Connecting magnetic micro-particles with DNA G-quadruplexes†
Vineeth Thachappilly
Mukundan
a,
Quang Minh
Nhat Tran
a,
Yuanhua
Miao
ab and
Anh Tuân
Phan
*a
aDivision of Physics and Applied Physics, School of Physical and Mathematical Sciences, Nanyang Technological University, Singapore. E-mail: phantuan@ntu.edu.sg
bTechnical Institute of Physics and Chemistry, Chinese Academy of Sciences, Beijing 100190, P. R. China
First published on 16th October 2012
Abstract
Particle self-assembly using DNA has relied mainly on the interaction between complementary strands of double helix DNA. In contrast, G-quadruplex DNA has rarely been used in particle self-assembly. G-quadruplex DNA can adopt a wide range of structures with diverse topologies and stabilities controlled by cations. We report on the possibility of connecting micrometer-sized magnetic beads using DNA G-quadruplexes under a magnetic field. The average chain length of the resulting fragments was used to quantify the strength of the interaction, which was shown to depend on the nature and concentration of cations, the DNA sequences, the time duration and strength of the applied magnetic field. Through the use of different sequences, we propose that the length of bead chains could be quantitatively associated with the stability and structure of DNA G-quadruplexes formed between the beads. This work may allow us to control the interactions in colloidal assembly architectures using G-quadruplexes and also to study the structure, dynamics and other properties of the G-quadruplex “DNA glue”.
Introduction
The most well-known (or canonical) structure of DNA is the Watson–Crick double helix based on the A·T and G·C base pairs. Besides this canonical structure, DNA can also adopt other alternative structures. One important example of non-canonical DNA structures is the four-stranded helical structure called the G-quadruplex built from the stacking of G·G·G·G tetrads.1–4 G-quadruplexes are stabilized by cations, such as K+ or Na+, coordinating the carbonyl groups in the core of the structure. A G-quadruplex with only three G-tetrad layers could be more stable than a duplex with 10–20 base pairs. The stability of G-quadruplexes depends on the nature of cations, with K+ usually favoring a more stable structure than Na+.5,6Recently, DNA has attracted much attention as a versatile and convenient material for nanotechnological applications. Colloidal self-assembly using DNA is an attractive scheme to induce specific interactions between micro-particles or nano-particles in order to control colloidal architectures. The first attempt to use DNA as a “molecular glue” to link particles was reported in 1996,7,8 in which the authors induced the aggregation of gold nano-particles using duplex DNA with sticky ends. Following these pioneered works, a number of studies have been reported on the use of DNA to self-assemble nano-particles.9–12 Alternative DNA structures, such as G-quadruplexes and i-motif, have also been used to direct the self-assembly of gold nano-particles.13–15 However, the understanding about such systems is still limited due to the difficulties in their observation and analysis at nanometer scales. On the other hand, micro-particles offer an easier alternative where the colloidal systems can be visualized using a simple microscope. In the past decade, it has been reported that DNA duplex could be used to direct the assembly of micro-particles.16–26 To date, reported DNA-based assembly studies of micro-particles have relied mainly on the formation of the Watson–Crick duplex using complementary strands.
Formation of chains by magnetic particles in the presence of an external magnetic field has been used for different purposes. This method has been used to determine the force–distance law between colloidal particles.27,28 Several groups were able to form stable links between particles using polymers or biomolecules where the kinetics of adhesion has been studied.29,30
Here we study the interaction between micro-particles mediated by DNA G-quadruplexes. Magnetic beads coated with DNA were aligned and allowed to interact in the direction of magnetic field lines. Stable chains which remained in the solution after the removal of field were quantitatively analyzed. We introduced the average chain length as a measure to quantify the strength of interaction. We controlled the strength of inter-bead interaction using various parameters such as the nature and concentration of cations, DNA sequences, the time duration and strength of the applied magnetic field. We found that the length of magnetic bead chains could be quantitatively associated with the stability of the DNA structure and the number of such bonds formed between the beads. This work may allow us (i) to control the interactions in colloidal assembly architectures and also (ii) to study the structure, dynamics and other properties of the “DNA glue”.
Results and discussion
Formation of bead chains bridged by DNA G-quadruplexes
Fig. 1 shows the schematic of our strategy for the formation of magnetic bead chains bridged by DNA G-quadruplexes. Biotin-ended DNA sequences, e.g. GQ2 (Table 1), capable of forming G-quadruplexes were immobilized on the surface of streptavidin-coated magnetic beads; a magnetic field was applied for a certain period of time, causing alignments of magnetic beads; the magnetic field was removed and chains of beads bridged by DNA were observed on a microscope. An example image showing individual bead chains is given in the ESI, Fig. S1.† The average chain length depended on several parameters: the sequence and quantity of DNA, the nature and concentration of salt, and the strength and time duration of the magnetic field applied. In this work, only one parameter was varied at a time. The average chain length was measured for different areas of observation in different experiments and used to quantify the magnetic chain formation.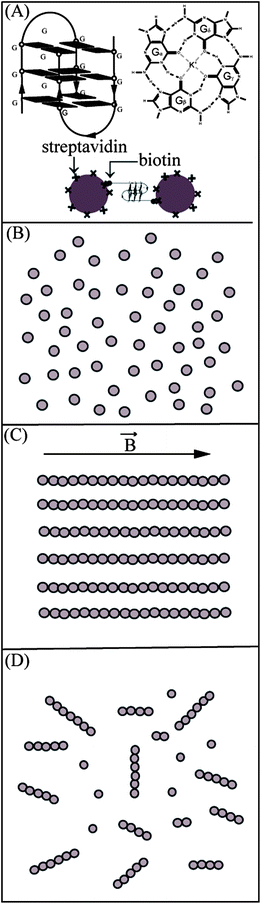 | ||
| Fig. 1 Schematic diagram showing our strategy for the formation of magnetic bead chains bridged by DNA G-quadruplexes. (A) A possible G-quadruplex formed by the two strands of GQ2 with three layers of G-tetrad. The structure of a G-tetrad, with four guanine bases forming hydrogen bonds, is shown. A scheme for the bonding between the beads is provided with streptavidin coated beads and DNA with biotin at the end. (B) Single isolated DNA-coated beads before the application of a magnetic field. (C) Magnetic beads aligned along a straight line under a magnetic field. (D) Permanent bead chains formed after the removal of the field. | ||
| Name | Sequence |
|---|---|
| GQ2 (two-G-tract) | 5′-Biotin-TTTTTTTTTT-GGGTTAGGGT-3′ |
| MQ2 (mutated) | 5′-Biotin-TTTTTTTTTT-GTGTTAGTGT-3′ |
| GQ1 (one-G-tract) | 5′-Biotin-TTTTTTTTTT-AGGGT-3′ |
| GQ3 (three-G-tract) | 5′-Biotin-TTTTTTTTTT-GGGTTAGGGTTAGGGT-3′ |
| GQ4 (four-G-tract) | 5′-Biotin-TTTTTTTTTT-GGGTTAGGGTTAGGGTTAGGGT-3′ |
| G4Q1 (one-G-tract) | 5′-Biotin-TTTTTTTTTT-GGGGT-3′ |
| G4Q1-ΔT (one-G-tract) | 5′-Biotin-TTTTTTTTTT-GGGG-3′ |
| DX (duplex) | 5′-Biotin-TTTTTTTTTT-CGCGATCGCG-3′ |
| T10 | 5′-Biotin-TTTTTTTTTT-3′ |
Magnetic beads coated with the sequence GQ2 were suspended in de-ionised water containing 100 mM of salt (KCl, NaCl or LiCl). After the application of a magnetic field of 30 mT for 20 min, 8 μl of the sample was transferred to a negatively-charged glass well, where the magnetic chains would float over the surface. Typical images of bead chains obtained for different types of salts are displayed in Fig. 2A–C; the distributions of chain lengths in typical images are shown in Fig. 3A–C. The corresponding quantitative analysis of the average chain lengths is plotted in Fig. 4A. In KCl, the average chain length varies from 3.75 to 5 whereas in NaCl, it varies from 2.5 to 4, and in LiCl, it ranges from 1.8 to 2.5. This cation-dependent bead chain formation is consistent with the beads being held together by DNA G-quadruplexes (Fig. 1A), because the stability of G-quadruplexes formed by GQ2 in the presence of different cations would be in the order K+ > Na+ > Li+.5,6 To confirm this, UV melting experiments were performed for the d(GGGTTAGGGT) sequence (i.e. GQ2 without the T10 part) in the presence of different cations (Fig. S2†). A decrease of the 295 nm absorbance with increasing temperature indicated the unfolding of a G-quadruplex.31 The melting temperature of this G-quadruplex at 10 μM DNA strand concentration was estimated to be around 40 and 32 °C in solution containing 100 mM K+ and Na+, respectively, while in Li+ solution the formation of G-quadruplexes was not detected.
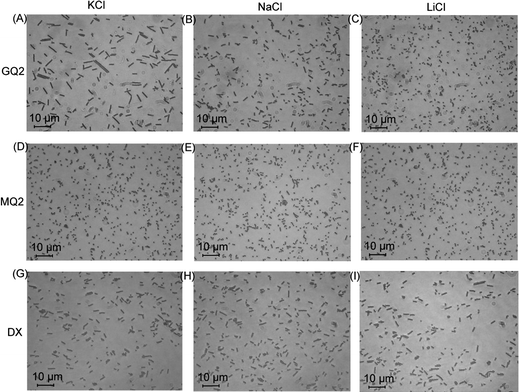 | ||
| Fig. 2 Images showing the chains after the removal of magnetic field under different salts at 100 mM concentration. GQ2 in (A) KCl, (B) NaCl and (C) LiCl. MQ2 in (D) KCl, (E) NaCl and (F) LiCl. DX in (G) KCl, (H) NaCl and (I) LiCl. Magnetic field was applied for 20 min at 30 mT. | ||
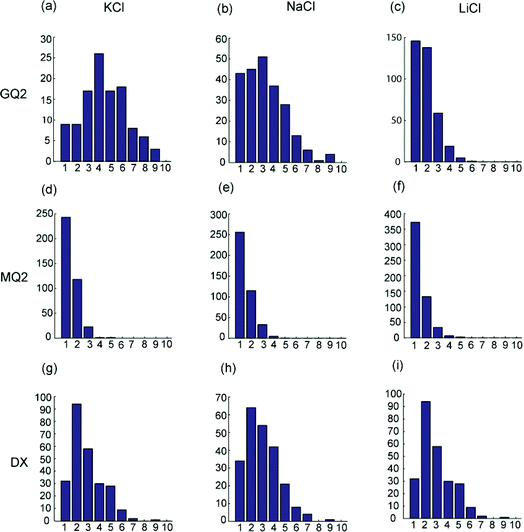 | ||
| Fig. 3 Chain length distributions under different salts at 100 mM concentration. The horizontal axis indicates the length of the chains (in terms of the number of beads) while the vertical axis shows the number of chains in the image. GQ2 in (a) KCl, (b) NaCl and (c) LiCl. MQ2 in (d) KCl, (e) NaCl and (f) LiCl. DX in (g) KCl, (h) NaCl and (i) LiCl. Magnetic field is applied for 20 min at 30 mT. | ||
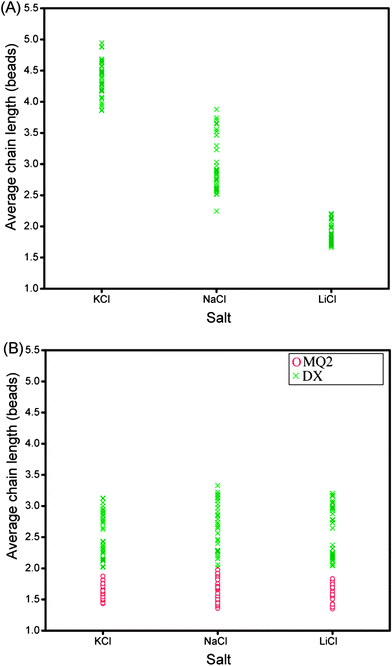 | ||
| Fig. 4 (A) Average chain length as a function of three different salts for GQ2. Each point represents the average length calculated from a particular image. (B) Red circles show the behavior of the mutated sequence (MQ2) in three different salts. Green crosses show the behavior of duplex DNA (DX) under the same salts. The salt concentration was 100 mM and magnetic field was applied for 20 min at 30 mT. | ||
The micrometer-sized magnetic beads were held together by intermolecular hydrogen bonds in the DNA. The process of transferring the sample from an Eppendorf tube to the microscope slide involved forces such as viscous drags, which could have disruptive effects and limit the chain length observed. Although the quantitative results of chain length measurements might somewhat depend on the experimental setup, particularly the sample transfer process, we were able to obtain consistent and reproducible results when repeating each experiment many times with a given protocol.
To further test our assumption that the chains were linked together by DNA G-quadruplexes, we used the MQ2 sequence (Table 1), which differed from the original GQ2 sequence by only one G-to-T substitution in each G-tract. This modification should disrupt the G-quadruplex formation and, indeed, the resulting experiments showed no long chains (Fig. 2D–F, 3D–F, and 4B, red circles).
DNA duplexes, which are widely used in both nanoparticle and microparticle assemblies, do not show the cation specificity as observed for G-quadruplexes. Chain formation mediated by 10-bp DNA duplexes (Fig. 2G–I, 3G–I, and 4B, see figure legends) formed by the self-complementary sequence DX (Table 1) was different from chain formation mediated by DNA G-quadruplexes (Fig. 2A–C, 3A–C, and 4A). In the case of duplex DNA, the average chain length was independent of the nature of cations and was found to be shorter than that mediated by G-quadruplexes in K+ solution.
Effect of the magnetic field strength
The number of successful bonds formed between beads might depend on the distance between them, which in turn could be controlled by adjusting the magnetic field strength when beads align in the direction of field lines. Here we study this effect by measuring the chain length as a function of the magnetic field strength applied. The GQ2 coated beads were subjected to a magnetic field of 5 to 50 mT in the presence of 100 mM KCl (Fig. 5 and S3†). The duration of the application of the magnetic field was 20 min, sufficient for the formation of a good number of bonds (see below). As evident from Fig. 5, even at the lowest field of 5 mT, we observed a good number of chains with the average length ranging from 2.5 to 3.5 beads. The average of the average chain length was increased up to 4 at a slightly higher field of 10 mT, and up to 4.5 at the field strength of 20 mT. From 30 to 50 mT, the average chain length remained nearly the same indicating saturation.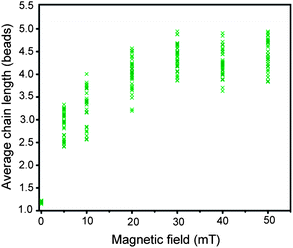 | ||
| Fig. 5 Average chain length as a function of magnetic field strength for GQ2 in the presence of 100 mM KCl. Magnetic field was applied for 20 min. | ||
Effect of the salt concentration
Previously, the effect of salt concentration on DNA-mediated colloidal assemblies has been studied primarily through the size or the melting temperature of the resulting aggregates formed.17,18 In most cases, these two quantities increase with the increase in salt concentration. In this study, the effect of salt on the length of bead chains was investigated. The chain formation of beads coated with GQ2 was performed at a magnetic field of 30 mT for 20 min in a solution containing 10 to 100 mM KCl. At low K+ concentration, the system was characterized by a large number of single beads with only a few small chains; the average chain length increased when the K+ concentration increased (Fig. 6 and S4†). The chain length showed a gradual increase when the K+ concentration increased from 10 to 60 mM, but nearly saturated from 60 to 100 mM (Fig. 6).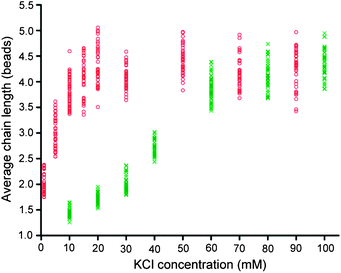 | ||
| Fig. 6 Average chain length as a function of KCl concentration for GQ2. Green crosses represent the effect of KCl alone while red circles represent the combined effect of KCl and LiCl. The total concentration of KCl and LiCl was 100 mM. Magnetic field was applied for 20 min at 30 mT. | ||
There could be two contributions for the increase in chain length: (i) the stability of the DNA G-quadruplex structures increased with the increase in K+ concentration and (ii) the repulsion between the beads decreased with the increase in salt concentration, bringing the beads closer and leading to an increase in the number of bonds. In experiments involving DNA duplexes, these effects are difficult to distinguish. However, given that Li+ could reduce the charge on the particles, hence, reducing the repulsion between the beads, while having little effect on G-quadruplex formation, we were able to distinguish between these two effects. Experiments were conducted in a mixture of different concentrations of K+ and Li+, while keeping the total salt concentration at 100 mM. The average chain length is plotted as a function of K+ concentration in Fig. 6 (red circles), with the saturated average chain length being achieved at 20 mM K+ and 80 mM Li+. Under this condition, K+ was mainly responsible for the stability of the DNA structure, while Li+ helped in reducing the charge on the particles and the repulsion between them.
Effect of the formation time
We investigated the effect of the time of application of a magnetic field on the chain formation for the sequence GQ2 (the magnetic field was kept at 30 mT, KCl concentration was 100 mM) (Fig. 7 and S5†). At 10 s, most of the beads were single with a few chains. After that, we observed a continuous increase in the average chain length which could be attributed to the gradual formation of multiple G-quadruplexes between the beads. The average chain length further increased for the time up to 5 minutes, and then reached a plateau. Fitting the average chain length as an exponential function of time gives a time constant of about 2 minutes, indicating a characteristic time necessary for the beads to come closer and form bonds. The experiment is repeated for the duplex forming sequence DX under the same conditions and results are represented by red circles in Fig. 7. Apart from a shorter chain length observed, the data show similar behaviors.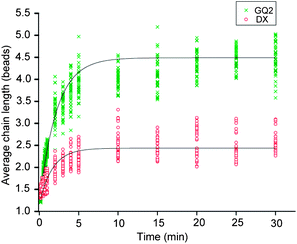 | ||
| Fig. 7 Average chain length as a function of time duration of the magnetic field for GQ2 (green crosses) and DX (red circles) in 100 mM KCl. The time constant for the chain formation as fitted by a single exponential is ∼2 min for both cases. The strength of the applied magnetic field was 30 mT. | ||
Effect of the G-DNA surface density
Another way of tuning the strength of interaction between the beads is to control the number of interacting DNA molecules between them. This can be achieved by diluting G-quadruplex-forming DNA on the surface of beads with a different DNA sequence which does not form any stable intermolecular structures. The number of bonds formed could be altered by changing the ratio between the two sequences. This way of controlling is relevant in situations where the use of constant environmental factors is needed.In this section, we study the effect of the surface density of G-quadruplex-forming DNA (G-DNA) concentration on the stability of the chains. Here we dilute the G-DNA (GQ2) with another sequence, T10 (Table 1), which does not form any stable intermolecular structures. The total amount of DNA used for immobilization on the bead surface was kept fixed, while the concentration ratio of GQ2 to T10 was varied. The same ratio between sequences would be attached on the surface of the beads. Here the role of T10 was to increase the separation between GQ2 molecules on the surface. Even at the fraction of 0.1 for G-DNA, there were already a number of short chains (Fig. S6†). The average chain length increased when the fraction of G-DNA increased and saturated when this fraction reached 0.6 (Fig. 8).
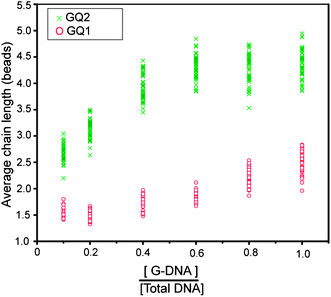 | ||
| Fig. 8 Average chain length as a function of G-DNA density on the surface of beads. Green crosses represent the data for GQ2 and red circles represent GQ1 in 100 mM KCl. Magnetic field was applied for 20 min at 30 mT. | ||
To make sure that bead surfaces were maximally covered (or saturated) with DNA, we performed an additional experiment with a doubled amount of DNA normally used for immobilization at 20% of G-DNA and 80% of T10, where the chain length was not yet saturated. We obtained an average chain length ranging from 2.80 to 3.40, which was the same as in the original case (Fig. S7†), confirming the saturation of DNA on the surface of beads.
On the other hand, the GQ1 sequence, which requires four strands to come together to form a G-quadruplex, leads to shorter chains and shows weaker dependence on the surface DNA density (Fig. 8 and S8†). From the G-DNA fraction of 0.1 to 0.6, the average chain length was short and remained nearly unchanged. At the G-DNA fractions of 0.8 and 1.0, a small increase in the chain length was observed. The reasons, for this short chain length, are discussed in the next section.
Effect of the DNA sequence
In contrast to duplex DNA, G-quadruplex structures could be of different topologies. In this section, we are comparing four different sequences which have the potential to form different structures. The four sequences, GQ1 (one-G-tract), GQ2 (two-G-tract), GQ3 (three-G-tract), and GQ4 (four G-tract), contain one, two, three, and four (GGG) tracts, respectively (Table 1), and have the ability to form tetrameric, dimeric, dimeric, and monomeric G-quadruplexes, respectively, in solution.32–35 However, it is possible that some of them could form more than one structure, e.g. the four-G-tract sequence can form monomeric, dimeric or tetrameric G-quadruplexes.Experiments were conducted in the presence of 100 mM KCl and a magnetic field was applied for 20 min at 30 mT. Results are summarized in Fig. 9 and S9.† The chains formed by the GQ1 and GQ4 are the shortest with the average chain length ranging from 2.25 to 3.25 approximately. The chains formed by the GQ2 sequence are the longest with the average length ranging from 4 to 5, followed by GQ3 whose average length varies from 3.5 to 4. This could be partially understood based on the nature of the G-quadruplexes formed by these sequences in solution. GQ1 needs four strands to come together to form a G-quadruplex, limiting the maximum number of G-quadruplexes that could be formed between two beads. A single strand of GQ4 has the ability to form an intramolecular G-quadruplex on itself, inhibiting the formation of inter-bead G-quadruplexes. In the cases of GQ2 and GQ3, the structures formed could be different,33,34 even though both have the ability to form a structure from two strands.
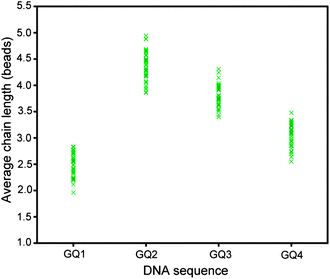 | ||
| Fig. 9 Average chain length observed for sequences with different number of G-tracts in 100 mM KCl. Magnetic field was applied for 20 min at 30 mT. | ||
The stability of G-quadruplex can be greatly affected by even small changes in the sequence and this can be detected using our method. To illustrate this, we performed experiments with three different DNA sequences: the G4Q1 sequence (5′-T10-GGGGT-3′) has four guanines in the G-tract, while the GQ1 sequence (5′-T10-AGGGT-3′) has only three; the G4Q1-ΔT sequence (5′-T10-GGGG-3′) differs from G4Q1 by a deletion of the thymine at the 3′ end. The average chain length for G4Q1 ranges from 3 to 3.7, as compared to the range of 2 to 3 observed for GQ1 (Fig. 10). The presence of an extra G in the G-tract could lead to an increase in the number G-tetrads formed, which might be responsible for the increase in the chain length. The higher average chain length (3.7 to 5) observed for G4Q1-ΔT as compared to that of G4Q1 might be due to additional stacking or interlocking interactions arising from the removal of the thymine at the 3′ end.
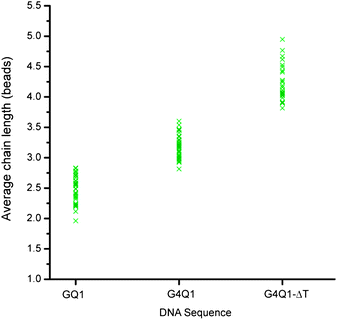 | ||
| Fig. 10 Average chain length observed for three different DNA sequences GQ1, G4Q1, and G4Q1-ΔT (Table 1) in 100 mM KCl. Magnetic field was applied for 20 min at 30 mT. | ||
Conclusions
We have successfully formed DNA G-quadruplexes between micrometer-sized beads for the first time, as demonstrated by the formation of magnetic bead chains. This DNA structure offers a wide range of possibilities for tuning the strength of interaction through the use of a comprehensive list of parameters. The average chain length was used to quantify the chain formation process, which depended on the nature and concentration of cations, DNA sequences, the time duration and strength of the applied magnetic field. These results indicate that G-quadruplexes are good candidates for both micro- and nano-particle assemblies. Our G-quadruplex-mediated particle-connection approach can also be applied to characterize G-quadruplex ligands.Methods
Chemicals and DNA
The magnetic beads used were 1.05 μm polystyrene Dynabeads (MyOne Streptavidin C1) purchased from Invitrogen. These polystyrene particles were embedded with magnetic material inside and functionalized with streptavidin on the surface through covalent coupling. Different salts were purchased from either Sigma or Merck and used for making solutions of the required ionic strength. Biotinylated DNAs were purchased from 1st BASE in dried form and diluted with de-ionised water to obtain the required concentration. All DNA sequences (Table 1) have a continuous stretch of ten thymines after biotin, used to separate the recognition part of DNA molecules away from the bead surface, thereby, preventing their interactions with the surface.Attachment of DNA
Typically, 6 μl of the original bead solution, expected to contain 6 × 107 beads, was used. The beads were washed with de-ionised water to remove the surfactant and other preservatives, and reduced to minimum volume. Based on the manufacturer's manual (each bead can attach to up to 106 small oligonucleotides), we used 12 μl of 10 μM DNA solution for 6 μl of beads. To facilitate the binding of biotin to streptavidin, 100 mM of NaCl was added, which could be removed in the later stages. After adding the DNA, the sample was gently shaken for 1 hour continuously. After that, it was washed two times using de-ionised water to remove salt and excess DNA which was not attached to beads. The DNA coated beads were then suspended in 360 μl water to make the required concentration.Formation of bead chains
60 μl of the above solution was taken in an Eppendorf tube and a required amount of salt was added. Immediately, it was placed at the centre of two square Nd–Fe–B magnets whose magnetic field was measured using a Gauss meter. The magnetic field was applied at different field strengths by varying the distance between the magnets and for different time durations. After that, the magnetic field was removed and the solution was gently shaken to disperse the beads uniformly. This process could affect the length of the chains by breaking the weak links in the chains, thereby, leading to shorter chains than originally formed. 8 μl of the resulting solution was transferred to a negatively-charged glass well for observation in a Nikon TE2000-U inverted microscope. Each experiment was repeated four times and 10 images were taken from each experiment.Imaging and processing
Images were captured using the Nikon NIS-elements software and were processed using the National Instruments Vision Assistant software. This created a table containing information such as area for each chain in the image. This table was processed using a program written in Matlab giving the length of different chains in the image which was used for calculating the average chain length corresponding to a particular image. This was repeated for 40 pictures from 4 identical experiments and plotted in the graph.UV melting experiments
The stability of G-quadruplexes was measured in UV melting experiments conducted on a JASCO V-650 spectrophotometer. Absorbance at 295 nm was recorded as a function of temperature from 90 to 20 °C. Heating and cooling were done at the rate of 0.2 °C min−1. Experiments were performed in quartz cuvettes, with 1 cm path length. The concentration of DNA was 10 μM and the solution contained 100 mM respective chloride salt.Acknowledgements
This research was supported by Nanyang Technological University grants to A.T.P.References
- J. T. Davis, Angew. Chem., Int. Ed., 2004, 43, 668–698 CrossRef CAS.
- D. J. Patel, A. T. Phan and V. Kuryavyi, Nucleic Acids Res., 2007, 35, 7429–7455 CrossRef CAS.
- S. Neidle, Curr. Opin. Struct. Biol., 2009, 19, 239–250 CrossRef CAS.
- A. T. Phan, FEBS J., 2010, 277, 1107–1117 CrossRef CAS.
- D. Sen and W. Gilbert, Nature, 1990, 344, 410–414 CAS.
- C. C. Hardin, T. Watson, M. Corregan and C. Bailey, Biochemistry, 1992, 31, 833–841 CrossRef CAS.
- A. P. Alivisatos, K. P. Johnsson, X. G. Peng, T. E. Wilson, C. J. Loweth, M. P. Bruchez and P. G. Schultz, Nature, 1996, 382, 609–611 CrossRef CAS.
- C. A. Mirkin, R. L. Letsinger, R. C. Mucic and J. J. Storhoff, Nature, 1996, 382, 607–609 CrossRef CAS.
- R. C. Jin, G. S. Wu, Z. Li, C. A. Mirkin and G. C. Schatz, J. Am. Chem. Soc., 2003, 125, 1643–1654 CrossRef CAS.
- D. Nykypanchuk, M. M. Maye, D. van der Lelie and O. Gang, Nature, 2008, 451, 549–552 CrossRef CAS.
- S. Y. Park, A. K. R. Lytton-Jean, B. Lee, S. Weigand, G. C. Schatz and C. A. Mirkin, Nature, 2008, 451, 553–556 CrossRef CAS.
- R. J. Macfarlane, B. Lee, M. R. Jones, N. Harris, G. C. Schatz and C. A. Mirkin, Science, 2011, 334, 204–208 CrossRef CAS.
- Z. Li and C. A. Mirkin, J. Am. Chem. Soc., 2005, 127, 11568–11569 CrossRef CAS.
- F. Seela, S. Budow and P. Leonard, Org. Biomol. Chem., 2007, 5, 1858–1872 RSC.
- W. X. Wang, H. J. Liu, D. S. Liu, Y. R. Xu, Y. Yang and D. J. Zhou, Langmuir, 2007, 23, 11956–11959 CrossRef CAS.
- C. M. Soto, A. Srinivasan and B. R. Ratna, J. Am. Chem. Soc., 2002, 124, 8508–8509 CrossRef CAS.
- V. T. Milam, A. L. Hiddessen, J. C. Crocker, D. J. Graves and D. A. Hammer, Langmuir, 2003, 19, 10317–10323 CrossRef CAS.
- P. H. Rogers, E. Michel, C. A. Bauer, S. Vanderet, D. Hansen, B. K. Roberts, A. Calvez, J. B. Crews, K. O. Lau, A. Wood, D. J. Pine and P. V. Schwartz, Langmuir, 2005, 21, 5562–5569 CrossRef CAS.
- P. L. Biancaniello, A. J. Kim and J. C. Crocker, Phys. Rev. Lett., 2005, 94, 058302 CrossRef.
- M. P. Valignat, O. Theodoly, J. C. Crocker, W. B. Russel and P. M. Chaikin, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 4225–4229 CrossRef CAS.
- A. J. Kim, P. L. Biancaniello and J. C. Crocker, Langmuir, 2006, 22, 1991–2001 CrossRef CAS.
- R. Dreyfus, M. E. Leunissen, R. Sha, A. V. Tkachenko, N. C. Seeman, D. J. Pine and P. M. Chaikin, Phys. Rev. Lett., 2009, 102, 048301 CrossRef.
- D. C. Li, J. Rogers and S. L. Biswal, Langmuir, 2009, 25, 8944–8950 CrossRef CAS.
- M. E. Leunissen, R. Dreyfus, F. C. Cheong, D. G. Grier, R. Sha, N. C. Seeman and P. M. Chaikin, Nat. Mater., 2009, 8, 590–595 CrossRef CAS.
- M. E. Leunissen, R. Dreyfus, R. J. Sha, T. Wang, N. C. Seeman, D. J. Pine and P. M. Chaikin, Soft Matter, 2009, 5, 2422–2430 RSC.
- W. B. Rogers and J. C. Crocker, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 15687–15692 CrossRef CAS.
- F. L. Calderon, T. Stora, O. Mondain Monval, P. Poulin and J. Bibette, Phys. Rev. Lett., 1994, 72, 2959–2962 CrossRef CAS.
- A. Koenig, P. Hebraud, C. Gosse, R. Dreyfus, J. Baudry, E. Bertrand and J. Bibette, Phys. Rev. Lett., 2005, 95, 128301 CrossRef CAS.
- E. M. Furst, C. Suzuki, M. Fermigier and A. P. Gast, Langmuir, 1998, 14, 7334–7336 CrossRef CAS.
- L. Cohen-Tannoudji, E. Bertrand, L. Bressy, C. Goubault, J. Baudry, J. Klein, J. F. Joanny and J. Bibette, Phys. Rev. Lett., 2005, 94, 038301 CrossRef.
- J. L. Mergny, A. T. Phan and L. Lacroix, FEBS Lett., 1998, 435, 74–78 CrossRef CAS.
- Y. Wang and D. J. Patel, Biochemistry, 1992, 31, 8112–8119 CrossRef CAS.
- A. T. Phan and D. J. Patel, J. Am. Chem. Soc., 2003, 125, 15021–15027 CrossRef CAS.
- N. Zhang, A. T. Phan and D. J. Patel, J. Am. Chem. Soc., 2005, 127, 17277–17285 CrossRef CAS.
- K. N. Luu, A. T. Phan, V. Kuryavyi, L. Lacroix and D. J. Patel, J. Am. Chem. Soc., 2006, 128, 9963–9970 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c2sm26652k |
| This journal is © The Royal Society of Chemistry 2013 |