 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
One pathway, many compounds: heterologous expression of a fungal biosynthetic pathway reveals its intrinsic potential for diversity†
Zahida Wasil‡a, Khomaizon A. K. Pahirulzaman‡b, Craig Buttsa, Thomas J. Simpsona, Colin M. Lazarus*b and Russell J. Cox*ac
aSchool of Chemistry, University of Bristol, Cantock's Close, Bristol BS8 1TS, UK. E-mail: r.j.cox@bris.ac.uk; Fax: +44 (0) 117 925 1295; Tel: +44 (0) 117 928 9184
bSchool of Biological Sciences, University of Bristol, Woodland Road, Bristol, BS8 1UG, UK. E-mail: C.M.Lazarus@bristol.ac.uk
cInstitut für Organische Chemie, Leibniz Universität Hannover, Schneiderberg 1B, 30167 Hannover, Germany. E-mail: russell.cox@oci.uni-hannover.de
First published on 8th August 2013
Abstract
Heterologous expression of genes from the proposed aspyridone biosynthetic cluster from Aspergillus nidulans in the host Aspergillus oryzae led to the production of eight different compounds in addition to aspyridone A 1, one of the previously observed products. The pathway genes were incapable of producing aspyridone B 2, the previously accepted final product of the pathway. Expression of restricted sets of genes in addition to the core polyketide synthase – nonribosomal peptide synthetase (PKS-NRPS) genes apdA and apdC revealed: that apdE encodes a cytochrome P450 enzyme with ring-expanding and unprecedented dephenylation activity; that apdB encodes an N-hydroxylase, an activity not previously suspected; that the productivity of ApdA and ApdC proteins appears to be significantly enhanced in the presence of the downstream ApdE oxidase; and no obvious chemical roles for ApdD and ApdG. Furthermore, the ApdC enoyl reductase appears to operate with different stereoselectivity in different PKS cycles. All of these features illustrate the inherent diversity of compounds potentially produced by the apd pathway and the high utility of a whole pathway expression strategy for investigating and revealing new biosynthetic chemistry in fungi.
Introduction
The advent of rapid and economical genome sequencing of bacteria and fungi has revealed thousands of biosynthetic gene clusters encoding the synthesis of secondary metabolites. In the majority of cases the gene clusters can be rapidly annotated (e.g. using bioinformatic tools such as AntiSmash1) and the class of compound they specify can be identified, but it is not yet possible to predict the precise structure of the encoded metabolite for any given natural product biosynthetic gene cluster. Fungi, in particular, present a problem because they can often contain more than 50 biosynthetic gene clusters and because the core synthases2 and synthetases3 are often iterative, displaying complex and cryptic programming. Gene clusters which have not been linked to the biosynthesis of a particular compound are referred to as orphan clusters. The vast majority of fungal biosynthetic gene clusters currently fall into this category.Attempts have been made to link fungal biosynthetic gene clusters with particular compounds. For example knockout or silencing strategies can quickly achieve this if the compound of interest is observable.4–6 However most fungal biosynthetic gene clusters appear to be silent – that is, not transcribed at a sufficient level to produce an observable chemical product. In the case of such silent clusters, knockout and silencing strategies cannot be used. A further significant problem lies in the complex way in which fungal gene clusters are regulated.7 Fungi are eukaryotes and thus each structural gene (for example encoding an individual biosynthetic enzyme) is controlled by its own promoter. These promoters are activated under unique (often unknown) circumstances.
Some silent fungal biosynthetic gene clusters encode a transcriptional regulator. In a limited number of cases, for example that of the aspyridone A 1 biosynthetic gene cluster in Aspergillus nidulans, it has proven possible to ‘switch on’ biosynthesis by overexpressing this transcription factor.8 This has the effect of activating the promoters of the biosynthetic genes and thus causing transcription of the structural genes, in turn resulting in the production of the biosynthetic proteins themselves and the synthesis of a new compound. However, because not all silent biosynthetic gene clusters contain a transcription factor this method is limited.
Furthermore, switching on a range of genes simultaneously does not allow the effect (or the chemical programme) of each individual biosynthetic protein to be observed, and it is also possible that the induction of transcription factors may activate unknown genes outside the biosynthetic gene cluster, producing unpredictable chemical effects. Numerous other methods of activating silent fungal gene clusters have been reported, including chemical epigenetic modification,9 manipulation of signalling cascades10 and alteration of growth conditions (e.g. the OSMAC11 approach),12 but none are systematic, general, widely applicable in a range of fungi or, most crucially, predictably applicable to a gene cluster of interest.
We have been seeking to develop a systematic and general method for investigating the biosynthetic chemistry of fungal gene clusters based on heterologous expression – i.e. rapidly moving genes into a tractable host organism in a cumulative manner and observing chemical changes. We recently reported the development of a generally applicable plasmid system in which structural biosynthetic genes can be cloned downstream of strong promoters, thus overcoming the cryptic native transcription systems.13 The cloning vectors (Fig. 1A) feature a fungal selection marker (e.g. argB14 for use in the arginine auxotroph of Aspergillus oryzae M-2-3) as well as a Gateway cassette downstream of the strong inducible amyB promoter (PamyB) into which large fungal synth(et)ase genes can be rapidly cloned using in vitro recombination. The vector also contains colE1 and ampR for replication and selection in E. coli and a 2μ origin of replication and URA3 for selection in yeast. Three further strong fungal promoters (Padh, PgpdA and Peno) are also included so that up to three tailoring genes can be expressed in parallel from the same system (Fig. 1B). Here we demonstrate how this system can be used to obtain detailed chemical information from a silent fungal gene cluster and provide insights into new and unprecedented reactivity.
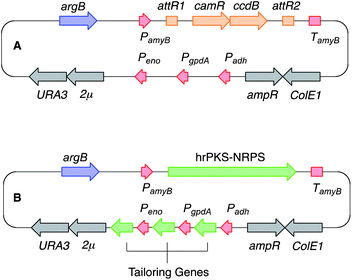 | ||
| Fig. 1 Schematic representation of the fungal expression vector system used in this study. See text for abbreviations. (A) Native vector; (B), with genes for expression; red, fungal promoter and terminator sequences; blue, fungal selection marker; grey, genes for use in yeast and E. coli; orange, in vitro recombination cassette; green, biosynthetic genes. | ||
Results
Hertweck and coworkers reported the discovery of a potential biosynthetic gene cluster in the filamentous fungus A. nidulans centred on a large open reading frame (apdA) encoding an iterative highly-reducing polyketide synthase (hrPKS) fused to a single module nonribosomal peptide synthetase (NRPS).8 Such hrPKS-NRPS genes, first described for fusarin C15 are now known to be a common feature in the genomes of many fungi.16 Also clustered with apdA were seven other ORFs encoding cytochrome P450 enzymes (apdB and apdE), an enoyl reductase (apdC), a FAD-dependent monooxygenase (apdD), an exporter (apdF), a dehydrogenase (apdG) and a transcription factor (apdR). Ectopic expression of apdR downstream of the inducible alcohol dehydrogenase promoter (PalcA) in the A. nidulans strain SB4.1 led to the production of 1 and 2 and the up-regulation of genes apdA, apdB, apdC, apdD, apdE and apdG implicating these in biosynthesis. While these results showed that the apd cluster was most likely involved in aspyridone biosynthesis the precise chemical steps and intermediates could not be established. More recently Tang and coworkers proved that apdA and apdC encode proteins which work together to form the precursor preaspyridone A by in vitro reconstitution of the proteins,17 but these experiments did not reveal information about the later steps of biosynthesis. We reasoned that the apd cluster would make a useful testbed for our heterologous expression technology, and the results could also allow us to delineate the precise chemical intermediates and steps of biosynthesis (Fig. 2).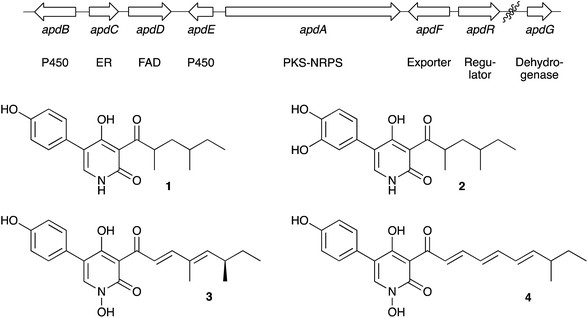 | ||
| Fig. 2 Aspyridone A 1 and B 2, related 2-pyridones tenellin 3 and desmethylbassianin 4 and the proposed apd biosynthetic gene cluster. | ||
The apdA gene encodes a hrPKS-NRPS. In the case of many fungal hrPKS-NRPS it is known that the enoyl reductase (ER) domain is catalytically inactive (for example the cases of tenellin 3 and desmethylbassianin 4).18 Vederas and coworkers have also demonstrated similar behaviour in the hrPKS system responsible for lovastatin biosynthesis.19 In all these cases the missing ER functionality is supplied by a trans-acting protein usually encoded within the same gene cluster as the PKS itself. The apdA-encoded hrPKS-NRPS falls into this category and the apdC gene is known to encode the trans-acting ER.17 Because hrPKS lacking functional ER domains are known to function poorly and erratically on their own20 we expressed apdA and apdC together in the fungal host Aspergillus oryzae. Transformants and untransformed controls were grown in nutrient broth medium, acidified, extracted with EtOAc, and concentrated residues were examined by LCMS. Three new peaks were observed in transformants as compared to controls. These compounds were purified by automated mass-directed reverse-phase chromatography (Table 1).
| Transformant (total crude) | 1 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | Total yield | Approx. fold increase |
|---|---|---|---|---|---|---|---|---|---|---|---|
| apdAC (430) | — | 60.7 | 0.6 | tr. | — | — | — | — | — | 61.3 | — |
| apdACE (1126) | 119.8 | — | — | 15.0 | 183.6 | 72.2 | — | — | — | 390.6 | 6.5 |
| apdABC (805) | — | 141.0 | 2.2 | tr. | — | — | — | — | — | 143.2 | 2 |
| apdAC + tenA (653) | — | 121.0 | 2.2 | tr. | — | — | — | — | — | 123.2 | 2 |
| apdACEB (300) | 1.8 | 34.6 | 5.7 | tr. | 2.6 | — | 6.2 | 2.7 | 4.0 | 57.6 | 1 |
| apdACED (448) | 5.1 | — | 0.3 | 8.2 | 49.9 | — | — | 8.0 | 71.5 | 1 | |
| apdACEBG (622) | 12.6 | 98.0 | — | tr. | 42.6 | — | 15.8 | 8.2 | — | 177.2 | 3 |
The major component (61 mg L−1) was shown to possess molecular formula C19H25NO4 by HRMS (observed 332.1852; calculated 332.1856 for M[H]+). 1H NMR indicated the presence of an aliphatic group consistent with the 2,4-dimethylhexanoyl sidechain of aspyridone and HSQC and HMBC spectra confirmed the product to be the expected 5 based on the in vitro work of Tang and coworkers.17 The minor second component (6, 0.6 mg L−1) possessed an identical molecular formula and a near identical set of NMR data to 5. 1H NMR in CDCl3 revealed fractional differences (<0.05 ppm) in chemical shifts for the side-chain protons, but 1H NMR in DMSO-d6 revealed a significant difference in the coupling between methine H-5 and methylene H-14 protons consistent with 5 and 6 being epimeric at C-5. Minor component 6 forms crystals on standing, while 5 does not, and X-ray analysis of 6 showed it to be the 7,9-anti isomer. Previous papers have shown a syn-dimethyl side chain for aspyridone. Finally, a minor component with formula C19H25NO3 (observed m/z 338.1735, calculated 338.1727 for [M]Na+) was observed in low yield. The same compound was produced in higher titre in a subsequent experiment and NMR analysis of this showed it to be the non-hydroxylated analogue 7.
In a second experiment apdA, apdC and apdE were coexpressed. The apdE gene encodes a cytochrome P450 enzyme, but its function is unknown. Transformants expressing these three genes produced three new compounds. The major component (184 mg L−1) possessed a molecular formula of C19H25NO5 – a single oxygen more than 5 (observed HRMS 370.1625, calculated 370.1625 for [M]Na+). 1D and 2D NMR analysis showed this compound to be 14-hydroxypreaspyridone A 8. The second major compound (120 mg L−1) had a molecular formula of C19H23NO4 (observed HRMS 330.1692, calculated 330.1700 for [M]H+) and a distinctive 2-pyridone proton in the 1H NMR. Inspection of the 1D and 2D NMR, and comparison with the literature values, showed this to be aspyridone A 1 itself. Crystals of 1 were prepared and the X-ray structure showed that the side-chain pendant methyls matched those of 6 in being arranged anti. The third compound (72 mg L−1) recovered from this experiment had a molecular formula of C13H19NO3 (observed HRMS 238.1429, calculated 238.1438 for [M]H+), corresponding to the loss of phenoxide from 1. This was confirmed by 1D and 2D NMR and the structure was shown to be the dephenylated 2-pyridone 9. Crystals of 9 could be prepared, and the X-ray crystal structure confirmed the structure determined by NMR and again showed an anti arrangement of the side-chain pendant methyl groups (Fig. 3). Experiments to determine the origin of the amino acid component of aspyridone A 1 were conducted by supplementing growing cultures of A. oryzae + apdACE with [1-13C]-tyrosine during the fermentation. Isolated aspyridone A 1 was specifically 13C-labelled at C-4 (5% incorporation), while isolated 9 was also labelled at C-4 (13% incorporation).
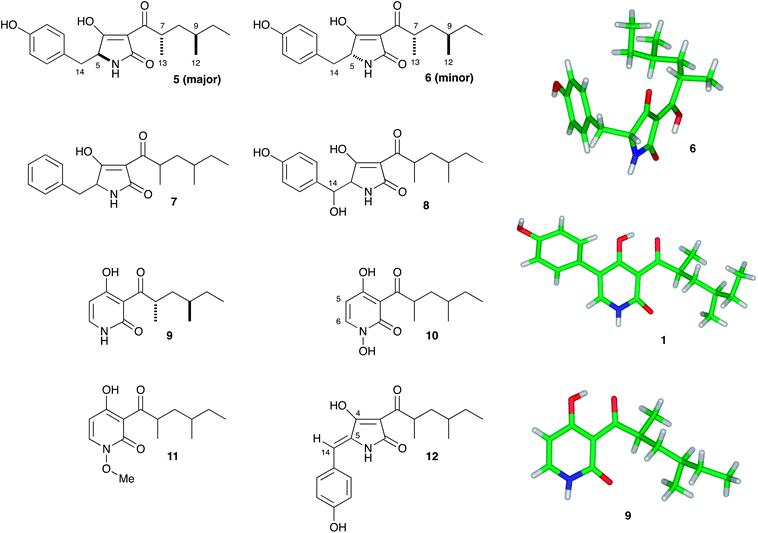 | ||
| Fig. 3 Compounds produced from heterologous expression experiments. | ||
Next, apdA, apdC and apdB were coexpressed. Once again apdB encodes a cytochrome P450 monooxygenase with no known function. In this experiment the same three compounds were produced as by expression of apdA and apdC alone, although the overall titre was around 2.5-fold higher. The gene tenA also encodes a cytochrome P450 enzyme known to be involved in the oxidative ring expansion of acyl tetramic acids to 2-pyridones. However, expression of tenA with apdA and apdC led to the production of the same compounds as the expression of apdA and apdC alone, although again, the overall titre was around 2-fold higher than expression of the two genes alone.
Next, a series of 4-way coexpression experiments were performed. We reasoned that apdB might function after apdE and so in the first of these experiments apdA, apdC and apdE were coexpressed with apdB. This resulted in many of the previously-observed compounds being produced, together with three new compounds in low titre. The major of these 10 (6 mg L−1) had a molecular formula of C13H19NO4 (observed HRMS of 276.1219, calculated 276.1206 for [M]Na+), one oxygen more than the dephenylated 2-pyridone 9. 1H NMR indicated that none of the C–H protons of 9 were missing, and significant chemical shift perturbations of H-5 and H-6 showed that the oxidation must be on the pyridone nitrogen. This conclusion was confirmed by 2D NMR. The second compound 11 (3 mg L−1) had a similar 1H NMR to 10 featuring an additional 3-proton singlet at 3.93 ppm. HRMS indicated a molecular formula of C14H22NO4 consistent with a methylation of 10 (observed HRMS of 290.1368, observed 290.1363 for [M]Na+). Further 1D and 2D NMR experiments indicated that the new methyl was attached to the N-hydroxyl.
Finally another minor compound was purified (4 mg L−1). This proved to be the dehydrated compound 12 (observed HRMS of 352.1528, calculated 352.1520 for [M]Na+). In order to determine the geometry of the C-5/C-14 olefin of 12 several methods were attempted. Although crystals of 12 could be prepared, they did not diffract X-rays sufficiently well to be able to determine the structure. NOESY and ROESY NMR experiments were also performed. These suggested that the NH was proximal to the phenyl protons but the exchangeability of the NH meant that unambiguous evidence for the geometry could not be gained this way. Finally, the 3JHC coupling constant between H-14 and C-4 was measured using an EXSIDE experiment.21 The observed value of 3.5 Hz is consistent with a cis relationship of these atoms, and DFT calculations of the 3JHC values for the two possible E/Z isomers of 12 reinforced this conclusion with calculated values of 4.5 Hz and 7.5 Hz for the cis and trans geometries respectively.
The apdD gene encodes an FAD-dependent monooxygenase and Hertweck and coworkers speculated that this might be involved in hydroxylation of aspyridone A 1 to form aspyridone B 2. We thus expressed apdA, apdC and apdE along with apdD. Once again this experiment resulted in no new compounds, and the overall titre was again lowered, this time to ca 70 mg L−1.
The role of apdG was investigated in a five-way experiment featuring expression of apdA, apdC and apdE (which make aspyridone A 1) along with apdB and apdG. This experiment was achieved by expressing apdABCE as previously described, and introducing a second vector carrying apdG driven by PgpdA. This vector carries a selection marker consisting of the bleomycin resistance gene ble downstream of PtrpC. However, this experiment did not result in the production of any new compounds, giving approximately the same distribution of compounds as expression of apdABCE alone.
Finally we compared the productivity of the A. nidulans SB4.1 strain with that of the A. oryzae apdACEB strain. A. nidulans SB4.1 was grown in fermentation broth and extracted with EtOAc (affording 1069 mg L−1 of crude extract). LCMS analysis was performed and the results compared to the A. oryzae extracts. Comparison of the LCMS traces showed that in our hands the A. nidulans SB4.1 strain produced a similar range of compounds to the A. oryzae apdACEB strain. The major components were the two preaspyridone A diastereomers 5 and 6, with lesser amounts of the 14-hydroxy compound 8 and the anhydro compound 12. Hertweck and coworkers reported aspyridone A 1 as the major component, but in our hands the A. nidulans SB4.1 strain produced this compound in low titre, and did not appear to produce aspyridone B 2 at all. The dephenylated compounds were also not observed by us or the previous investigators in the SB4.1 strain (Fig. 4).
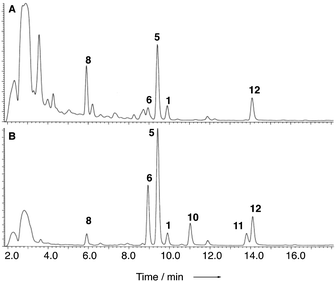 | ||
| Fig. 4 Comparison of the productivity of A. nidulans SB4.1 (trace A) and A. oryzae apdACEB (trace B). | ||
Discussion
Combinations of the putative aspyridone biosynthetic genes and a heterologous combination of aspyridone genes with tenA from the tenellin biosynthetic cluster were easily constructed in our expression vector. Transcription of each gene was confirmed by qRT-PCR,† confirming our previous observation that all promoters are active in this system.13 Expression of apdA and apdC alone led to the formation of preaspyridone A diastereomers 5 and 6 in agreement with the in vitro results of Tang and coworkers.17 Similar biosynthetic chemistry has been shown to operate during the biosynthesis of pretenellin A 13 and many other related compounds including the cytochalasans.22,23 In brief, the hrPKS-NRPS builds a polyketide, in this case a dimethyltetraketide which is linked to L-tyrosine by the NRPS, which then catalyses a Dieckmann cyclisation to form the acyl tetramic acid ring and release it from the synthetase.20,24 In the case of the aspyridone synthetase Tang and coworkers have shown in in vitro experiments that the NRPS module displays extended substrate selectivity, utilising L-phenylalanine, L-p-fluorophenylalanine and L-tryptophan in addition to L-tyrosine.17 Our results tend to confirm this observation, insofar as the formation of 7 is most easily explained as the result of utilisation of phenylalnine in vivo. The incorporation of D-tyrosine most simply explains the formation of 6.Preaspyridone A 5 is the substrate for oxidative modification catalysed by the cytochrome P450 monooxygenase ApdE. Again, similar enzymes are known – for example TenA converts the tetramic acid pretenellin A 13 to the 2-pyridone pretenellin B 14 (Scheme 1). In the case of apdE, however, the chemistry is more diverse with three products being formed. The expected 2-pyridone 1 is observed, but so are the 14-hydroxylated compound 8 and the 5-dephenylated pyridone 9. The formation of both compounds can be explained by oxidative mechanisms. In the case of TenA it is thought that hydrogen atom abstraction from the benzylic carbon leads to a benzylic radical 16 which can rearrange and eliminate to form the observed 2-pyridone nucleus (Scheme 2).4 Benzylic alcohols such as 8 are not thought to be intermediates in the ring expansion in the case of TenA.4 In the case of ApdE it appears that a benzylic radical could also be formed. One reaction pathway would lead to the observed 2-pyridone by the same rearrangement that occurs for tenellin, but the benzylic radical could also be hydroxylated directly (rebound mechanism) to form the observed alcohol 8. It appears unlikely that this oxidation could be catalysed by an adventitious enzyme in A. oryzae as the 14-hydroxylated compound 8 is not formed when apdA and apdC are expressed alone. Furthermore, the equivalent compound in the tenellin series, prototenellin-D 15 is not observed when tenS, tenC and the P450-encoding tenA genes are expressed in A. oryzae, but is only observed in extracts of the native host Beauveria bassiana, again suggesting that B. bassiana contains a similar adventitious enzyme not present in A. oryzae.
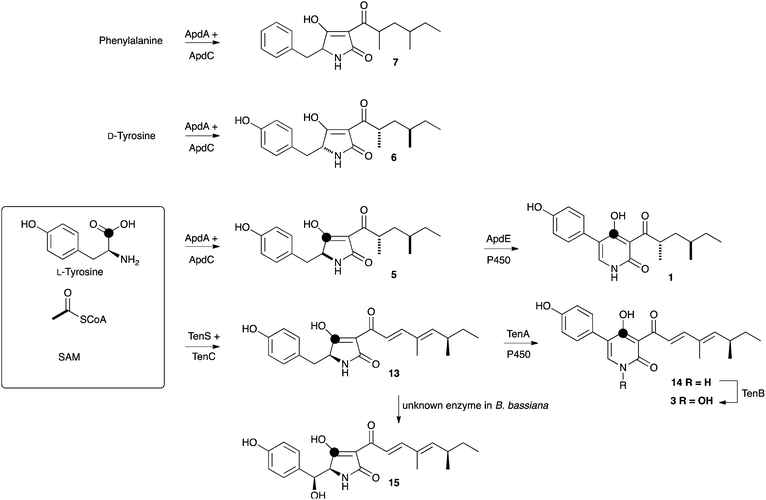 | ||
| Scheme 1 Biosynthetic pathways of aspyridone A 1 and tenellin 3. | ||
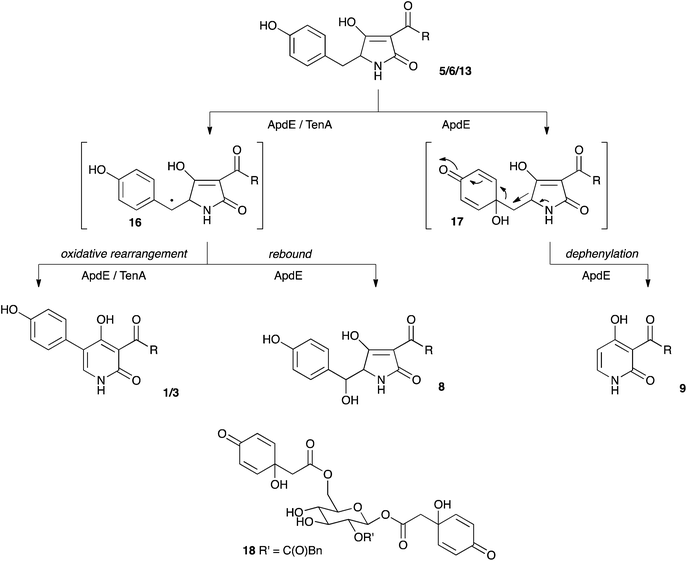 | ||
| Scheme 2 Oxidative mechanisms leading to 2-pyridones and β-hydroxy tetramic acids. | ||
It also appears that the enzyme ApdE is responsible for the oxidative dephenylation of preaspyridone A 5. Hydroxylation at the phenyl-bridgehead carbon, leads to a putative hydroxyquinone 17 (Scheme 2) which could undergo a coupled ring-expansion and dephenylation. Such hydroxyquinones are known in natural products such as jacaglabroside B 18.25 Similar compounds have also been synthesised from para-hydroxyaromatic species by mild oxidants including bisacetoxyiodobenzene and oxone.26 The product of the dephenylation was shown to be derived from tyrosine, like its precursor preaspyridone A 5, because of a high incorporation of label from [1-13C]-tyrosine. Thus, unlike TenA which is a highly selective ring-expandase, ApdE appears to be much less selective in its oxidation chemistry. The experiments also showed that TenA appears to be highly selective for its substrate as it could not act on the preaspyridone A diastereomers 5/6 which are very similar to pretenellin A 13.
The production of 9 and its derivatives also sheds light on the biosynthesis of a class of fungal metabolites known as the atpenins 19–22 which are potent and specific inhibitors of mitochondrial succinate-ubiquinone oxidoreductase (Fig. 5) and important tools for studying respiration.27 Although clearly related to other fungal 2-pyridones, the biosynthesis of the atpenins has not been reported. Our results suggest that they are likely to be the products of PKS-NRPS systems very similar or identical to the apd cluster, but containing additional genes for oxidation, methylation and chlorination of intermediate 9.
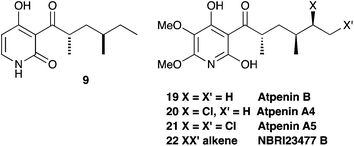 | ||
| Fig. 5 Structural relationship between 9 and the atpenins. | ||
The polyketide moieties of 1, 6, 9 and all the atpenins 19–22 have been determined to feature pendant methyl groups which are arranged anti. Pendant methyl groups are derived from S-adenosyl methionine (SAM) during polyketide biosynthesis in fungi, catalysed by a C-methyltransferase (CMeT) domain of the PKS. However, the stereochemistry of the carbon bearing the methyl is set by the enoyl reductase (ER, Scheme 3). In the case of ApdA the ER of the PKS is known to be inactive, but ER activity is provided in trans by ApdC. Fungal PKS are iterative, that is a single set of catalytic domains acts again and again to produce the polyketide chain. In the case of aspyridone 1 the acetate starter unit is extended three times to produce the observed tetraketide. Methylation and enoyl reduction must occur during both the first and second cycle of extension and modification (Scheme 3). Remarkably it appears that the trans-acting ER catalyses the reduction with opposite stereoselectivity in each case to produce the observed anti dimethylation pattern. This mirrors the recently reported activity of the hypothemycin KR of the hrPKS Hpm8 from the fungus Hypomyces subiculosus which also appears to be able to catalyse reactions in different PKS cycles with opposite stereoselectivities.28
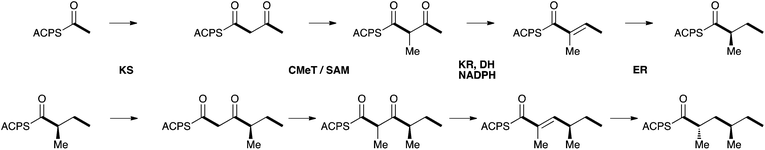 | ||
| Scheme 3 Proposed biosynthesis of the aspyridone polyketide. | ||
The role of apdB was also demonstrated. This gene encodes another cytochrome P450 monooxygenase. Initial experiments showed that ApdB appears not to act on the tetramic acids 5 or 6. However, it does appear to N-hydroxylate the dephenylated 2-pyridone 9, forming 10 and 11. These compounds were only observed in experiments where apdB was expressed and so this is very unlikely to be a native reaction of the A. oryzae host. Interestingly, although northern blotting experiments suggested that apdB was expressed during the biosynthesis of aspyridones in the native host A. nidulans, N-oxygenated compounds were not observed and our experiments also appear to rule out a role for apdB during aromatic ring hydroxylation to form aspyridone B 2. Furthermore, the formation of the N–O-methyl compound 11 must be catalysed by an endogenous A. oryzae O-methyl transferase as none of the expressed genes was predicted to encode proteins with methyltransferase activity.
We were then left with the possibility of either ApdD or ApdG being involved in ring-hydroxylation of 1 to form aspyridone B 2. However expression of apdD or apdG with combinations of the other genes which produce 1 did not lead to the formation of 2 or related compounds. It thus appears possible that aspyridone B 2 is formed in A. nidulans by an enzyme encoded outwith the predicted apd gene cluster.
Precise titres of all purified compounds were measured by making standard solutions of the purified metabolites and using these to calibrate a standard HPLC method (Table 1). Individual fungal expression clones were grown in triplicate and the average titres of each metabolite determined by HPLC of exhaustive organic extracts of each fermentation. Consideration of the overall productivity of each clone (Table 1) shows that as well as the chemical make-up of the extract varying by expression clone, the total amount of produced material also varies. Previous work in Bristol, as well as by Vederas and coworkers has already shown that hrPKS enzymes are more productive when expressed together with their cognate trans-acting ER, suggesting a direct protein–protein interaction between the hrPKS and the ER.19,20 The results of the experiments reported here show that the presence of a downstream cytochrome P450 can also boost the productivity of the system, from 60 mg L−1 to nearly 400 mg L−1, again suggesting a higher-order protein–protein organisation. Interestingly this phenomenon also appears to occur when proteins are present which are not acting as chemical catalysts. For example, the probable presence of ApdB appears to increase the productivity of ApdA + ApdC approximately 2-fold, despite the fact that ApdB has no substrate to act on. Conversely, the probable presence of ApdB appears to lower the influence of ApdE, perhaps indicating an effect of competition. However such arguments must remain inconclusive in the absence of information about actual protein concentrations in vivo.
Conclusion
Heterologous expression of biosynthetic genes from the aspyridone pathway has allowed the individual catalytic activities of each encoded enzyme to be investigated and the intermediates to aspyridone A 1 to be determined. It has also allowed the unprecedented oxidative chemistry catalysed by ApdE to be observed. ApdE shares 48% amino acid identity and 64% similarity to TenA, but nevertheless these enzymes show different substrate selectivities and different fidelities. The activity of ApdE gives clues about the biosynthesis of the atpenins and based on our results it should now be possible to rapidly discover a putative atpenin biosynthetic gene cluster in Penicillium sp. FO125, the producing organism.Our results also raise questions about the identity and definition of fungal biosynthetic gene clusters, their control and their chemical products. Fungal biosynthetic gene clusters can be defined by a number of means – including cotemporal transcription, as demonstrated by Hertweck and coworkers in the case of the apd genes,8 and by many bioinformatic means (some even claiming accuracy29). It is clear that in the case of the apd genes, apdA to apdG are cotemporally transcribed, seemingly in response to the expression of apdR. However, not all of these genes encode proteins which are catalytically active – only apdA, apdC, and apdE are required for the synthesis of aspyridone A 1. There appear to be insufficient genes in the cluster to encode the synthesis of aspyridone B 2, and transcription and bioinformatic analyses are inadequate for determining the role of apdB, or for predicting the new chemistry enacted by ApdE. However, all these features were revealed by our heterologous expression experiments.
Furthermore, our experiments show that heterologous expression of genes using strong constitutive promoters can significantly enhance titres of the produced compounds – for example expression of apdA and apdC alone affords one major compound at 61 mg L−1, but coexpression with apdE increases this significantly to 390 mg L−1 (but for three compounds). The origin of this effect is not directly discernible from our experiments, but suggests that the productivity of the aspyridone biosynthetic enzymes are enhanced when they work together. The ability of genes from the apd cluster to encode the synthesis of diverse compounds is exemplified by our coexpression of apdABCE which produced at least seven different compounds attributable to the apd genes.
This result emphasises our previous observation that differential gene expression levels during tenellin biosynthesis in the native host Beauveria bassiana can affect both titre and biosynthetic programming,30 in turn indicating that fine control over gene expression can affect the chemical structures of the compounds produced from a single biosynthetic gene cluster. This can occur in eukaryotes where each structural gene is controlled by its own promoter and where complex networks of control elements are involved in the activation of genes within each pathway. In our experiments where this fine control has been bypassed we assume that the relative ratios of the produced proteins is different from that produced under native conditions. It has long been known that the production of individual secondary metabolites by filamentous fungi is very sensitive to growth and media conditions, but our results also suggest that changes in gene expression could also change the compounds produced from a single gene cluster.
Clear lessons emerge from this analysis: first, that the definition of fungal biosynthetic gene clusters, however achieved, is at best speculative; second, that heterologous expression can reveal a significant number of the details of secondary metabolic pathways in fungi; third, that the ability of fungal pathways to produce (many) more than one compound is inherent – and that a single compound should not be regarded as the sole product of a fungal biosynthetic gene cluster; fourth, that genome mining in fungi will remain problematic while so many gene expression pathways, enzymatic activities and protein–protein interactions remain highly unpredictable; and finally that heterologous expression offers a systematic and broadly applicable method for investigating the chemistry of silent and orphan fungal biosynthetic gene clusters, in addition to providing access to new natural products.
Experimental
1 Cloning procedures
All primer sequences are given in the ESI.†![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 699, 1071, 3955 bp confirming the removal of apdB, which contains an additional HindIII site. This plasmid was used to transform A. oryzae M-2-3. Metabolites were extracted from 11 of the 12 transformants recovered.
699, 1071, 3955 bp confirming the removal of apdB, which contains an additional HindIII site. This plasmid was used to transform A. oryzae M-2-3. Metabolites were extracted from 11 of the 12 transformants recovered.The apdA gene was amplified from A. nidulans genomic DNA as three overlapping fragments of ∼4 kb each using primer pairs: apdA1-F and apdA1-R; apdA2-F and apdA2-R; apdA3-F and apdA3-R. The forward primer for fragment 1 and reverse primer for fragment 3 contained 30 bases homologous to the vector region for recombination. Homologous recombination in S. cerevisiae was used to reassemble apdA between GATEWAY attL sites to allow easy transfer into any expression vector that contains a GATEWAY destination cassette. pEYAtenSeGFP13 was digested with Not1 and Xho1 and used as the vector. The tenS gene was replaced by apdA, while the eGFP remained to facilitate identification of A. oryzae transformants that successfully expressed the new hybrid gene. Plasmids extracted from S. cerevisiae colonies were shuttled into E. coli TOP10 cells. Four colonies were extracted and the plasmids digested with Not1 plus Kpn1 to prove that apdA had been correctly assembled to create pEYAapdAeGFP. GATEWAY LR recombination between pEYAapdAeGFP and pTAYAGSargPageASP resulted in production of pTAYAargASP ready to transform A. oryzae.
Fifty nine colonies were recovered following transformation of A. oryzae with pTAYAargASP. All transformants were subcultured onto selective medium. In order to confirm all the genes in the pTAYAargASP were expressed, qRT-PCR was performed (see ESI†). Fourteen transformants were cultured for metabolite extraction. All 14 had a yellow pigmented morphology, but one was discarded due to contamination. In all 13 extracts analysed, new compounds were detected.
The ble gene was amplified and inserted into NgoMIV-cut pTAYAGSbarPage by homologous recombination in yeast to create pTAYAGSblePage. The plasmid was confirmed to carry the ble-resistance gene by restriction analysis with NgoMIV + Asc1.
pTAYAGSblePage was then used as a vector to co-express apdG together with pTAYAargASP in A. oryzae. The apdG gene was amplified using the primers apdG-F and apdG-R and placed under control of PgpdA in Asc1-cut pTAYAGSblePage. Two promoter fragments (PgpdA + Padh and Peno) were also amplified (PgpdA + Padh, adh/asc-F and newgpdA-R; Peno, eno/asc/page-F and eno/asc/page-R) and inserted to ‘patch’ the Asc1 sites downstream of Padh and Peno in Asc1-cut pTAYAGSblePage. The resulting plasmid, pTAYAGSbleG was recovered in E. coli and confirmed by restriction digestion with NgoMIV.
Twelve transformants were obtained by co-transformation of A. oryzae with pTAYAargASP (carrying apdABCE) and pTAYAGSbleG (carrying apdG). Transformants were selected on a minimal medium containing 100 μg mL−1 zeocin (a cheaper alternative to bleomycin or phleomycin). Metabolites were extracted from all 12 transformants, but novel peaks were detected in only 5.
2 Fermentation conditions
The Aspergillus oryzae transformants were first grown on Czapek Dox Agar (minimal media, see ESI†) and for further production of spores, they were inoculated and grown on DPY solid media for 7–10 days at 25 °C.For purification of compounds the crude extract was made into a solution of 50 mg mL−1 in HPLC grade methanol and 200 μL aliquots were injected in each run of mass-directed HPLC preparative purification.
3 Analytical LCMS
Samples as prepared above (20 μL) were analysed using a Waters 2795HT HPLC system. Detection was achieved by uv between 200 and 400 nm using a Waters 998 diode array detector, and by simultaneous electrospray (ES) mass spectrometry using a Waters ZQ spectrometer detecting between 150 and 600 m/z units. Chromatography (flow rate 1 mL min−1) was achieved using a Phenomenex Kinetex column (2.6 μ, C18, 100 Å, 4.6 × 100 mm) equipped with a Phenomenex Security Guard precolumn (Luna C5 300 Å). Solvents were: A, HPLC grade H2O containing 0.05% formic acid; B, HPLC grade MeOH containing 0.045% formic acid; and C, HPLC grade CH3CN containing 0.045% formic acid. Gradients were as follows.Method 1. MeOH: 0 min, 10% B; 10 min, 90% B; 12 min, 90% B; 13 min, 10% B; 15 min, 10% B.
Method 2. CH3CN: 0 min, 10% C; 10 min, 90% C; 12 min, 90% C; 13 min, 10% C; 15 min, 10% C.
4 Purification and characterization of compounds
Purification of compounds was generally achieved using a Waters mass-directed autopurification system comprising of a Waters 2767 autosampler, Waters 2545 pump system, a Phenomenex LUNA column (5μ, C18, 100 Å, 10 × 250 mm) equipped with a Phenomenex Security Guard precolumn (Luna C5 300 Å) eluted at 4 mL min−1. Solvent A, HPLC grade H2O + 0.05% formic acid; Solvent B, HPLC grade MeOH + 0.045% formic acid; solvent C, HPLC grade CH3CN + 0.045% formic acid. The post-column flow was split (100:1) and the minority flow was made up with solvent A to 1 mL min−1 for simultaneous analysis by diode array detector (Waters 2998), evaporative light scattering (Waters 2424) and ESI mass spectrometry in positive and negative modes (Waters Quatro Micro).Solvent used were: A, HPLC grade water containing 0.05% formic acid; and Solvent B, HPLC grade CH3CN containing 0.045% formic acid. Extracts from (apdACEB) were purified with Method 1, (apdACED) with Method 2, (apdACE) with Method 3 and (apdACEBG) with Method 4.
Method 1: 0 min, 50% B; 22 min, 75% B; 24 min, 95% B; 26 min, 95% B; 27 min, 50% B; 30 min, 50% B.
Method 2: 0 min, 55% B; 22 min, 60% B; 24 min, 95% B; 26 min, 95% B; 27 min, 55% B; 30 min, 55% B.
Method 3: 0 min, 55% B; 22 min, 75% B; 24 min, 95% B; 26 min, 95% B; 27 min, 55% B; 30 min, 55% B.
Method 4: 0 min, 50% B; 22 min, 65% B; 24 min, 95% B; 26 min, 95% B; 27 min, 50% B; 30 min, 50% B.
Detected peaks were collected into glass test tubes. Combined fractions were evaporated under a flow of dry N2 gas and residues weighed and dissolved directly in NMR solvent for NMR analysis.
5 3JCH coupling constant calculations
The values of 3JCH for the cis and trans isomers of 12 were established by computation of a truncated analogue with the polyketide sidechain removed (see ESI†). The geometries were optimised with DFT/B3LYP functional using 631g basis set before calculation of coupling constants using the Gauge Independent Atomic Orbital (GIAO) approach with a 631g+(d,p) basis set. All calculations were performed in Gaussian09.31 Geometry optimisation and 3JCH calculations were repeated for the corresponding enol forms and found to give coupling constant values in agreement with the corresponding enone forms, so no further account of enolisation was considered in this analysis.Acknowledgements
ZW Thanks the Government of Pakistan (HEC) for funding. KAKP thanks Majlis Amanah Rakyat (MARA) Malaysia for funding. EPSRC is thanked for LCMS equipment (EP/F066104/1). We also thank Professors C. Hertweck and A. Brakhage for the provision of A. nidulans SB4.1.Notes and references
- M. H. Medema, K. Blin, P. Cimermancic, V. de Jager, P. Zakrzewski, M. A. Fischbach, T. Weber, E. Takano and R. Breitling, Nucleic Acids Res., 2011, 39, W339–W346 CrossRef CAS.
- R. J. Cox, Org. Biomol. Chem., 2007, 5, 2010–2026 CAS.
- A. Munawar, J. W. Marshall, R. J. Cox, A. M. Bailey and C. M. Lazarus, ChemBioChem, 2013, 14, 388–394 CrossRef CAS.
- L. M. Halo, M. N. Heneghan, A. A. Yakasai, Z. Song, K. Williams, A. M. Bailey, R. J. Cox, C. M. Lazarus and T. J. Simpson, J. Am. Chem. Soc., 2008, 130, 17988–17996 CrossRef CAS.
- M. L. Nielsen, J. B. Nielsen, C. Rank, M. L. Klejnstrup, D. M. K. Holm, K. H. Broggard, B. J. Hansen, J. C. Frisvad, T. O. Larsen and U. H. Mortensen, FEMS Microbiol. Lett., 2011, 321, 157–166 CrossRef CAS.
- J. F. Sanchez, R. Entwistle, J.-H. Hung, J. Yaegashi, S. Jain, Y.-M. Chiang, C. C. C. Wang and B. R. Oakley, J. Am. Chem. Soc., 2011, 133, 4010–4017 CrossRef CAS.
- A. A. Brakhage, Nat. Rev. Microbiol., 2012, 11, 21–32 CrossRef.
- S. Bergmann, J. Schümann, K. Scherlach, C. Lange, A. A. Brakhage and C. Hertweck, Nat. Chem. Biol., 2007, 3, 213–217 CrossRef CAS.
- R. B. Williams, J. C. Henrikson, A. R. Hoover, A. E. Lee and R. H. Cichewicz, Org. Biomol. Chem., 2008, 6, 1895–1897 CAS.
- R. M. Perrin, N. D. Fedorova, J. W. Bok, R. A. Cramer, J. R. Wortman, H. S. Kim, W. C. Nierman and N. P. Keller, PLoS Pathog., 2007, 3, e50 Search PubMed.
- H. Bode, B. Bethe, R. Höfs and A. Zeeck, ChemBioChem, 2002, 3, 619–627 CrossRef CAS.
- G. Bills, G. Platas, A. Fillola, M. Jiménez, J. Collado, F. Vicente, J. Martín, A. González, J. Bur-Zimmermann, J. Tormo and F. Peláez, J. Appl. Microbiol., 2008, 104, 1644–1658 CrossRef CAS.
- A. K. Pahirulzaman, K. Williams and C. M. Lazarus, Methods Enzymol., 2012, 517, 241–260 Search PubMed.
- K. Gomi, Y. Iimura and S. Hara, Agric. Biol. Chem., 1987, 51, 2549–2555 CrossRef CAS.
- Z. Song, R. J. Cox, C. M. Lazarus and T. J. Simpson, ChemBioChem, 2004, 5, 1196–1203 CrossRef CAS.
- D. Boettger and C. Hertweck, ChemBioChem, 2013, 14, 28–42 CrossRef CAS.
- W. Xu, X. Cai, M. E. Jung and Y. Tang, J. Am. Chem. Soc., 2010, 132, 13604–13607 CrossRef CAS.
- M. N. Heneghan, A. A. Yakasai, K. Williams, K. A. Kadir, Z. Wasil, W. Bakeer, K. M. Fisch, A. M. Bailey, T. J. Simpson, R. J. Cox and C. M. Lazarus, Chem. Sci., 2011, 2, 972–979 RSC.
- J. Kennedy, K. Auclair, S. Kendrew, C. Park, J. Vederas and C. Hutchinson, Science, 1999, 284, 1368–1372 CrossRef CAS.
- L. M. Halo, J. W. Marshall, A. A. Yakasai, Z. Song, C. P. Butts, M. P. Crump, M. Heneghan, A. M. Bailey, T. J. Simpson, C. M. Lazarus and R. J. Cox, ChemBioChem, 2008, 9, 585–594 CrossRef CAS.
- V. V. J. Krishnamurthy, J. Magn. Reson., Ser. A, 1996, 121, 33–41 CrossRef CAS.
- J. Schümann and C. Hertweck, J. Am. Chem. Soc., 2007, 129, 9564–9565 CrossRef.
- K. Scherlach, D. Boettger, N. Remme and C. Hertweck, Nat. Prod. Rep., 2010, 27, 869–886 RSC.
- X. Liu and C. T. Walsh, Biochemistry, 2009, 48, 8746–8757 CrossRef CAS.
- M. Salomé Gachet, O. Kunert, M. Kaiser, R. Brun, R. A. Muñoz, R. Bauer and W. Schühly, J. Nat. Prod., 2010, 73, 553–556 CrossRef.
- Z. You, A. H. Hoveyda and M. L. Snapper, Angew. Chem., Int. Ed., 2009, 48, 547–555 CrossRef CAS.
- H. Miyadera, K. Shiomi, H. Ui, Y. Yamaguchi, R. Masuma, H. Tomoda, H. Miyoshi, A. Osanai, K. Kita and S. Omura, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 473–477 CrossRef CAS.
- H. Zhou, Z. Gao, K. Qiao, J. Wang, J. C. Vederas and Y. Tang, Nat. Chem. Biol., 2012, 8, 331–333 CrossRef CAS.
- M. R. Andersen, J. B. Nielsen, A. Klitgaard, L. M. Petersen, M. Zachariasen, T. J. Hansen, L. H. Blicher, C. H. Gotfredsen, T. O. Larsen and K. F. Nielsen, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, E99–E107 CrossRef CAS.
- A. A. Yakasai, J. Davison, Z. Wasil, L. M. Halo, C. P. Butts, C. M. Lazarus, A. M. Bailey, T. J. Simpson and R. J. Cox, J. Am. Chem. Soc., 2011, 133, 10990–10998 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision A.1, Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Containing oligonucleotide primer sequences and additional experimental details. CCDC 941137–941139. For ESI and crystallographic data in CIF or other electronic format. See DOI: 10.1039/c3sc51785c |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2013 |