Synthesis of new enzymatically degradable thermo-responsive nanogels
Garbiñe
Aguirre
,
Jose
Ramos
and
Jacqueline
Forcada
*
POLYMAT, Bionanoparticles Group, Departamento de Química Aplicada, UFI 11/56, Facultad de Ciencias Químicas, Universidad del País Vasco UPV/EHU, Apdo. 1072, 20080 Donostia-San Sebastián, Spain. E-mail: jacqueline.forcada@ehu.es; Fax: +34-943-015-270; Tel: +34-943-018-182
First published on 17th October 2012
Abstract
Two new families of thermo-responsive and enzymatically degradable nanogels were synthesized by batch emulsion polymerization of N-vinylcaprolactam (VCL) with dextran methacrylates (Dex-MA) with different degrees of substitution (DS). The first family was prepared using different amounts of Dex-MA with high DS forming highly cross-linked nanogel particles with the typical thermal behavior of PVCL-based nanogels: below the volume phase transition temperature (VPTT) nanogel particles were swollen and above it they were collapsed. After their enzymatic degradation with dextranase, nanogel particles swelled due to the cleavage of some glucopyranosyl bonds of dextran, but preserved their identity. On the other hand, the second family was prepared using different amounts of Dex-MA with low DS forming slightly cross-linked nanogel particles with an anomalous thermal behavior. Surprisingly, above the VPTT of the nanogel particles monodisperse interparticle reversible aggregates were formed. In addition, after their enzymatic degradation, a release of reducing sugars together with an intense de-swelling due to the fragmentation of the nanogel structure was observed. Both nanogel families could be suitable for drug delivery in tissues or organs where dextranase is present.
1. Introduction
In recent years, micro and nanogels have received considerable attention because of their unique capability to swell rapidly in a thermodynamically good solvent.1,2 They are cross-linked polymer particles in the colloidal range being able to vary their volume in response to external stimuli such as temperature, pH, pressure, ionic strength, solvent composition, electric or magnetic field, and light intensity, among others.3–5 This stimuli-responsive behavior gives rise to a wide range of pharmaceutical, therapeutical, and biomedical applications.In this sense, special attention is paid to temperature-sensitive nanogels based on polymers having a lower critical solution temperature (LCST) near the physiological one, such as poly(N-isopropylacrylamide) (PNIPAM) and poly(N-vinylcaprolactam) (PVCL). In the case of PNIPAM- or PVCL-based nanogels, the phase transition of these nanoparticles is observed as a volume phase transition temperature (VPTT), since together with the increase in the hydrophobicity of the polymer chains, the dissociated water molecules are expelled from the nanogel interior, thus causing a decrease in volume occurring above the LCST of the polymer. Although PNIPAM is the building block of a plethora of temperature-sensitive nanogels,6–8 its use as a biomaterial may be limited because of its higher cytotoxicity and its lower cell viability with respect to PVCL.9 Thus, nowadays, PVCL, which is a biocompatible polymer,10 is being extensively studied as a better alternative in the design of temperature-sensitive nanogels for in vitro and in vivo applications.10–12
It is well known that the swelling of nanogels is mainly controlled by the type and amount of the cross-linker used. Until now, all the cross-linkers used during the synthesis of PVCL-based nanogels are bifunctional, N,N′-methylenebisacrylamide (MBA) being the most common one.13–17 However, only in few works, PVCL-based nanogels were prepared using bifunctional cross-linkers with a higher molecular weight such as poly(ethylene glycol) diacrylate 200 (PEGDA), 200 being the molecular weight of the PEG chain.18,19 The use of PEGDA instead of MBA as the cross-linker increased the hydrophilicity of the nanogels, altered their swelling behavior and decreased their final size. On the other hand, when PVCL-based nanogels are to be used as drug delivery nanocarriers, biodegradability of the network is desired. In this scenario, the use of multifunctional degradable cross-linkers with high molecular weights would be beneficial. The nanogel network of these new types of thermo-responsive PVCL-based nanogels would be able to disintegrate under controlled conditions releasing entrapped compounds.
A challenging approach to prepare degradable hydrogels is based on the use of dextran as a naturally occurring polysaccharide whose backbone can be cleaved upon incubation with dextranase. In the first step, the covalent functionalization of dextran chains with polymerizable vinyl groups was carried out via their multiple hydroxyl groups. Thus, it was possible to synthesize dextran chains with different degrees of substitution (DS) which are able to suffer different degradation mechanisms (hydrolytic or enzymatic).20–24 In the second step, hydrogels were obtained by a free radical polymerization in aqueous solution.25,26 However, the synthesis of dextran-based nanogels requires more complex polymerization processes. In the first attempt, lipid coated dextran nanogels were obtained by polymerization of an aqueous dextran hydroxyethyl methacrylate (Dex-HEMA) solution entrapped in liposomes. Naked Dex-HEMA nanogels were prepared by removing the lipid coating by Triton X100.27 More recent works highlight the inverse miniemulsion polymerization as a very promising process to obtain biodegradable nanogels.28–30 On the one hand, Raemdonck et al.28 synthesized biodegradable cationic dextran nanogels by inverse miniemulsion photopolymerization of dextran methacrylate (Dex-MA) or Dex-HEMA with [2-(methacryloyloxy)ethyl]trimethylammonium chloride (TMAEMA). On the other hand, Klinger et al.29,30 prepared enzymatically cleavable nanogels by free radical inverse miniemulsion copolymerization of acrylamide with Dex-MA. As a drawback, nanogels obtained by this polymerization process required an exhaustive purification to transfer them to the aqueous phase.
Related to the synthesis of dextran-based thermo-responsive nanogels, only two works have been published.31,32 In both of them, nanogels were prepared by emulsion polymerization using NIPAM as a temperature-sensitive monomer and maleilated dextran or Dex-lactateHEMA as cross-linkers.
In this work, taking advantage of the biocompatibility and thermo-sensitivity of PVCL and biodegradability of dextran, biocompatible, thermo-sensitive, and degradable nanogels based on PVCL and Dex-MA are reported for the first time. The syntheses were carried out via free radical emulsion polymerization of VCL with different types and amounts of Dex-MA as macro-cross-linkers in a batch reactor. Colloidal characteristics, such as the evolution of the average hydrodynamic particle diameter and swelling ratio at different temperatures, were measured by Photon Correlation Spectroscopy (PCS). In order to understand the internal morphology of nanogels, high-resolution 1H transverse relaxation NMR measurements were carried out and analyzed. In addition, to assess the biodegradability, dextranase enzyme was encapsulated into nanogels and the enzymatic degradation of the system was analyzed.
2. Experimental section
2.1. Materials
N-Vinylcaprolactam (VCL, Sigma-Aldrich), N,N′-methylenebisacrylamide (MBA, Sigma-Aldrich), dextran T40 (Mr ≈ 40![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000) (Sigma-Aldrich), glycidyl methacrylate (GMA, Sigma-Aldrich), 4-(N,N′-dimethylamino)pyridine (DMAP, Sigma-Aldrich), hydrochloric acid (HCl, Fluka), dimethyl sulfoxide (DMSO, Scharlab), 2,2′-azobis (N,N′-dimethyleneisobutyramidine) dihydrochloride (ADIBA, Wako Chemical Gmbh), hexadecyltrimethylammonium bromide (HDTAB, Sigma-Aldrich), dextranase D-5884 from Penicillium (EC 3.2.1.11, specific activity 400–800 U mg−1 protein) (Sigma-Aldrich), and glucose (Sigma-Aldrich) were used as supplied. Double deionized (DDI) water was used throughout the work.
000) (Sigma-Aldrich), glycidyl methacrylate (GMA, Sigma-Aldrich), 4-(N,N′-dimethylamino)pyridine (DMAP, Sigma-Aldrich), hydrochloric acid (HCl, Fluka), dimethyl sulfoxide (DMSO, Scharlab), 2,2′-azobis (N,N′-dimethyleneisobutyramidine) dihydrochloride (ADIBA, Wako Chemical Gmbh), hexadecyltrimethylammonium bromide (HDTAB, Sigma-Aldrich), dextranase D-5884 from Penicillium (EC 3.2.1.11, specific activity 400–800 U mg−1 protein) (Sigma-Aldrich), and glucose (Sigma-Aldrich) were used as supplied. Double deionized (DDI) water was used throughout the work.
2.2. Synthesis and characterization of macro-cross-linkers
Different dextran methacrylates (Dex-MA) were prepared and characterized according to Van Dijk-Wolthuis et al.20,21 In brief, 25 g of dextran with an average molecular weight (Mw) of 40000 g mol−1 (Dex40) were dissolved in 245 mL of DMSO under nitrogen atmosphere. After dissolution of 4.95 g of DMAP, a variable amount (1.3 and 11.3 g) of GMA was added. The solution was stirred at 30 °C for 48 h, after which adding an equimolar amount of concentrated HCl to neutralize DMAP stopped the reaction. The reaction mixture was transferred to dialysis tubes (Mw cut-off of 12000–14000 g mol−1) and extensively dialyzed for 2 weeks against DDI water at 4 °C. Then, Dex-MA was lyophilized and stored at 20 °C before use. The degree of substitution (DS, i.e., the number of methacrylate groups per dextran chain taking into account that each chain is composed of 225 glucopyranosyl groups) was determined by proton nuclear magnetic resonance spectroscopy (1H NMR) in D2O with a Bruker AVANCE spectrometer. In this work, two macro-cross-linkers were prepared with a DS of 9 and 86, respectively (i.e., Dex40MA9 and Dex40MA86).
2.3. Synthesis of nanogels
Biodegradable PVCL-based nanogels were synthesized by emulsion polymerization in a batch reactor. 2 g of VCL as the main monomer, 0.04 g of HDTAB as the cationic emulsifier and a variable amount (from 0.08 to 1 g) of Dex-MA as the macro-cross-linker were dissolved in 185 g of DDI water and placed into a 250 mL jacketed glass reactor, fitted with a reflux condenser, stainless steel stirrer, sample device, and nitrogen inlet tube reactor. The reaction mixture was heated at 70 °C, stirred at 300 rpm and purged with nitrogen for 1 h before starting the polymerization reaction. After adding the cationic initiator (0.02 g of ADIBA) in 15 g of DDI, the polymerization reaction was allowed to continue with stirring for 5 h under nitrogen atmosphere at 70 °C. The reaction mixture was subsequently cooled to 25 °C and the final dispersion was filtered through glass wool. Prior to colloidal characterization nanogels were dialyzed against distilled water.Two families of nanogels were synthesized using different amounts (4, 6, 16, 24, 50 wt% with respect to VCL) of the two Dex-MA macro-cross-linkers previously synthesized (Dex40MA9 and Dex40MA86). As commented before, the difference between these two macro-cross-linkers consists of the amount of methacrylate groups per dextran chain. That is, Dex40MA86 contains 86 methacrylate groups randomly distributed along the 225 glucopyranosyl groups of the dextran chain, while Dex40MA9 contains only 9 methacrylate groups per dextran chain. Furthermore, a nanogel with 0.08 g of MBA was synthesized following the same reaction conditions.
2.4. Characterization of nanogels
Transmission Electron Microscopy (TEM, TECNAI G2 20 TWIN 200 kV LaB6) was used for the direct observation of the nanogel particles. The sample was prepared by adding two drops of a 1% aqueous solution of phosphotungstic acid (PTA) as negative staining into the diluted sample at 0.05 wt%. Then, a drop of the dilute sample was placed on copper grids covered with Formvar® (polyvinyl formal) and dried under UV light.
2.5. Uptake of dextranase into nanogels
Nanogel dispersions were incubated, with known concentrations of dextranase, for 2 days at 20 °C and pH 8. These conditions were used to inhibit enzyme activity and avoid the early degradation of the nanogels.34 The dispersions were then centrifuged at 10000 rpm for 2 h, using a Centrikon T-2140 (Kontron Instruments) ultracentrifuge. 1 mL of supernatant was incubated at 37 °C and pH 6 for 12 hours after the addition of 19 mL of dextran solution (10 mg mL−1). After this, 2 mL of the supernatant was incubated with 3 mL of Sumner reagent and 200 μL of sodium sulfite solution (30 mg mL−1) as a color stabilizer for 15 min at 95 °C.35,36 After cooling, the dextranase concentration in the supernatant was determined by measuring the absorbance at 575 nm, and therefore, the absorbed amounts of dextranase by nanogels.24,37
2.6. Enzymatic degradation of nanogels
Final dispersions were incubated in an imidazole buffer (1 mM, pH 8) with two concentrations of dextranase (5 and 20 U per gnanogel) at 20 °C for two days. One unit of enzymatic activity (U) is defined as the amount of enzyme that produces 1 μmol min−1 of reducing sugars at pH 5.5 and 37 °C. Once dextranase was encapsulated, the pH of the samples was modified to pH 6 adding the suitable amount of HCl and the samples were incubated at 37 °C.In order to follow enzymatic degradation, 2 mL of the sample was taken at different times. These samples were heated at 95 °C for 10 min to inactivate the enzyme.38 The concentration of reducing oligosaccharides was determined spectrophotometrically with the Sumner reagent. Degraded samples were incubated with 3 mL of Sumner reagent for 15 min at 95 °C. After cooling, the absorbance was measured at 575 nm and the concentration of reducing oligosaccharides was calculated using glucose solutions as reference.35,36
In addition, the swelling ratio of nanogels after degradation was calculated following the next expression: (dpf/dp0)3, in which dpf is the average hydrodynamic diameter after 10 days of degradation and dp0 is the average hydrodynamic diameter of the dialyzed nanogel.
3. Results and discussion
It is well known that biocompatibility and thermo sensitivity highlight PVCL-based nanogels as promising nanocarriers in drug delivery. Furthermore, the incorporation of degradable cross-linking points into the PVCL-based nanogel network would improve the characteristics of these nanocarriers. In this case, the degradation of the cross-linker leads to the disintegration of the nanogel network structure yielding free polymer chains, mainly biocompatible PVCL. In addition, in the case of having entrapped drugs inside these degradable nanogels, the rupture of the network causes the release of the drug. The controlled release of the drug inside PVCL-based nanogels will depend on the rate of network degradation, which is a function of the type of degradable cross-linker. In this work, Dex-MA is chosen as the macro-cross-linker because it can be only degraded by the action of dextranase enzyme. By this way, nanogels prepared with Dex-MA can be dispersed at any pH without any degradation, and only the presence of dextranase causes the cleavage of the dextran chains.25 The parameters controlling the effectiveness of Dex-MA as the macro-cross-linker are (i) the molecular weight of the dextran chain and (ii) the degree of substitution (DS), which is defined as the number of methacrylate groups per dextran chain. Maintaining the Mw of Dex-MA constant, the degree of cross-linking can be only tuned by changing DS. In that way, dextran having a Mw of 40000 g mol−1 was chosen and modified with different amounts of GMA to obtain two macro-cross-linkers with different DS (Dex40MA9 and Dex40MA86). Dex40MA9 is a low substituted Dex-MA having one methacrylate group every 25 glucopyranosyl groups and Dex40MA86 is a high substituted Dex-MA having one methacrylate group every 2 or 3 glucopyranosyl groups.
3.1. Temperature sensitivity of nanogels
Depending on the DS of the macro-cross-linker, different nanogel particle structures can be obtained. The first family of enzymatically degradable PVCL-based nanogels was prepared using Dex40MA86 as the macro-cross-linker. When this macro-cross-linker is used highly cross-linked nanogel particles are formed. Fig. 1 shows the effect of the macro-cross-linker concentration on the thermal behavior (temperature sensitivity) of the final nanogel particles. In addition, the thermal behavior of a reference nanogel produced by using 4 wt% of MBA as the bifunctional cross-linker is also shown. Only the heating cycles of the nanogels are shown because all the thermal behaviors obtained were reversible, i.e., no hysteresis between heating and subsequent cooling cycles was observed. In all the cases, the thermal behavior of nanogel particles is similar to that of other PVCL-based nanogels reported in our previous works.15,16,18,19 Below the VPTT nanogel particles were highly swollen, and above that critical temperature particles were shrunken reducing their size due to the increase in the hydrophobic interactions between non-polar groups. In the case of the nanogel synthesized using MBA as the cross-linker, the hydrodynamic diameters are smaller, probably due to the smaller size of this cross-linker in comparison with the Dex40MA86 macro-cross-linker. In addition, increasing the amount of the macro-cross-linker from 4 to 16 wt%, the hydrodynamic diameter of nanogel particles at high temperatures increases due to the higher content of Dex40MA86 chains per particle. But, from 16 wt% to 50 wt% the hydrodynamic diameter of the nanogel particles at high temperatures is maintained almost constant. Moreover, it is well known that the VPTT of PVCL can be modulated through incorporation of hydrophilic or hydrophobic comonomers.16,19 However, in this case, the VPTT is independent of the macro-cross-linker concentration. It seems that the type of interactions between the PVCL chains and the water molecules does not change in the presence of the macro-cross-linker at least at the studied range of concentration. | ||
Fig. 1 Average hydrodynamic particle diameter as a function of temperature for nanogels synthesized using the Dex40MA86 macro-cross-linker. ■ 4 wt%;  6 wt%; 6 wt%;  16 wt%; 16 wt%;  24 wt%; 24 wt%;  50 wt%; and 50 wt%; and  4 wt% of the MBA bifunctional cross-linker. 4 wt% of the MBA bifunctional cross-linker. | ||
Apart from being a macro-cross-linker, Dex-MA is a hydrophilic polymer. Therefore, the increase in Dex-MA chains inside the final nanogel particles implies an increase in their hydrophilicity. Due to this, apart from the effect of cross-linking, the hydrophilic–hydrophobic balance inside nanogel particles must be taken into account when the effect of the Dex40MA86 macro-cross-linker on the swelling ratio is analyzed.
Fig. 2 shows the effect of the macro-cross-linker concentration on the swelling ratio of the final nanogel particles. Increasing the amount of Dex40MA86 macro-cross-linker from 4 wt% to 6 wt%, and from 24 wt% to 50 wt%, the swelling ratio decreases. This expected result was also observed in the case of using bifunctional cross-linkers as MBA and PEGDA,16,18 because an increase in the concentration of macro-cross-linker causes an increase in the degree of cross-linking of the final nanogel particles. Surprisingly, this expected behavior is not maintained in all the range studied. As can be seen, from 6 wt% to 24 wt% the swelling behavior is similar. At this point, it is important to take into account the hydrophilicity of the final nanogel particles, which is related with the amount of Dex40MA86 macro-cross-linker.
 | ||
Fig. 2 Swelling ratio as a function of temperature for nanogels synthesized using the Dex40MA86 macro-cross-linker. ■ 4 wt%;  6 wt%; 6 wt%;  16 wt%; 16 wt%;  24 wt%; 24 wt%;  50 wt%; and 50 wt%; and  4 wt% of the MBA bifunctional cross-linker. 4 wt% of the MBA bifunctional cross-linker. | ||
In Table 1, the number of nanogel particles per cm3 of water, Dex40MA86 chains per particle, the initial derived count rate values obtained by PCS and the maximum swelling ratio of the nanogels are shown. It must be taken into account that, when the amount of macro-cross-linker in the nanogel particles increases, their hydrophilicity also increases. This effect can be observed in the initial values of the derived count rate. The initial value of the derived count rate is a function of the solids content of the nanoparticles and it is related with the hydrophilicity of the nanogels. As can be seen in Table 1, by fixing the initial solids content at 0.005 wt%, when the concentration of macro-cross-linker increases, the values of the derived count rate decrease. From 4 wt% to 6 wt% of Dex40MA86, the number of particles increases but the derived count rate slightly decreases. This means that the cross-linking degree rather than the hydrophobic–hydrophilic balance of the nanogels, decreasing the maximum swelling ratio, controls the swelling ratio of these nanogels. On the other hand, from 6 wt% to 16 wt%, the number of nanogel particles and the derived count rate value decrease four and two times, respectively. However, the number of Dex40MA86 chains per particle increases more than 10 times meaning that these nanogel particles are much more hydrophilic. There is a competition between the hydrophilic Dex40MA86 chains, which tend to be extended to the aqueous phase, and the hydrophobic PVCL chains, which tend to collapse above the VPTT. In this case, the hydrophilic contribution of the huge amount of Dex40MA86 chains per nanogel particle is predominant and in this way, their swelling ratio is controlled by the hydrophobic–hydrophilic balance inside the nanogel rather than by the cross-linking ability of Dex40MA86. This high amount of Dex40MA86 chains per nanogel particle tends to be extended, hindering the total collapse of nanogel particles and maintaining the maximum swelling ratio constant. In the case of the nanogel synthesized with a 24 wt% of Dex40MA86 the same behavior is observed and the maximum swelling ratio is maintained constant. From 24 wt% to 50 wt%, the derived count rate decreases again and the number of particles increases. As in the case of 6 wt%, the number of Dex40MA86 chains per nanogel particle increases in agreement with the amount of macro-cross-linker added, but the swelling ratio decreases. Therefore, at such high macro-cross-linker concentrations the cross-linking ability of Dex40MA86 returns to control the thermal behavior of these nanogels.
| Dex40MA86 (wt%) | Number of nanogel particles per cm3 water | Dex40MA86 chains per particle | Derived count rate (kcps) | Maximum swelling ratio |
|---|---|---|---|---|
| 4 | 4.4447 × 1012 | 1384 | 63.4 | 6.43 |
| 6 | 6.2614 × 1012 | 1445 | 56.3 | 5.45 |
| 16 | 1.3358 × 1012 | 18526 |
32.6 | 5.37 |
| 24 | 2.0576 × 1012 | 17685 |
29.4 | 5.33 |
| 50 | 2.7716 × 1012 | 27509 |
16.6 | 3.73 |
For the sake of comparison, in Fig. 2 the swelling ratio of the nanogel prepared with 4 wt% of MBA as the bifunctional cross-linker is also shown. As can be seen, when the same amounts of bifunctional cross-linker and multifunctional macro-cross-linker are used (4 wt%), the same swelling ratio as a function of temperature is obtained. However, when the same amount of reactive groups able for the cross-linking of PVCL chains is used (4 wt% and 24 wt% in the case of MBA and Dex40MA86, respectively) the swelling ratio is lower in the case of using the macro-cross-linker. The reason for this lower swelling ability, assuming that Dex40MA86 reacts completely, is the different distribution of the cross-linking points inside nanogel particles. Taking into account that the main features of Dex40MA86 as a macro-cross-linker are: (i) big size together with high hydrophilicity in water and (ii) multiple methacrylate groups randomly distributed along its chain, it is easy to expect a nanogel with more homogeneously distributed cross-linking points inside by using this macro-cross-linker.
The second family of enzymatically degradable PVCL-based nanogels was prepared using Dex40MA9 as the macro-cross-linker. Having in mind that using the same amount of macro-cross-linker, the effectiveness of Dex40MA9 as a cross-linking agent is lower than that of Dex40MA86 because the former has only one methacrylate group every 25 glucopyranosyl groups. Therefore, when the Dex40MA9 macro-cross-linker is used slightly cross-linked nanogel particles are formed. The effect of the cross-linker concentration on temperature sensitivity of the final nanogel particles is shown in Fig. 3. As can be seen, the average hydrodynamic diameters of these nanogels slightly decrease around the VPTT, and then increase instantaneously just at the critical temperature of the PVCL chains. Comparing with the highly cross-linked nanogel particles produced using Dex40MA86, the thermal behavior is inverted in terms of the hydrodynamic diameter, i.e., the nanogel particles have smaller diameters below than those above the VPTT. This anomalous behavior suggests that during the increase in hydrophobicity of the PVCL chains with the temperature, a first period of water expulsion from the nanogel inner part causes the decrease in hydrodynamic diameter, followed by a second period consisting in the aggregation of the nanogel particles, which is responsible for the increase in particle size. In addition, a sharp increase of the derived count rate is observed above the VPTT due to the hydrophobicity of the PVCL chains. To verify this assumption, nanogel particles were observed by TEM. In Fig. 4, a TEM micrograph of the final nanogel particles obtained using a 16 wt% of Dex40MA9 is shown. As can be seen, these nanogel particles are monodisperse with diameters of around 20–30 nm due to their dry state while observing by TEM. The diameters obtained above the VPTT measured by PCS and those analyzed by TEM are clearly very different. These larger diameters together with the increase of the derived count rate suggest the formation of interparticle reversible aggregates. Moreover, increasing the amount of Dex40MA9 macro-cross-linker larger aggregates are formed. The formation of aggregates was also observed by other authors for thermo-sensitive graft copolymers based on PVCL and PEO.39–41 In these works, PEO chains were grafted to PVCL chains and the thermal behavior of these amphiphilic copolymers was studied. Below their LCST, PVCL-g-PEO copolymers in water were di-hydrophilic. Above their LCST, these graft copolymers became amphiphilic and stable aggregates were formed consisting of a hydrophobic PVCL core and a hydrophilic PEO shell. Aggregates with high colloidal stability were obtained due to steric stabilization conferred by the PEO chains grafted on the PVCL chains.
 | ||
Fig. 3 Average hydrodynamic particle diameter as a function of temperature for nanogels synthesized using the Dex40MA9 macro-cross-linker. ■ 6 wt%;  16 wt%; 16 wt%;  24 wt%; and 24 wt%; and  50 wt%. 50 wt%. | ||
 | ||
| Fig. 4 TEM micrograph of a nanogel obtained using 16 wt% of Dex40MA9. | ||
To study in depth the formation of stable nanogel aggregates, the polydispersity indices (PDIs) of the slightly cross-linked nanogel particles were analyzed. As can be seen in Fig. 5, below the VPTT the PDI values of these nanogels are higher than those above. These PDI values decrease to reach typical values of monodisperse conventional latex particles. Below the VPTT, the slightly cross-linked nanogel particles could be considered as single macromolecules of different chain lengths dissolved in water as a consequence of the free-radical polymerization process used to produce them. This means that their swollen structure is loose and thereby their PDI value is high and comparable to that of single macromolecules dissolved in water. However, above their VPTT, an amphiphilic structure is obtained, the PVCL chains being more hydrophobic when increasing the temperature and the Dex40MA9 chains continue being hydrophilic and extended. For this reason, it is supposed that a reorganization of the nanoparticle structure takes place and interparticle aggregation occurs. The structure of the aggregates is depicted in Fig. 5 as a core–shell aggregate, consisting of a core formed by the PVCL hydrophobic chains and a shell made of the Dex40MA9 chains, which confers steric stabilization to the aggregates. Furthermore, due to the very low values of PDI above the VPTT, these aggregates are monodisperse and spherical. It is interesting to point out that these aggregates are reversible (in other words, the aggregation process is completely reversible) and they suffer fragmentation when the temperature drops below the VPTT of the PVCL chains.
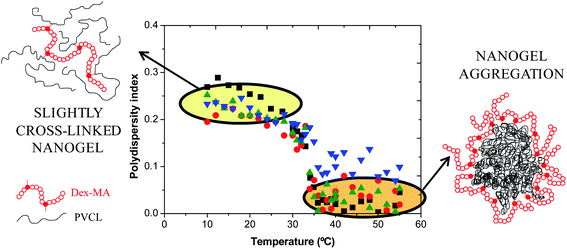 | ||
Fig. 5 Polydispersity indices as a function of temperature for nanogels synthesized using the Dex40MA9 macro-cross-linker. ■ 6 wt%;  16 wt%; 16 wt%;  24 wt%; and 24 wt%; and  50 wt%. 50 wt%. | ||
3.2. Morphology and inner structure of nanogels
The general concept for nanogel structures is a core–shell model in which core and shell have different structures and cross-linking degree. To confirm this heterogeneous nanogel structure and to study the distribution of the PVCL chains in the nanogel, 1H NMR measurements were performed. The heterogeneity in the nanogel microstructure would be reflected in the 1H transverse magnetization relaxation due to different degree of the PVCL proton mobility provoked by the different cross-linking densities.17,42 From transverse magnetization (T2) decay it is possible to show a bimodal distribution of the PVCL chain mobility inside the nanogel particles from which the morphological core–shell model can be derived. The proton high-resolution spectrum of the nanogel prepared with a 6 wt% of Dex40MA86 measured in D2O at 23 °C is shown in Fig. 6. Different resonances have been identified with the following chemical shifts: R1 = 1.8 ppm, R2 = 2.5 ppm, R3 = 3.3 ppm, R4 = 4.4 ppm, and R5 = 4.8 ppm. R5 corresponds to deuterium oxide, while the others were assigned to different protons of the polymeric chain. Resonance R1 of methylene protons was used in the following measurements to investigate the heterogeneity of the different nanogels. Biexponential decay for the integral of R1 resonance for the different nanogels was assumed.where S(t)/S(0) is the normalized NMR signal integral as a function of spin-echo time t and Ci (i = S and L) are the relative contribution of the decays characterized by short (T2S) and long (T2L) transverse relaxation times. The coefficients CS and CL are related to the number of protons in the methylene fragments of PVCL describing quantitatively the bimodal heterogeneity of the polymer network in nanogel particles. Once short and long transverse relaxation times are calculated, the ratio of the cross-linking density of the nanogel in the core and shell could be obtained from the following relationship:

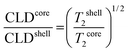
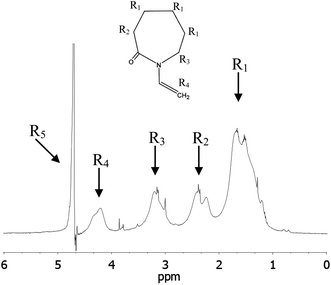 | ||
| Fig. 6 Proton high-resolution spectrum of a nanogel prepared using 6 wt% of Dex40MA86 (2 wt% in D2O) at 23 °C. | ||
The values of the 1H transverse relaxation times (T2S and T2L), relative amount of methylene protons of PVCL (CS and CL) and the ratio of the cross-linking density (CLDcore/CLDshell) of different nanogel particles are shown in Table 2. For the nanogel synthesized using MBA as the cross-linker, a heterogeneous core–shell microstructure is obtained and the ratio of the average cross-linking density between the core and shell was 3.03, which is in agreement with the value obtained by Balaceanu et al.17 This means that the core is three times more cross-linked than the shell and therefore more PVCL chains are located in the shell than in the core.
| Macro-cross-linker | T 2S [ms] | C S | T 2L [ms] | C L | CLDcore/CLDshell | |
|---|---|---|---|---|---|---|
| Type | Amount | |||||
| MBA | 4 wt% | 0.42 | 0.30 | 3.85 | 0.68 | 3.03 |
| Dex40MA86 | 6 wt% | 1.28 | 0.42 | 4.14 | 0.61 | 1.79 |
| Dex40MA86 | 50 wt% | 1.36 | 0.38 | 4.09 | 0.64 | 1.73 |
| Dex40MA9 | 6 wt% | 3.50 | 0.45 | 3.50 | 0.45 | 1 |
| Dex40MA9 | 50 wt% | 3.57 | 0.45 | 3.57 | 0.45 | 1 |
In the case of the highly cross-linked nanogel particles (by using Dex40MA86) the expected core–shell microstructure is also confirmed. The values were similar for both nanogels studied (6 wt% and 50 wt% of the Dex40MA86 macro-cross-linker). This means that the ratio between the core and shell, in terms of mobility, did not change with the increase of the Dex40MA86 amount. The same behavior was observed by Balaceanu et al.17 for microgels prepared with different amounts of MBA as the cross-linker. In both nanogels synthesized using Dex40MA86 more percentage of PVCL chains are in the shell than in the core, probably due to the higher reactivity of the Dex40MA86 macro-cross-linker compared to that of VCL. Imaz and Forcada18 reported that during the emulsion polymerization of VCL using PEGDA as the cross-linker, this was consumed faster compared to VCL, and hence, PEGDA was preferentially incorporated into the core. The reactivities of the acrylic double bond of PEGDA and the methacrylic group of Dex-MA could be considered similar in comparison with the reactivity of VCL. Therefore, Dex-MA is preferentially incorporated into the core. On the other hand, the same cross-linking density ratio is obtained in both highly cross-linked nanogels (6 wt% and 50 wt% of Dex40MA86). This ratio is lower than that obtained in the case of the nanogel synthesized with MBA. This means that more homogeneous nanogels are obtained using the Dex40MA86 macro-cross-linker, and that core and shell regions are not so marked. As it was explained before, the reason for this is the large size of this macro-cross-linker together with its high hydrophilicity in water and the multiple methacrylate groups randomly distributed along the Dex40MA86 chain.
In the case of slightly cross-linked nanogel particles (by using Dex40MA9) the cross-linking density ratio was 1, which means that the PVCL chains are homogeneously distributed inside the nanogel particles and the morphology of the nanogel particles is not core–shell. As discussed before, because of the low DS and the high hydrophilicity of Dex40MA9, these slightly cross-linked nanogel particles show the same behavior as those of PVCL-g-PEO and they can be considered as slightly cross-linked macromolecules dissolved or swollen by water.
3.3. Enzymatic degradation of nanogels
Nanogel particles must be able to absorb dextranase in order to ensure their degradation. The enzyme must bind to the dextran chains to degrade them. During the uptake of dextranase, it is important to inhibit the activity of the enzyme in order to avoid early degradation of nanogels. For this purpose the uptake was carried out at pH 8 and 20 °C.34Fig. 7 shows the effect of the type and amount of macro-cross-linker on the uptake of dextranase by poly(VCL-co-DexMA) nanogels. As can be seen, in all the cases dextranase is absorbed into nanogels. It is interesting to point out that neither the amount of Dex-MA (6 wt% or 50 wt%) nor the degree of substitution (9 or 86) affect the uptake of the enzyme. In the case of highly cross-linked nanogel particles (by using Dex40MA86) their pore sizes are large enough to enable the penetration of dextranase, but the enzyme probably only enters the periphery of the nanogel particles, i.e., the shell. However, in the case of slightly cross-linked nanogel particles (by using Dex40MA9), their structure is looser and their pores are much larger, allowing dextranase to penetrate deeper.
 | ||
Fig. 7 Absorbed amount of dextranase (%) at pH 8 and 20 °C inside poly(VCL-co-DexMA) nanogels after two days of incubation. Using Dex40MA86: ■ 6 wt% and  50%; using Dex40MA9: 50%; using Dex40MA9:  6 wt% and 6 wt% and  50 wt%. 50 wt%. | ||
The uptake of the enzyme increases with the amount of dextranase until 7.5 U per gnanogel when a plateau is obtained. The absorption capacity of all the nanogels is very high, achieving 95% of uptake using 100 U per gnanogel. In this way, the capacity of different nanogels to absorb dextranase is reported for the first time, the control of the degradation process of nanogel particles being possible by varying the activity of dextranase, thanks to their high uptake capacity.
Once the uptake of dextranase was analyzed for different nanogel particles, the pH and the temperature of the medium were modified to 6 and 37 °C, respectively. The dispersions were incubated with the enzyme dextranase for 10 days, using two different dextranase activities (5 and 20 U per gnanogel). Dextranase cleaves the linkages between glucose molecules of dextran, releasing shorter oligosaccharides, which are known as reducing sugars. The most common products after degradation are isomaltose and glucose, with small amounts of larger oligosaccharides. In addition, it was also reported that Dex-MA can be degraded to a similar extent as native dextran.24,26 In this case, dextranase is capable of hydrolyzing a bond between a substituted and an unsubstituted glucopyranose residue, but bonds between two substituted glucopyranose residues cannot be cleft by the enzyme. The degradation process is mainly controlled by the DS of Dex-MA. Therefore, degradation of low substituted dextrans is favored and release of reducing sugars is possible. In the case of the degradation process of high substituted dextrans, the degradation is possible, but not the release of reducing sugars because of the lack of long unsubstituted chain segments.
The swelling ratios after the degradation process are shown in Table 3. As can be seen, in the case of using a 6 wt% of Dex40MA86, nanogel particles are swollen after the incubation process, the swelling being higher when the dextranase activity increases from 5 to 20 U per gnanogel. In the case of using a 50 wt% of Dex40MA86, a lower swelling is observed and the swelling ratio is higher when the activity of dextranase increases. Lower swellings could be due to two facts: (i) the first one could be the high amount of dextran used for the same dextranase amount, more time being necessary to achieve the same swelling ratio, and (ii) the second one could be the low penetration of dextranase inside the core of the nanogel particles due to the higher amount of macro-cross-linker used, more time being needed to cleave dextran and penetrate deeper inside the core. On the other hand, in the case of slightly cross-linked nanogel particles (by using Dex40MA9), a de-swelling of the nanogel particles is observed using 6 and 50 wt% of the Dex40MA9 macro-cross-linker, being higher when increasing the amount of dextranase and dextran macro-cross-linker inside nanogel particles. Slightly cross-linked nanogels are easily degraded almost to dissolve completely their structure.
| Macro-cross-linker | Swelling ratio after adding dextranase | ||
|---|---|---|---|
| Type | Amount | 5 U per gnanogel | 20 U per gnanogel |
| Dex40MA86 | 6 wt% | 4.68 | 6.84 |
| Dex40MA86 | 50 wt% | 1.05 | 1.87 |
| Dex40MA9 | 6 wt% | 0.68 | 0.51 |
| Dex40MA9 | 50 wt% | 0.08 | 0.03 |
These results can be explained analyzing the release of reducing sugars from the two families of nanogels. The release of reducing sugars during the enzymatic degradation of hydrogels and microgels based on Dex-MA was studied by other authors.22,35–38 It was observed that the release of reducing sugars took place only when long unsubstituted chain segments are available in Dex-MA chains. The cumulative release of oligosaccharides from nanogels as a function of the DS for nanogels using 6 and 50 wt% of macro-cross-linkers is shown in Fig. 8 and 9, respectively. As can be seen in both figures, in the case of highly cross-linked nanogel particles (by using 6 and 50 wt% of Dex40MA86), reducing sugars are not released. The reason is that dextranase needs longer unsubstituted chain segments to allow multiple scissions, at least 9, which result in the release of reducing sugars.27 In the case of slightly cross-linked nanogel particles (by using 6 and 50 wt% of Dex40MA9) reducing sugars are released due to the presence of long unsubstituted chain segments. In both cases, the release of reducing sugars is higher when the dextranase activity increases from 5 to 20 U per gnanogel, the released amount of reducing sugars being higher in the case of the nanogel synthesized using 50 wt% of the Dex40MA9 macro-cross-linker. This higher release observed is in agreement with the higher de-swelling obtained (see Table 3). Therefore, the extent of nanogel degradation can be controlled by the amount of Dex40MA9 inside the nanogel.
 | ||
Fig. 8 Cumulative release of reducing sugars as a function of time in a nanogel having 6 wt% of macro-cross-linkers. Family of Dex40MA86: ■ 5 U per gnanogel and □ 20 U per gnanogel; family of Dex40MA9:  5 U per gnanogel and 5 U per gnanogel and  20 U per gnanogel. 20 U per gnanogel. | ||
 | ||
Fig. 9 Cumulative release of reducing sugars as a function of time in a nanogel having 50 wt% of macro-cross-linkers. Family of Dex40MA86: ■ 5 U per gnanogel and □ 20 U per gnanogel; family of Dex40MA9:  5 U per gnanogel and 5 U per gnanogel and  20 U per gnanogel. 20 U per gnanogel. | ||
As a conclusion of the results on enzymatic degradation of nanogels presented, two degradation mechanisms are proposed for high and low cross-linked nanogel particles (Fig. 10). Both types of nanogel particles are enzymatically degraded. In the case of the highly cross-linked nanogel particles (by using Dex40MA86), after the incubation process, nanogel particles are swollen. This swelling is due to the cleavage of some glucopyranosyl bonds. However, reducing sugars are not released. This can be attributed to the fact that the hydrolysis of a glucopyranosyl bond does not necessarily have to result in the release of a reducing sugar. On the other hand, in the case of slightly cross-linked nanogel particles (by using Dex40MA9), as dextranase is able to bind to macro-cross-linker chains, reducing sugars are released. Due to the release of reducing sugars, these nanogel particles show a de-swelling after the incubation process, which is higher when increasing the amount of dextranase and dextran macro-cross-linker inside nanogel particles.
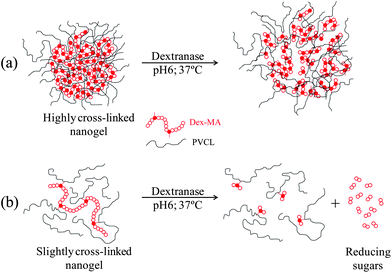 | ||
| Fig. 10 Proposed degradation mechanisms for (a) a highly cross-linked nanogel synthesized with the Dex40MA86 macro-cross-linker and (b) a slightly cross-linked nanogel synthesized with the Dex40MA9 macro-cross-linker. | ||
4. Conclusions
Two new families of enzymatically degradable PVCL-based nanogels were synthesized by batch emulsion polymerization using dextran methacrylates with different DS (Dex40MA86 and Dex40MA9). In both families, the type of Dex-MA macro-cross-linker did not affect the VPTT of the PVCL-based nanogel particles.The first family of enzymatically degradable PVCL-based nanogels consisted of highly cross-linked nanogel particles (by using Dex40MA86). Below the VPTT, nanogel particles were highly swollen and above this critical temperature, nanoparticles were shrunken. The swelling ratio of these nanogel particles can be explained, on the one hand, taking into account the ability of cross-linking (from 4 wt% to 6 wt% and from 24 wt% to 50 wt% of the macro-cross-linker) and, on the other hand, taking into account the degree of hydrophilicity of the nanogels and the tendency of the Dex40MA86 chains to be extended to the aqueous phase, hindering the collapse of the final nanogel particles (from 6 wt% to 24 wt%). In addition, it was observed that the nanogel particles synthesized with the Dex40MA86 macro-cross-linker had a more homogeneous structure than the nanogel particles synthesized using MBA as a bifunctional cross-linker. The enzymatic degradation of these nanogel particles was studied using dextranase enzyme. After the enzymatic degradation, highly cross-linked nanogel particles were swollen due to the cleavage of some glucopyranosyl bonds of dextran without releasing reducing sugars.
The second family of enzymatically degradable PVCL-based nanogels consisted of slightly cross-linked nanogel particles (by using Dex40MA9). In this case, an unexpected behavior was observed, the thermal behavior was inverted in terms of the hydrodynamic diameter, i.e., nanogel particles had smaller diameters below than above the VPTT. Due to the low DS of the macro-cross-linker used, the nanogel particles behaved as amphiphilic copolymers and an interparticle aggregation process occurs when the VPTT was reached. In terms of enzymatic degradation, dextranase was easily able to bind to Dex40MA9 chains releasing reducing sugars and causing a de-swelling of the nanogel particles.
Regarding the future use of these new nanogels in drug delivery, after degradation, highly cross-linked nanogels became looser, making the release of drug easier but maintaining their identity. However, slightly cross-linked nanogels were fragmented to several parts, losing their identity and allowing an abrupt release of drug.
Acknowledgements
This work has been supported by the Spanish Plan Nacional de Materiales (MAT2012-36,270-C04-01). Technical support provided by SGIker (UPV/EHU, MICINN, GV/EJ, ESF) is gratefully acknowledged.References
- B. R. Saunders and B. Vincent, Adv. Colloid Interface Sci., 1999, 80, 1–25 CrossRef CAS.
- J. K. Oh, R. Drumright, D. J. Siegwart and K. Matyjasezewski, Prog. Polym. Sci., 2008, 33, 448–477 CrossRef CAS.
- A. Pich and W. Richtering, Adv. Polym. Sci., 2010, 234, 1–37 CrossRef CAS.
- H. Senff and W. Richtering, J. Chem. Phys., 1999, 111, 1705–1711 CrossRef CAS.
- M. J. Murray and M. J. Snowden, Adv. Colloid Interface Sci., 1995, 54, 73–91 CrossRef CAS.
- T. Hoare and R. Pelton, Langmuir, 2008, 24, 1005–1012 CrossRef CAS.
- C. M. Nolan, C. D. Reyes, J. D. Debord, A. J. García and L. A. Lyon, Biomacromolecules, 2005, 6, 2032–2039 CrossRef CAS.
- M. Bradley, J. Ramos and B. Vincent, Langmuir, 2005, 21, 1209–1215 CrossRef CAS.
- H. Vihola, A. Laukkanen, L. Valtola, H. Tenhu and J. Hirvonen, Biomaterials, 2005, 26, 3055–3064 CrossRef CAS.
- A. Imaz and J. Forcada, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 1173–1183 CrossRef CAS.
- J. Ramos, A. Imaz, J. Callejas-Fernández, L. Barbosa-Barros, J. Estelrich, M. Quesada-Pérez and J. Forcada, Soft Matter, 2011, 7, 5067–5082 RSC.
- J. Ramos, A. Imaz and J. Forcada, Polym. Chem., 2012, 3, 852–856 RSC.
- V. Boyko, A. Pich, Y. Lu, S. Richter, A. Friedrich and H. J. P. Adler, Polymer, 2003, 44, 7821–7827 CrossRef CAS.
- A. Pich, A. Tessier, V. Boyko, Y. Lu and H. J. P. Adler, Macromolecules, 2006, 39, 7701–7707 CrossRef CAS.
- A. Imaz and J. Forcada, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 2510–2524 CrossRef CAS.
- A. Imaz and J. Forcada, Eur. Polym. J., 2009, 45, 3164–3175 Search PubMed.
- A. Balaceanu, D. E. Demco, M. Möller and A. Pich, Macromolecules, 2011, 44, 2161–2169 CrossRef CAS.
- A. Imaz and J. Forcada, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 2766–2775 CrossRef CAS.
- A. Imaz and J. Forcada, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 3218–3227 CrossRef CAS.
- W. N. E. Van Dijk-Wolthuis, O. Fransen, H. Talsma, M. J. van Steenbergen, J. J. Kettenes-van den Bosch and W. E. Hennink, Macromolecules, 1995, 28, 6317–6322 CrossRef CAS.
- W. N. E. Van Dijk-Wolthuis, J. J. Kettenes-van den Bosch, A. van der Kerk-van Hoof and W. E. Hennink, Macromolecules, 1997, 30, 3411–3413 CrossRef CAS.
- W. N. E. Van Dijk-Wolthuis, M. J. van Steenbergen, W. J. M. Underberg and W. E. Hennink, J. Pharm. Sci., 1997, 86, 413–417 CrossRef CAS.
- W. N. E. Van Dijk-Wolthuis, S. K. Y. Tsang, J. J. Kettenes-van den Bosch and W. E. Hennink, Polymer, 1997, 38, 6235–6242 CrossRef CAS.
- O. Franssen, R. D. van Ooijen, D. de Boer, R. A. A. Maes, J. N. Herron and W. E. Hennink, Macromolecules, 1997, 30, 7408–7413 CrossRef CAS.
- W. N. E. Van Dijk-Wolthuis, J. A. M. Hoogeboom, M. J. van Steenbergen, S. K. Y. Tsan and W. E. Hennink, Macromolecules, 1997, 30, 4639–4645 CrossRef CAS.
- O. Franssen, R. D. van Ooijen, D. de Boer, R. A. A. Maes and W. E. Hennink, Macromolecules, 1999, 32, 2896–2902 CrossRef CAS.
- T. G. Van Thienen, B. Lucas, F. M. Flesch, C. F. van Nostrum, J. Demeester and S. C. De Smedt, Macromolecules, 2005, 38, 8503–8511 CrossRef CAS.
- K. Raemdonck, B. Naeye, K. Buyens, R. E. Vandenbroucke, A. Hogset, J. Demeester and S. C. De Smedt, Adv. Funct. Mater., 2009, 19, 1406–1415 CrossRef CAS.
- D. Klinger, M. Aschenbrenner, C. K. Weiss and K. Landfester, Polym. Chem., 2012, 3, 204–216 RSC.
- D. Klinger and K. Landfester, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 1062–1075 Search PubMed.
- X. Huang, G. P. Misra, A. Vaish, J. M. Flanagan, B. Sutermaster and T. L. Lowe, Macromolecules, 2008, 41, 8339–8345 CrossRef CAS.
- F. Li, H. Wu, L. Fan, H. Zhang, H. Zhang and C. Gu, Polym. Int., 2009, 58, 1023–1033 Search PubMed.
- J. Ramos, A. Costoyas and J. Forcada, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 4461–4478 Search PubMed.
- M. Shamsolahrar, A. Kheirolomoom and M. Farshbaf Dadjour, Sci. Iran., 2002, 9, 39–46 Search PubMed.
- G. L. Miller, Anal. Chem., 1959, 31, 426–428 CrossRef CAS.
- M. Beesh, P. Majewska and Th. F. Vandamme, Int. J. Drug Delivery, 2010, 2, 22–31 Search PubMed.
- O. Franssen, O. P. Vos and W. E. Hennink, J. Controlled Release, 1997, 44, 237–245 CrossRef CAS.
- D. Reischl and A. Zimmer, Nanomedicine: NBM, 2009, 5, 8–20 Search PubMed.
- S. Verbrugghe, A. Laukkanen, V. Aseyev, H. Tenhu, F. M. Winnik and F. E. Du Prez, Polymer, 2003, 44, 6807–6814 Search PubMed.
- S. Verbrugghe, K. Bernaerts and F. E. Du Prez, Macromol. Chem. Phys., 2003, 204, 1217–1225 CrossRef CAS.
- A. Laukkanen, L. Valtola, F. M. Winnik and H. Tenhu, Polymer, 2005, 46, 7055–7065 CrossRef CAS.
- S. Schachschal, A. Balaceanu, C. Melian, D. E. Demco, T. Eckert, W. Richtering and A. Pich, Macromolecules, 2010, 43, 4331–4339 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2013 |