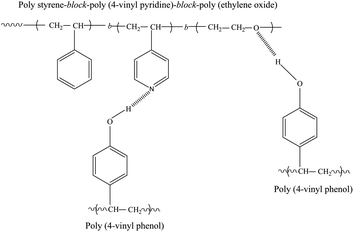 | ||
| Fig. 1 Schematic representation of possible hydrogen bonding interactions between the SVPEO triblock copolymer and PVPh homopolymer. | ||
The hydroxyl stretching region in the infrared spectra of PVPh/SVPEO complexes at room temperature is shown in Fig. 2. It can be seen that the hydroxyl region of pure PVPh consists of two bands; a broad band at 3354 cm−1 which can be attributed to the hydrogen bonded hydroxyl groups (self-associated) and a relatively narrow band at 3525 cm−1 assigned to the free hydroxyl groups (non-associated).35 In the figure the free hydroxyl absorption band can be observed as a shoulder indicating that a relatively small amount of free hydroxyl groups compared with the extensively distributed self-associated hydroxyl groups. Upon the addition of the triblock copolymer, the peak corresponding to the free hydroxyl groups decreases in intensity and eventually disappears, whereas the hydrogen bonded hydroxyl band shifts towards the lower wavenumber region with increase in the SVPEO content. The peak of 20 wt% PVPh at 3159 cm−1 corresponds to the hydrogen bonding interactions of hydroxyl groups of PVPh with P4VP and/or PEO blocks of the block copolymer. The shift in peak position indicates that the inter-associated hydrogen bonds between PVPh and SVPEO are stronger than the self-associated hydrogen bonds in pure PVPh or pure SVPEO (peak at 3400 cm−1). Coleman et al.36 have explained the average strength of the intermolecular interactions (Δν) between the free hydroxyl region and those of the hydrogen bonded species in polymer blends. Table 1 summarizes the Δν values of the complexes and the results imply that the hydrogen bonding strength of the PVPh/SVPEO complexes is intermediate between those values for the PVPh/P4VP and PVPh/PEO binary systems. By analysing Δν values, the average strength of hydrogen bonding can be measured, both the PEO and P4VP blocks can form hydrogen bonds with PVPh, but the interaction between PVPh and P4VP is stronger than the interaction between PVPh and PEO.
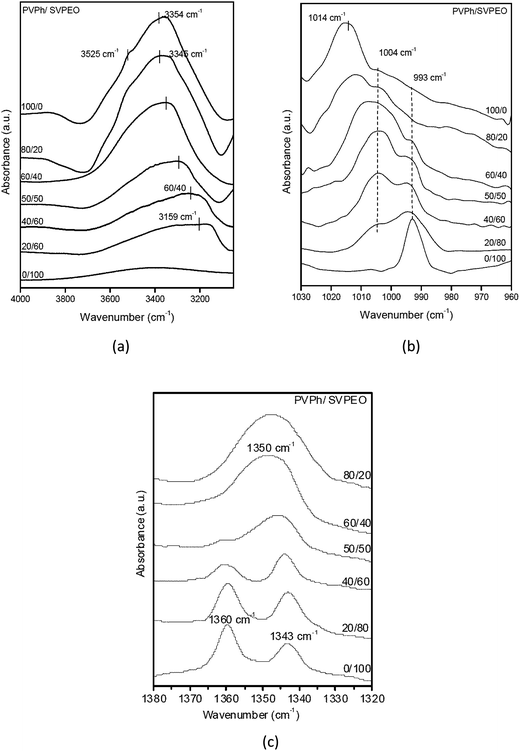 | ||
| Fig. 2 FTIR spectra of PVPh/SVPEO complexes at room temperature: (a) hydroxyl region; (b) pyridine region; (c) ether region. | ||
The hydrogen bonding interactions between PVPh and P4VP can be identified by examining the pyridine region in the spectra of the complexes. The characteristic bands of the pyridine ring at 1590, 1050, 993, and 625 cm−1 are sensitive to hydrogen-bonding interactions.38,39 However, the P4VP bands at 1590 cm−1 are difficult to analyse due to overlapping with the absorption band of PVPh in the 1600 cm−1 region. Therefore, only the band at 993 cm−1 can be used to identify the existence of hydrogen bonding interactions between the hydroxyl group of PVPh and the pyridine group of P4VP. FTIR spectra in the range of 1030–960 cm−1 of PVPh/SVPEO complexes with different compositions are plotted in Fig. 2b. The bands at 993 and 1014 cm−1 correspond to the aryl CH bending of the pure pyridine ring and PVPh phenol ring, respectively. Another absorption band observed in the complexes at 1004 cm−1 is attributed to the hydrogen-bonding interaction between the pyridine ring of P4VP and the phenol group of PVPh. The spectral changes in both wavenumber regions suggest that strong hydrogen bonding interactions exist between pendant pyridine groups of P4VP and the phenol group of PVPh in all complex compositions. This interaction is very significant in the formation of a stable complex. Moreover, this hydrogen bonding can contribute much to the positive deviation of Tg observed in the Tg–composition plot of the complex which is described in the later part of this article.
Fig. 2c represents the FTIR spectra of the CH2 wagging region of PEO in SVEPO and its complexes with PVPh ranging from 1380 to 1320 cm−1. The pure PEO has two bands at 1360 and 1343 cm−1 corresponding to the crystalline phase of PEO.40 When the PVPh content in the complexes increases, these two bands are replaced by a broad band centered at 1350 cm−1 which represents the amorphous phase, suggesting a retardation of PEO crystallization by the addition of PVPh. From Fig. 2c it can be seen that the retardation of PEO crystallization peaks begins in complexes with 40 wt% of PVPh, which means that a considerably strong interaction between PVPh and PEO starts when the PVPh concentration is ∼40 wt%. Therefore it can be assumed that PEO ether groups have only weak interaction with PVPh hydroxyl groups at concentrations below 40 wt% of PVPh. This is due to the strong hydrogen bonding interaction between pyridine groups of P4VP with all the available hydroxyl groups of PVPh at low PVPh concentrations.
3.2 Phase behavior of complexes
The thermal properties of PVPh/SVPEO complexes were investigated by DSC, and the results are presented in Fig. 3 and Table 2. It can be seen from Fig. 3a that the plain shows a Tg at 165 °C whereas SVPEO exhibits two distinct Tgs at 107 °C and 150 °C corresponding to immiscible PS and P4VP blocks, respectively. The Tg of the PEO block could not be observed under the current experimental conditions. The melting temperature (Tm) of the PEO block can be observed at 52 °C. There is little change in the Tg of PS blocks again indicating that there are no interactions between PS and PVPh throughout all of the compositions.41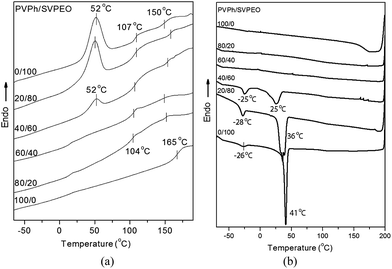 | ||
| Fig. 3 DSC thermograms of PVPh/SVPEO complexes: (a) second scan at a heating rate of 10 °C min−1; (b) cooling scan at a rate of −10 °C min−1. | ||
| PVPh/SVPEO | T m (°C) | T g (PVPh) (°C) | T g (PS) (°C) | T f (°C) | |
|---|---|---|---|---|---|
| 0/100 | 52 | 150 | 107 | −26 | 41 |
| 20/80 | 50 | 158 | 107 | −28 | 36 |
| 40/60 | 52 | 154 | 106 | −25 | 25 |
| 60/40 | 150 | 105 | |||
| 80/20 | 152 | 104 | |||
| 100/0 | 165 | ||||
It has been proven that the formation of binary complexes of PVPh/P4VP and PVPh/PEO is due to the intermolecular hydrogen bonding interactions.42 In PVPh/SVPEO complexes, a single Tg corresponding to the PVPh and P4VP components was detected, which is attributable to the miscibility between the components.43 As shown in Fig. 3a, the Tg of the P4VP/PVPh phase is substantially higher than the Tg of P4VP at lower PVPh contents (20 wt% PVPh complexes). This positive deviation can be attributed to the strong intermolecular interaction between P4VP and PVPh, which contributes significantly to the free energy of mixing and hence to the miscibility of polymer blocks. Above 40 wt% of PVPh, there is a reduction in the Tg value of the complexes due to the miscibility of PEO blocks with PVPh at these compositions.
The crystallizable PEO component in the block copolymer SVPEO exhibits a melting temperature at 52 °C. It can be clearly seen that the Tm of PEO blocks in PVPh/SVPEO complexes remains almost unchanged at very low PVPh concentrations. The melting peak corresponding to the crystalline phases reduces in its intensity and eventually disappears in 40–50 wt% PVPh complexes, which is due to the miscibility of PEO with PVPh at higher PVPh contents.
Fig. 3b shows the crystallization curves during the cooling scan of PVPh/SVPEO complexes obtained at a cooling rate of −10 °C min−1. The appearance of two separate crystallization peaks of pure SVPEO and PVPh/SVPEO complexes indicates fractionated crystallization with increase of the PVPh content. The existence of more than one crystallization exotherm is termed as fractionated crystallization.44 This behavior has been reported for other semicrystalline block copolymers.45–47 Fractionated crystallization of a pure block copolymer takes place either due to morphological heterogeneity, i.e. heterogeneous nucleation and homogeneous nucleation or a slow crystallization rate due to a structural factor. Usually, homogeneous nucleation occurs in confined or unconnected crystalline domains and that preserves the spherulite morphology. However in connected domains, heterogeneous nucleation takes place leading to rupture of the morphology to form mixed morphologies. In the case of block copolymers with slow cooling rate, partial crystallization due to heterogeneous nucleation occurs in the higher temperature region and crystallization initiated by homogeneous nucleation also takes place in the lower temperature region. The peak of the crystallization exotherm is termed as freezing temperature (Tf). In the pure SVPEO block copolymer, at low cooling rates, a large part of the PEO block crystallizes at 41 °C whereas a minor fraction of the PEO can only crystallize at lower temperatures (−26 °C). In such cases both homogeneous nucleation and heterogeneous nucleation result in a lamellar morphology after crystallization, where the PEO is dispersed into droplets in an immiscible matrix,48 but no mixed morphologies are observed in the small angle X-ray scattering profile either.49 The exotherm at −26 °C can be explained as the crystallization of the PEO block originated from the homogeneous nucleation.
The fractionated crystallization behavior was also observed in PVPh/SVPEO complexes up to 40 wt% of PVPh. From FTIR analyses it was confirmed that PVPh interacts weakly with PEO compared to the strong interaction between PVPh and P4VP. Therefore the appearance of two exotherms can be explained by the two different crystallization mechanisms of PEO domains within the PVPh/P4VP mixed phase. The high temperature exotherm is from the first crystallization process due to heterogeneous nucleation of the continuous domains and the low temperature exotherm is produced by the homogeneous nucleation (non-connecting) PEO domains in the PVPh/P4VP mixed phase.
3.3 Nanostructured morphology of PVPh/SVPEO complexes
TEM examination provided the insight into the morphology of PVPh/SVPEO complexes. Based on the electron density of various groups, PS, P4VP, PVPh and PEO appear as deep, intermediate, light, and very light contrasts when stained with RuO4. The morphological transformations of PVPh/SVPEO complexes with 20 to 80 wt% of PVPh compositions are given in Fig. 4. It is seen that all the complexes exhibit heterogeneous morphology at the nanoscale level. | ||
| Fig. 4 TEM micrographs of PVPh/SVPEO complexes. (a) 0/100, (b) 20/80, (c) 40/60, (d) 50/50, (e) 60/40, and (f) 80/20 PVPh/SVPEO. | ||
The pure block copolymer shows a cylindrical morphology as shown in Fig. 4a. In fact, a pseudo “hexagonally packed cylinder” was observed for the SVPEO block copolymer, where some percolated microdomains coexist with this cylindrical structure in some areas (inset of Fig. 4a). The SAXS experiments also prove the existence of cylindrical morphology in the SVPEO block copolymer (Fig. 5), though a lateral view of these cylinders was not observed by TEM. A similar cylindrical morphology has been observed for the PS-b-poly(2-vinylpyridine)-b-poly(tert-butyl methacrylate) triblock copolymer as reported by Liedel et al.50
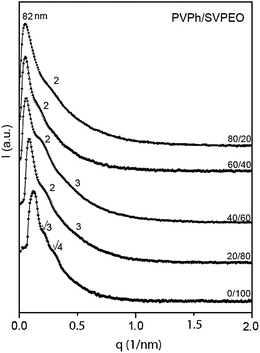 | ||
| Fig. 5 SAXS profiles of PVPh/SVPEO complexes. | ||
The 20 wt% PVPh complexes exhibit a twisted lamellar structure as shown in Fig. 4b. Here, the very dark region corresponds to PS and a mixed phase of PVPh and P4VP appears as grey and the PEO blocks appear as bright.51 At this composition, the concentration of PVPh is low compared to the block copolymer. Hence, the added PVPh strongly hydrogen bonded to P4VP and forms a single phase whereas the less-interacting PEO block phase separates within the matrix as spherical or elongated microdomains (Fig. 4b). At 40 wt% PVPh complexes, PEO also forms hydrogen bonds with PVPh since a higher number of hydroxyl units are available even after strong interaction with P4VP. This induces a bicontinuous structure for 40 wt% PVPh complexes as shown in Fig. 4c. This competitive hydrogen bonding destroys the ordered structure of the system and leads to the decrease in the interfacial area, which results in the planar interfaces and thereby the formation of a disordered bicontinuous phase. On further increasing the PVPh content to 50 wt%, the complexes adopt a highly disordered morphology with some wormlike structures dispersed in the matrix as shown in Fig. 4d. The complexes with 60 wt% PVPh show disordered morphologies in which PS domains are dispersed in the continuous PVPh/P4VP and PVPh/PEO matrix (Fig. 5e). As the content of PVPh increases to 80 wt%, the complexes show a completely homogenous phase of PVPh/P4VP and PVPh/PEO with phase separated spherical PS domains (Fig. 4f). Previously, Lee et al.52 have investigated the hydrogen bonding interactions and morphology in the blends of the P2VP-b-PEO block copolymer with the PVPh homopolymer. No self-assembly was observed and all the blends were homogenous at any compositions though the interactions between PVPh/P2VP and PVPh/PEO were different. The complete miscibility observed in this system was obviously due to the very low molecular weight of the blocks compared to the homopolymer. If the molecular weights of the homopolymer and each block were comparable or higher, self-assembled structures could have been formed through competitive hydrogen bonding interactions.
The SAXS profiles of PVPh/SVPEO complexes are shown in Fig. 5. Well defined scattering peaks can be observed for the plain SVPEO triblock copolymer. The first scattering peak is centered at the scattering vector q* for a long spacing of 35 nm. The peak positions in the SAXS profile indicate a hexagonal close-packed cylindrical lattice, situated at q:q* values of 1:√3:√4:√7… (where q* denotes the position of the first-order scattering maximum).53 The complexes give broad peaks, and the broadening of the peak indicates the reduction of the long-range order. However, the complexes with 10–50 wt% of PVPh show multiple scattering peaks, implying that they possess longer-range ordered nanostructures to some extent. The SAXS profile of the 20 wt% PVPh complex situated at q/q* values of 1:2:3, displaying characteristics of the lamellar structure. The complexes with 40 wt% PVPh give broad peaks, indicating the deterioration of long-range ordered structures. At 40 wt % PVPh, complexes only show small shoulders around 2 and 3, respectively, owing to the disordering of the lamellae present in the complexes and the average spacing between the neighboring microdomains is 51 nm. Complexes with high content of PVPh exhibit more disordered structures, which are revealed by the disappearance of higher order reflections in the SAXS profiles. The results show that there is a systematic increase in the size of the phase separated domain with the progressive incorporation of PVPh. Above 40 wt% PVPh, the complexes show only weak and broad peaks, displaying a disordered morphology as observed in 60 and 80 wt% PVPh complexes in Fig. 4e and f.
The formation mechanism of different self-assembled nanostructures in PVPh/SVPEO complexes at different compositions is schematically shown in Fig. 6. The complexes comprise an immiscible SVPEO triblock copolymer and a homopolymer PVPh, which is miscible with both P4VP and PEO blocks depending on the concentration. The pure triblock copolymer exhibited a cylindrical morphology as shown in Fig. 6a. Since the blocks in the triblock copolymers have the general tendency to separate, they exhibit amphiphilic characteristics which are caused by the restriction due to the presence of a covalent bond between the chemically different blocks, resulting in microphase separated structures. When a homopolymer is complexed with a triblock copolymer, involving competitive hydrogen bonding interactions, the weakly hydrogen bonded block is excluded from the homogenous region due to the high entropic penalty for conformational distortion. Here, by addition of more homopolymer, microphase separation takes place due to the self-assembly of the elementary block copolymer, i.e. it selectively swells the blocks due to the competitive hydrogen bonding which results in phase separation.
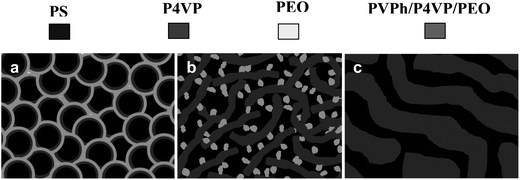 | ||
| Fig. 6 Schematic representation of phase morphologies in PVPh/SVPEO complexes: (a) cylindrical morphology of the SVPEO triblock copolymer, (b) twisted lamellae at 20 wt% PVPh concentration, and (c) bicontinuous phase at 40 wt% PVPh concentration. | ||
In 20 wt% of PVPh complexes, twisted lamellae with an average diameter of 40–50 nm were obtained which is schematically shown in Fig. 6b. At 20 wt%, the added PVPh and P4VP interact very strongly whereas PEO blocks, which are repelled by P4VP, have relatively weak hydrogen bonding with PVPh. In other words, PVPh acts as a selective amphiphilic solvent for the P4VP blocks of the SVPEO triblock copolymer. Therefore the added PVPh forms PVPh/P4VP single phase layers whereas the weakly interacting PEO phase separates as spherical or elongated microdomains. For the pure block copolymer, which is originally in the cylindrical phase, the addition of PVPh is thus expected to induce structural transformations, in analogy with block copolymer selective solvent systems. As the concentration of PVPh increases again, the microphase morphology varies, displaying a bicontinuous structure in 40 wt% PVPh complexes (Fig. 6c), whereas a matrix-dispersed wormlike morphology is obtained in 50 wt% of PVPh (Fig. 5d). As the concentration reaches 60–80 wt% PVPh, the interface between the PVPh/P4VP and PVPh/PEO microphases becomes less distinct. The interaction of PVPh between P4VP and PEO together with non-interacting PS leads to the formation of nanostructured spherical microdomains. The appearance of spherical morphology at high PVPh concentrations is due to the confinement of non-interacting PS blocks within the highly hydrogen bonded PVPh/P4VP and PVPh/PEO phases form the homogenous matrix. This is due to the hydrogen bonding interactions of PVPh with PEO along with P4VP due to the availability of free hydroxyl groups or in other words PVPh behaves as a common solvent for both P4VP and PEO blocks. The different nanostructures observed in these complexes are completely different from the cylindrical microphase of the pure SVPEO triblock copolymer, implying that the block copolymer blocks are actually swollen when PVPh is added. This is an ABC/D triblock copolymer/homopolymer system, where the triblock copolymer ABC is immiscible and the homopolymer D can interact with both B and C blocks, but unequally due to the competitive hydrogen bonding interaction between the B/D and C/D pairs, while the A block has no interactions with D and is entirely phase separated in all compositions. Here χAB and χBC are positive, but the values of χBD and χCD are negative, however the value of χBD is more negative than χCD. There is unequal hydrogen bonding interactions of PVPh with both P4VP and PEO, whereas the unreacted PS phase separated which altogether leads to form various nanostructures in PVPh/SVPEO complexes. The coexistence of three kinds of hydrogen bonds, i.e., inter-associated PVPh/P4VP, PVPh/PEO pairs and self-associated PVPh/PVPh, will bring about the competition of the PVPh distribution among P4VP and PEO microdomains. Moreover, the self-association of PVPh hydroxyl interactions tunes the possibility of adjusting the self-organized structures at the nanoscale through ordered–disordered transitions. The presence of hydrogen bonding interactions in the complexes enhances the miscibility of the blocks and facilitates the phase separation which in turn affects the properties of the complexes. The morphological variations of this system are shown to be influenced by the following factors: (1) intermolecular interaction between PVPh and P4VP is stronger than that between PVPh and PEO which indicates the existence of competitive hydrogen bonding, (2) strong interaction of PVPh/P4VP excludes microdomains of PEO at lower PVPh content, (3) formation of a homogenous phase of PVPh/P4VP and PVPh/PEO excludes microdomains of non-interacted PS at high PVPh content. So the geometry of the structures formed in the complexes is determined to a large extent by the competition between P4VP and PEO blocks in regards to hydrogen bonding with PVPh. Moreover it is also established that the addition of a homopolymer into to an ordered block copolymer will cause changes in the microdomain structure.
4 Conclusions
The microphase separation, mediated by competitive hydrogen bonding, in SVPEO triblock copolymer/PVPh homopolymer complexes was studied. The hydroxyl groups of PVPh can selectively interact with both the pyridine group of the P4VP block and ether groups of the PEO block, which leads to the formation of composition-dependent microphase separated morphologies in these blends. The disparity of weakly associated PVPh/PEO pairs and strongly associated PVPh/P4VP pairs results in compositionally dependent microphase separation and the formation of cylindrical, twisted lamellar, disordered bicontinuous and wormlike morphologies at lower PVPh concentrations. At higher concentrations, PVPh can act as a common solvent for both of the blocks which results in a homogeneous phase with PS as the only phase separated domain. The formation of various composition-dependent microphase separated morphologies in the PS–P4VP–PEO/PVPh complexes was explained based on the relative strength of hydrogen bonding between the different pairs in the system.Acknowledgements
The authors would like to thank AINSE Ltd for providing financial assistance to conduct SAXS experiments (ALNGRA 11157). This work benefitted from SasView software, originally developed by the DANSE project under NSF award DMR-0520547.References
- M. Muthukumar, C. K. Ober and E. L. Thomas, Science, 1997, 277, 1225 CrossRef CAS.
- L. A. Utracki, Polymer Alloys and Blends, Hanser Publishers, Munich, 1989 Search PubMed.
- D. R. Paul, in Polymers Blends, ed. D. R. Paul and S. Newman, Academic Press, New York, 1978, vol. 2, p. 35 Search PubMed.
- K. Kataoka, A. Harada and Y. Nagasaki, Adv. Drug Delivery Rev., 2001, 47, 113 CrossRef CAS.
- A. Rosler, G. W. M. Vandermeulen and H. A. Klok, Adv. Drug Delivery Rev., 2001, 53, 95 CrossRef CAS.
- I. W. Hamley, The Physics of Block Copolymers, Oxford University Press, Oxford, UK, 1998 Search PubMed.
- L. Liebler, Macromolecules, 1980, 13, 1602 CrossRef.
- B. Loewenhaupt, A. Steurer, G. P. Hellmann and Y. Gallot, Macromolecules, 1994, 27, 908 CrossRef CAS.
- F. S. Bates and G. H. Fredrickson, Phys. Today, 1999, 52, 32–38 CrossRef CAS.
- N. Hadjichristidis, H. Iatrou, M. Pitsikalis, S. Pispas and A. Avgeropoulos, Prog. Polym. Sci., 2005, 30, 725–782 CrossRef CAS.
- I. W. Hamley, Adv. Polym. Sci., 1999, 148, 113–137 CrossRef CAS.
- E. Helfand, Macromolecules, 1975, 8, 552–556 CrossRef.
- T. S. Baily, Morphological behavior spanning the symmetric AB and ABC block copolymer states, PhD thesis, University of Minnesota, 2001.
- H. Hueckstadt, A. Gopfert and V. Abetz, Polymer, 2000, 41, 2121 CrossRef.
- Y. Mogi, K. Mori, H. Kotsuji, Y. Matsushita, I. Noda and C. C. Han, Macromolecules, 1993, 26, 5169 CrossRef CAS.
- V. Abetz and T. Goldacker, Macromol. Rapid Commun., 2000, 21, 16 CrossRef CAS.
- C. A. Tyler, J. Qin, F. S. Bates and D. C. Morse, Macromolecules, 2007, 40, 4654 CrossRef CAS.
- T. H. Epps, E. W. Cochran, C. M. Hardy, T. S. Bailey, R. S. Waletzko and F. S. Bates, Macromolecules, 2004, 36, 2873 CrossRef.
- Y. Matsushita, M. Tamura and I. Noda, Macromolecules, 1992, 27, 3680 CrossRef.
- C. Auschra and R. Stadler, Macromolecules, 1993, 26, 2171 CrossRef CAS.
- S. Brinkmann, R. Stadler and E. L. Thomas, Macromolecules, 1995, 28, 6566 Search PubMed.
- V. Balsamo, F. V. Glydenfeldt and R. Stadler, Macromolecules, 1999, 32, 1226 CrossRef CAS.
- (a) S. Viswanathan and M. D. Dadmun, Macromolecules, 2002, 35, 5049 CrossRef CAS; (b) M. Jiang and H. Xie, Prog. Polym. Sci., 1991, 16, 977 CrossRef CAS.
- M. M. Coleman and P. C. Painter, Prog. Polym. Sci., 1995, 20, 1 CrossRef CAS.
- N. V. Salim, T. L. Hanley and Q. Guo, Macromolecules, 2010, 43, 7695 CrossRef CAS.
- N. V. Salim, N. Hameed and Q. Guo, J. Polym. Sci., Part B: Polym. Phys., 2009, 47, 1894 CrossRef CAS.
- N. Hameed and Q. Guo, Polymer, 2008, 49, 922 CrossRef CAS.
- N. Hameed, N. V. Salim and Q. Guo, J. Chem. Phys., 2009, 131, 214905 CrossRef.
- J. G. Li, Y. D. Lin and S. W. Kuo, Macromolecules, 2011, 44, 9295 CrossRef CAS.
- W. C. Chen, S. W. Kuo, U. S. Jeng and F. C. Chang, Macromolecules, 2008, 41, 1401 CrossRef CAS.
- W. C. Chen, S. W. Kuo, U. S. Jeng, C. J. Su and F. C. Chang, Macromolecules, 2010, 43, 1083 CrossRef.
- M. M. Coleman, X. Yang, P. C. Painter and J. F. Graf, Macromolecules, 1992, 25, 4414 CrossRef CAS.
- (a) H. Zheng, S. Zheng and Q. Guo, J. Polym. Sci., Part A: Polym. Chem., 1997, 35, 3169 CrossRef CAS; (b) Z. Zhong and Q. Guo, Polymer, 1998, 39, 517 CrossRef CAS; (c) Q. Guo and H. Zheng, Polymer, 1999, 40, 637 CrossRef CAS.
- (a) J. Huang, X. Li and Q. Guo, Eur. Polym. J., 1997, 33, 659 CrossRef CAS; (b) Z. Zhong and Q. Guo, J. Polym. Sci., Part A: Polym. Chem., 1998, 36, 401 CrossRef CAS.
- M. M. Coleman and P. C. Painter, Prog. Polym. Sci., 1995, 20, 1 CrossRef CAS.
- E. J. Moskala, D. F. Varnell and M. M. Coleman, Polymer, 1985, 26, 228 CrossRef CAS.
- W. C. Chen, S. W. Kuo, C. H. Lu, U. S. Jeng and F. C. Chang, Macromolecules, 2009, 42, 3580 CrossRef CAS.
- J. Dai, S. H. Gosh, S. Y. Lee and K. S. Siow, Polym. J., 1994, 26, 905 CrossRef CAS.
- J. Wang, M. K. Cheung and Y. Mi, Polymer, 2001, 42, 3087 CrossRef CAS.
- S. Chintapalli and R. Frech, Macromolecules, 1996, 29, 3499 CrossRef CAS.
- (a) Z. Zhong, S. Zheng, J. Huang, X. Cheng, Q. Guo and J. Wei, Polymer, 1998, 39, 1075 CrossRef CAS; (b) Q. Guo, Polymer, 1995, 36, 4753 CrossRef CAS; (c) Q. Guo, A. Habrard, Y. Park, P. J. Halley and G. P. Simon, J. Polym. Sci., Part B: Polym. Phys., 2006, 44, 889 CrossRef CAS; (d) Q. Guo, Polymer, 1993, 34, 70–76 CrossRef CAS.
- (a) S. W. Kuo, F. C. Chang and P. H. Tung, Macromolecules, 2006, 39, 9388 CrossRef CAS; (b) S. W. Kuo and F. C. Chang, Macromol. Chem. Phys., 2001, 202, 3112 CrossRef CAS.
- (a) Q. Guo, Makromol. Chem., Rapid Commun., 1990, 11, 279 CrossRef CAS; (b) Q. Guo, J. Huang, T. Chen, H. Zhang, Y. Yang, C. Hou and Z. Feng, Polym. Eng. Sci., 1990, 30, 44 CrossRef CAS; (c) S. Zheng, Q. Guo and Y. Mi, J. Polym. Sci., Part B: Polym. Phys., 1998, 36, 2291 CrossRef CAS; (d) H. Zheng, S. Zheng and Q. Guo, J. Polym. Sci., Part A: Polym. Chem., 1997, 35, 3161–3168 CrossRef CAS.
- (a) R. L. Cormia, F. P. Price and D. Turnbull, J. Chem. Phys., 1962, 37, 1333 CrossRef CAS; (b) Q. Guo, Thermochim. Acta, 2006, 451, 168 CrossRef CAS; (c) Q. Guo, R. Thomann and W. Gronski, Polymer, 2007, 48, 3925 CrossRef CAS.
- Y. L. Loo, R. A. Register, A. J. Ryan and G. T. Dee, Macromolecules, 2001, 34, 8968 CrossRef CAS.
- V. Balsamo, A. J. Muller, F. von Gyldenfeldt and R. Stadler, Macromol. Chem. Phys., 1998, 199, 1063 CAS.
- A. J. Muller, V. Balsamo, M. L. Arnal, T. Jakob, H. Schmalz and V. Abetz, Macromolecules, 2002, 35, 3048 CrossRef.
- (a) H. Frensch, P. Harnischfeger and B.-J. Jungnickel, Fractionated Crystallization in Incompatible Polymer Blends. In Multiphase Polymers: Blends and Ionomers, ed. L. A. Utracky and R. A. Weiss, ACS Symp. Ser., 1989, vol. 395, p. 101 Search PubMed.
- J. T. Xu, L. Xue, S. M. Mai and A. J. Ryan, J. Appl. Polym. Sci., 2004, 93, 870 CrossRef CAS.
- C. Liedel, M. Hund, V. Olszowka and A. Böker, Soft Matter, 2012, 8, 995 RSC.
- J. R. Kim, A. M. Jamieson, S. D. Hudson, I. Manas-Zloczower and H. Ishida, Macromolecules, 1998, 31, 5383 CrossRef CAS.
- L. T. Lee, E. M. Woo, S. S. Hou and S. Förster, Polymer, 2006, 47, 8350 CrossRef CAS.
- H. Hasegawa, H. Tanaka, K. Yamasaki and T. Hashimoto, Macromolecules, 1987, 20, 1651 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2013 |