 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The Ugi four-component reaction enables expedient synthesis and comparison of photoaffinity probes†
Jacob T. Busha,
Louise J. Walporta,
Joanna F. McGouranb,
Ivanhoe K. H. Leunga,
Georgina Berridgea,
Sander S. van Berkela,
Amit Basakc,
Benedikt M. Kesslerb and
Christopher J. Schofield*a
aDepartment of Chemistry, University of Oxford, Oxford OX1 3TA, UK. E-mail: christopher.schofield@chem.ox.ac.uk
bTarget Discovery Institute, University of Oxford, Roosevelt Drive, Oxford OX3 7FZ, UK
cDepartment of Chemistry, Indian Institute of Technology, Kharagpur 721302, India
First published on 18th July 2013
Abstract
Photoaffinity probes are increasingly being used for the study of biological interactions; however, the lack of structure–activity relationship studies has hindered their rational application. We describe the use of the Ugi four-component reaction (U-4CR) for the expedient and versatile assembly of photoaffinity scaffolds that can be linked to small molecule probes. The rates, yields and sites of crosslinking of five commonly used photoreactive groups comprising diazirines, aryl azides and a benzophenone, were compared using a human 2-oxoglutarate oxygenase as a model protein. The results reveal significant differences in the behavior of the probes and suggest that empirically guided optimization of probes for specific tasks is desirable. In the absence of such optimization it may be advisable to use a set of crosslinking probes/conditions; the U-4CR provides a convenient method for obtaining such a set.
Introduction
Photoaffinity probes (or capture compounds) show promise as useful tools for enabling the selective identification and quantification of proteins, as well as for mapping molecular interactions (for reviews see ref. 1–4). They can be used to profile multiple members of protein families simultaneously, offering potential for probing the biochemical origins of a phenotype.5,6 They can also be used to investigate small molecule–protein interactions providing insight into mechanisms of action and selectivity.7–12 Photoaffinity probes typically consist of three functional moieties: (i) a selectivity function, such as an enzyme inhibitor, (ii) a photoreactive crosslinking group, such as an aryl azide or diazirine, and (iii) a detection handle, such as biotin or a fluorescent dye.7 In a typical protocol the probe is incubated with cell lysate/cells, and binds to the target protein(s) (or other biopolymers) via its selectivity function (Fig. 1). The photoreactive group is activated by irradiation which enables reaction leading to covalent attachment to the target enzyme. The handle is then used to isolate and/or identify captured proteins, e.g. by mass spectrometry or antibody-based detection.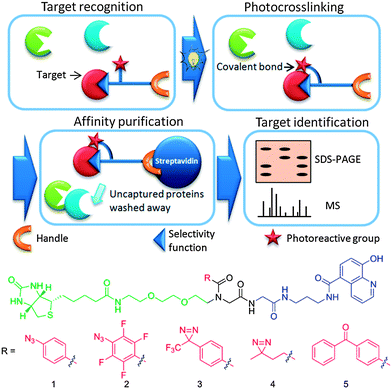 | ||
| Fig. 1 Above: schematic representation of the application of photoaffinity probes. Below: potential photoaffinity probes for 2-oxoglutarate oxygenases incorporating 5 different photoreactive groups. | ||
A challenge in the design of photoaffinity probes is the attachment of a detection handle and photoreactive group to the selectivity function in orientations that do not compromise target (protein) binding and enable sufficiently efficient crosslinking to the target(s) upon photoirradiation. Further, despite differences in the crosslinking properties and commercial/synthetic accessibility of popular photoreactive groups, there have been few studies to compare their efficiency.13,14 Thus, to maximize the likelihood of specific and efficient crosslinking, the use of a set of probes is desirable, with variation of the attachment point on the selectivity function and the nature of the photoreactive group. Although various tripodal templates and photoactivatable groups have been used in probe construction, their synthesis typically requires multiple steps, which has limited structure–activity relationship studies.
Here we report the use of the Ugi four-component reaction (U-4CR) for the straightforward synthesis of photoaffinity scaffolds, enabling the efficient conjugation of a range of photoreactive groups, purification/visualisation handles and functionalities for inhibitor attachment in a single step. The efficacy of 5 different photoaffinity probes 1–5, prepared by the U-4CR, was investigated using a 2-oxoglutarate (2-OG) oxygenase inhibitor (IOX1) as the selectivity function (Fig. 1).15 Substantially different reaction rates, yields and crosslinking sites were observed in studies on the human hypoxia sensing enzyme prolyl hydroxylase domain-containing protein 2 (PHD2/EGLN1).16
Results and discussion
The U-4CR,17 which couples a carboxylic acid, an amine, an aldehyde and an isonitrile in one pot to provide a peptoid structure, has been extensively used for the synthesis of inhibitor libraries and there are examples of its use for the conjugation of photoreactive groups or dyes with small molecules/inhibitors.18–21 We envisioned that the U-4CR would be well-suited for the synthesis of photoaffinity scaffolds since commonly used photoreactive groups can be obtained as carboxylic acids, purification handles are available as amines, and isonitriles can be used to provide an inhibitor attachment point. Initially, we tested the applicability of the U-4CR by reacting p-azidobenzoic acid (A), isonitrile methyl ester (I), paraformaldehyde and amine modified biotin (F), under microwave irradiation (100 °C, 20 min), producing the desired photoaffinity scaffold 6a in good yield (62%). The scope of the reaction was then explored by using other commonly applied photoreactive groups, tetrafluoroaryl azide B, aryl trifluoromethyl diazirine C, benzophenone D and alkyl diazirine E, pleasingly affording the corresponding photoaffinity scaffolds 6b–e in good yields (Table 1). |
We also investigated the incorporation of alternative detection handles in place of biotin, such as dyes and ‘clickable’ moieties. Dyes allow direct in-gel visualisation of captured proteins, whereas alkynes allow conjugation of a desired tag (e.g. biotin or dye) after photoreaction via a [3 + 2] cycloaddtion reaction with an azide.2,22 Thus, amine functionalised rhodamine H was reacted with p-azidobenzoic acid (A), isonitrile methyl ester (I), and paraformaldehyde to provide the desired photoaffinity scaffold 7 (56%). The use of propargylamine to incorporate an alkyne into the scaffold gave low yields of the desired product (<30%), likely due to the tendency of propargyl amides to undergo rearrangement;23 however, the use of 3-butynylamine resulted in formation of alkyne functionalised photoaffinity scaffold 8, (73%).
Finally, the scope for variation of the inhibitor attachment point was investigated. To this end, Boc-protected diamine-derived isonitrile J was used instead of an ester, which after deprotection provides a complementary handle for attachment of a selectivity function. The U-4CR provided 9 (53%), which was Boc-deprotected to give the desired amine 10. A “clickable” photoaffinity scaffold (11) suitable for strained cycloaddition reactions with azide functionalized inhibitors was obtained by reaction of 10 with a bicyclononyne-OSu ester (BCN-OSu) (Scheme 1).24
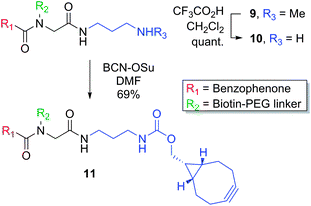 | ||
| Scheme 1 | ||
The U-4CR was subsequently used to synthesize photoaffinity probes containing various photoreactive groups for comparison of their crosslinking efficiency. We have previously reported that 5-carboxy-8-hydroxyquinoline (IOX1) can be utilized as a probe for 2-OG dependent oxygenases including PHD2 (EGLN1).9,15,25 We used the PHD2 catalytic domain (residues 181–426, hereafter PHD2) to investigate the rates, yields and sites of photocrosslinking obtained with IOX1 functionalised photoaffinity probes bearing commonly used photoreactive groups A–E. The biotinylated scaffolds 6a–e were hydrolysed prior to coupling with IOX1, to provide photoaffinity probes 1–5 (Scheme 2). Dissociation constants (KD) were determined by NMR spectroscopy using a solvent water relaxation technique and were found to be similar to the reported value for IOX1 (KD 60 μM).26 Of the five probes, 4 was found to bind to PHD2–MnII with the highest affinity (KD 9 μM). Probes 1, 2 and 5 bound with medium strength (KD 19, 28 and 25 μM respectively), and 3 bound more weakly (KD 126 μM) (Fig. S1†).
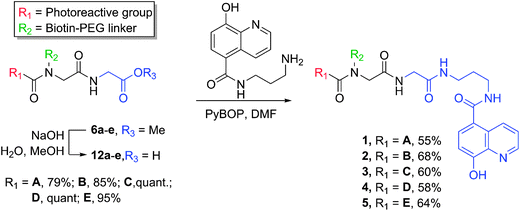 | ||
| Scheme 2 | ||
Crosslinking rates and yields were determined using 5 μM PHD2, 10 μM MnII (substituting FeII), with probes 1–5 at twice the determined KD values. Irradiation was carried out using a CaproBox (Caprotec, Berlin) at two different wavelengths, 310 nm, reported to be suited for the activation of aryl azides, and 350 nm, more suited for benzophenones.1 Probes 1–5 were incubated with PHD2 and MnII at room temperature (20 min) to allow equilibration prior to irradiation (4 °C). The extent of crosslinking was determined by electrospray ionization (ESI) mass spectrometry (MS) and plotted against time (Fig. 2 (310 nm), Fig. S2† (350 nm)). Control experiments demonstrated that crosslinking was competitively inhibited in the presence of 200 μM IOX1 (Table S11†). Additionally, irradiation of the unfunctionalised scaffolds 6a–e did not lead to modification (Table S12†), thus supporting the proposal of selective reaction and highlighting the potential use of the probes in inhibitor binding assays.
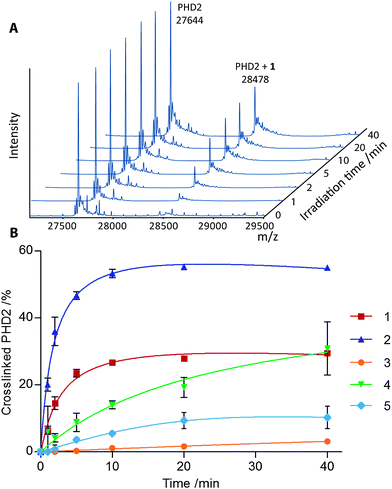 | ||
| Fig. 2 (A) Deconvoluted ESI MS of PHD2 after irradiation (310 nm) in the presence of MnII and 1. (B) Percentage of crosslinked PHD2 upon irradiation (310 nm) in the presence of probes 1–5, as determined by ESI MS. Data represent mean intensities of repeats (n = 3), error bars show 1 standard deviation. | ||
The best results in terms of initial rate and yield of crosslinking were achieved by irradiating aryl azide 2 at 310 nm, which yielded 55% crosslinked enzyme after just 10 min. Using this wavelength, aryl azide 1 and alkyl diazirine 4 also gave good overall crosslinking yields (30% and 28% respectively), although the alkyl diazirine was slower to react. Aryl azides 1 and 2 gave lower levels of crosslinking at 350 nm (13% and 11% respectively), consistent with literature reports on other proteins.1 At this wavelength, species crosslinked with the aryl azide 2 appeared to degrade with increased irradiation time, possibly due to further photoreactions. Interestingly, the commonly used aryl trifluoromethyl diazirine and benzophenone groups, probes 3 and 5, resulted in relatively low levels of crosslinking at both wavelengths (3–12%). In the case of 3 this appeared to be due, at least in part, to competitive quenching of the carbene with water, as determined by MS (Fig. S3†).
The rate of activation of the probes at 310 nm was inferred from the rate of decay of the parent peak in the mass spectrum and found to increase in the order 4 < 1 < 3 < 2 (Fig. S3†). The rate of activation of 5 could not be determined in this manner since both intramolecular reactions and reaction with water can afford products with the same mass as the parent compound, including via hemi-acetal/ketone equilibrium. Probe molecules that did not crosslink to enzyme were found to give a variety of products, depending on the nature of the photoreactive group. Irradiation of 3 predominantly formed a species with a mass corresponding to reaction with water, while 1 and 4 gave products with masses corresponding to intramolecular insertion of the nitrene/carbene. Azide 2 formed primarily a species with a mass that suggested reduction of the nitrene to the amine. These differences may arise from the relative reactivity of the activated species towards water/buffer versus intramolecular reactions, or from conformational constraints.
The crosslinking site(s) of probes 1, 2 and 4 on PHD2 were investigated by partial trypsinolysis followed by matrix-assisted laser desorption/ionization (MALDI) MS. We have found that trypsinolysis of the PHD2 catalytic domain results in rapid cleavage of N-(∼7 kDa) and C-(∼3 kDa) terminal PHD2 fragments to give a ‘core-fold’ (∼Ser245–Arg396), which is digested more slowly (Fig. 3).27 1, 2 and 4 (50 μM) were irradiated (12 min, 310 nm, 4 °C) in the presence of PHD2 (20 μM) and MnII (20 μM), after which reaction mixtures were incubated with trypsin. Aliquots were removed at intervals, quenched with acid and analyzed (Fig. 3). The MALDI mass spectra of the undigested samples (black lines, Fig. 3) exhibited 2 peaks at ∼28 kDa corresponding to unmodified and probe conjugated PHD2 respectively. After three hours of trypsin digestion the 2 peaks shifted to ∼18 kDa (red lines, Fig. 3), corresponding to complete loss of N- and C-terminal fragments to give the modified and unmodified ‘core-fold’. The ratio of the 2 peaks remained unchanged within the limit of detection for 1 and 4, indicating crosslinking predominantly to the core-fold region. In the case of 2, the ratio of crosslinking to the core-fold was lower than on the undigested enzyme, suggesting that crosslinking for 2 was divided over the core and the N- or C-terminal fragments. The MALDI mass spectrum for probe 2 at t = 5 min (green line, Fig. 3) exhibited 2 additional pairs of peaks at ∼25 kDa and ∼21 kDa corresponding to loss of the C- or N-terminal fragment, respectively. The decrease in the ratio of modified to unmodified PHD2 upon loss of the C-terminus, but not upon loss of the N-terminus, indicates that 2 crosslinks to both the core-fold and the C-terminus.
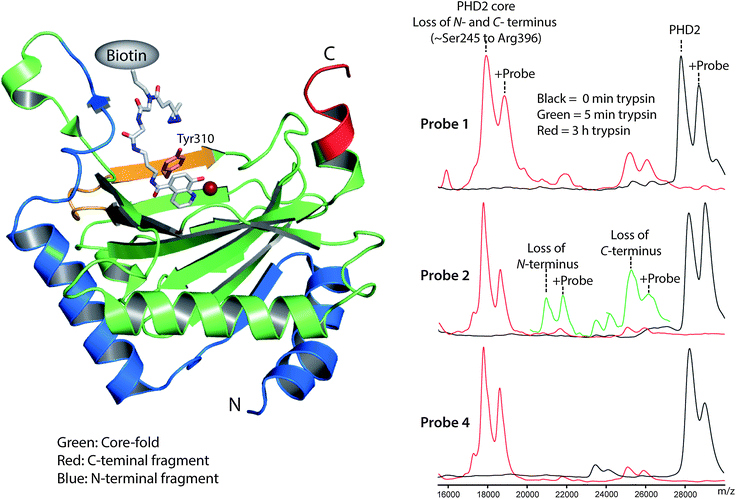 | ||
| Fig. 3 Limited trypsinolysis of PHD2 identifies the region of crosslinking. Above: view derived from a crystal structure of PHD2 (PDB ID: 3HQU)16 docked with diazirine 4 (grey) indicating the sites of photocrosslinking. The ‘core-fold’ (green), N-terminal (blue) and beginning of the C-terminal (red) region as defined by partial trypsinolysis are shown. The trypsin-derived peptide and tyrosine-residue identified as crosslinking to 4 by LCMS/MS in yellow and pink. Below: MALDI mass spectra derived from partial trypsinolysis of PHD2 crosslinked to probes 1, 2 and 4. Black line: before trypsin digest. Red line: 3 h trypsin digest. Green line: 5 min trypsin digest. | ||
Finally, we performed MS/MS fragmentation analyses of extended trypsin digestions to further investigate the sites of photocrosslinking. Probes 1, 2 and 4 were crosslinked to PHD2 under the conditions described for the MALDI studies followed by overnight trypsinolysis and MS/MS. No masses corresponding to trypsinolysed peptides plus probe were observed for 1 and 2, possibly either due to poor stability of the crosslinked peptides under our MS conditions, or due to dilution of the crosslinking over a number of peptides. However, with compound 4, crosslinked peptides were detected which consistently contained the peptide fragment 298AMVACYPGNGTGYVR312 (Table S13†). MS/MS analysis afforded peaks corresponding to probe conjugated peptide for y fragments from y3 to y10 and peaks for unmodified fragments y1 and y2 (Fig. S4†), indicating crosslinking to tyrosine 310. Tyrosine residues have recently been shown to react with diazirine generated carbenes via insertion into both O–H and ortho-C–H bonds of the phenol ring.28 Manual docking of the selectivity function of 1–5 with the active site of PHD2 (Fig. S5†), indicates that tyrosine 310, located at the edge of the PHD2 active site, is well situated for crosslinking (Fig. 3 and S5†). Collectively, the MS studies provide insights into the selectivity of photocrosslinking; notably they demonstrate that the use of different photoreactive groups can substantially alter the efficiency and site(s) of crosslinking.
Conclusions
In conclusion, the U-4CR provides a useful route for the synthesis of photoaffinity probes, enabling the expedient assembly of photoaffinity scaffolds with a variety of photoreactive groups, detection handles and inhibitor attachment points. The U-4CR was used for the synthesis of a set of photoaffinity probes for 2-OG oxygenases, containing different photoreactive groups. The photocrosslinking rates, yields and sites were investigated using PHD2 as a model system, revealing substantial differences between the probes. Aryl azides 1 and 2 and alkyl diazirine 4 were found to crosslink efficiently, while aryl trifluoromethyl diazirine 3 and benzophenone 5 gave low crosslinking yields. The relatively poor results observed for 3 and 5 contrast with other studies where these photoreactive groups have been reported to enable good crosslinking,14,15 suggesting that the optimum photoreactive group varies depending on the ‘intrinsic’ photochemical properties of the probe and the nature of its interaction with the target protein. The proximity and orientation of the photoreactive group to an appropriately functionalized group on the target, and the rate of reaction with enzyme versus quenching reactions, are likely to be important and context dependent factors in determining the crosslinking yield. The available evidence suggests that in most cases experimentally guided tailoring of the crosslinking reagent/protocol to particular targets is desirable.Acknowledgements
JTB was supported by the EPSRC and Pfizer Ltd. LJW was supported by the BBSRC. BMK and JFM were supported by the Biomedical Research Centre (NIHR), Oxford, UK. IKHL is supported by the British Heart Foundation (BHF). SSVB was supported by CRUK. We thank David Pryde and Rachel Grimley for helpful discussions.References
- P. P. Geurink, L. M. Prely, G. A. van der Marel, R. Bischoff and H. S. Overkleeft, Top. Curr. Chem., 2012, 324, 85–113 CrossRef CAS
.
- D. J. Lapinsky, Bioorg. Med. Chem., 2012, 20, 6237–6247 CrossRef CAS
- L. Dubinsky, B. P. Krom and M. M. Meijler, Bioorg. Med. Chem., 2012, 20, 554–570 CrossRef CAS
- T. Lenz, J. J. Fischer and M. Dreger, J. Proteomics, 2011, 75, 100–115 CrossRef CAS
- D. K. Nomura, J. Z. Long, S. Niessen, H. S. Hoover, S. W. Ng and B. F. Cravatt, Cell, 2010, 140, 49–61 CrossRef CAS
- P. Ranjitkar, B. G. Perera, D. L. Swaney, S. B. Hari, E. T. Larson, R. Krishnamurty, E. A. Merritt, J. Villen and D. J. Maly, J. Am. Chem. Soc., 2012, 134, 19017–19025 CrossRef CAS
- H. Koster, D. P. Little, P. Luan, R. Muller, S. M. Siddiqi, S. Marappan and P. Yip, Assay Drug Dev. Technol., 2007, 5, 381–390 CrossRef
- D. Rotili, M. Altun, R. B. Hamed, C. Loenarz, A. Thalhammer, R. J. Hopkinson, Y. M. Tian, P. J. Ratcliffe, A. Mai, B. M. Kessler and C. J. Schofield, Chem. Commun., 2011, 47, 1488–1490 RSC
- D. Rotili, M. Altun, A. Kawamura, A. Wolf, R. Fischer, I. K. Leung, M. M. Mackeen, Y. M. Tian, P. J. Ratcliffe, A. Mai, B. M. Kessler and C. J. Schofield, Chem. Biol., 2011, 18, 642–654 CrossRef CAS
- K. Liu, H. B. Shi, H. G. Xiao, A. G. L. Chong, X. Z. Bi, Y. T. Chang, K. S. W. Tan, R. Y. Yada and S. Q. Yao, Angew. Chem., Int. Ed., 2009, 48, 8293–8297 CrossRef CAS
- C. M. Salisbury and B. F. Cravatt, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 1171–1176 CrossRef CAS
- T. Kinoshita, A. Cano-Delgado, H. Seto, S. Hiranuma, S. Fujioka, S. Yoshida and J. Chory, Nature, 2005, 433, 167–171 CrossRef CAS
- J. J. Tate, J. Persinger and B. Bartholomew, Nucleic Acids Res., 1998, 26, 1421–1426 CrossRef CAS
- C. Dalhoff, M. Huben, T. Lenz, P. Poot, E. Nordhoff, H. Koster and E. Weinhold, ChemBioChem, 2010, 11, 256–265 CrossRef CAS
- O. N. F. King, X. S. Li, M. Sakurai, A. Kawamura, N. R. Rose, S. S. Ng, A. M. Quinn, G. Rai, B. T. Mott, P. Beswick, R. J. Klose, U. Oppermann, A. Jadhav, T. D. Heightman, D. J. Maloney, C. J. Schofield and A. Simeonov, PLoS One, 2010, 5, e15535 Search PubMed
- R. Chowdhury, M. A. McDonough, J. Mecinovic, C. Loenarz, E. Flashman, K. S. Hewitson, C. Domene and C. J. Schofield, Structure, 2009, 17, 981–989 CrossRef CAS
- A. Domling and I. I. Ugi, Angew. Chem., Int. Ed., 2000, 39, 3168–3210 CrossRef CAS
- J. Ge, X. Cheng, L. P. Tan and S. Q. Yao, Chem. Commun., 2012, 48, 4453–4455 RSC
- S. Brauch, M. Henze, B. Osswald, K. Naumann, L. A. Wessjohann, S. S. van Berkel and B. Westermann, Org. Biomol. Chem., 2012, 10, 958–965 CAS
- A. Domling, Chem. Rev., 2006, 106, 17–89 CrossRef
- M. Henze, O. Kreye, S. Brauch, C. Nitsche, K. Naumann, L. A. Wessjohann and B. Westermann, Synthesis, 2010, 2997–3003 CAS
- P. Sarathi Addy, B. Saha, N. D. Pradeep Singh, A. K. Das, J. T. Bush, C. Lejeune, C. J. Schofield and A. Basak, Chem. Commun., 2013, 49, 1930–1932 RSC
- J. Huang, H. Xiong, R. P. Hsung, C. Rameshkumar, J. A. Mulder and T. P. Grebe, Org. Lett., 2002, 4, 2417–2420 CrossRef CAS
- J. Dommerholt, S. Schmidt, R. Temming, L. J. Hendriks, F. P. Rutjes, J. C. van Hest, D. J. Lefeber, P. Friedl and F. L. van Delft, Angew. Chem., Int. Ed., 2010, 49, 9422–9425 CrossRef CAS
- R. J. Hopkinson, A. Tumber, C. Yapp, R. Chowdhury, W. Aik, K. H. Che, X. S. Li, J. B. L. Kristensen, O. N. F. King, K. K. Y. Mun Chiang Chan, H. Choi, L. J. Walport, C. C. Thinnes, J. T. Bush, C. Lejeune, A. M. Rydzik, N. R. Rose, E. A. Bagg, M. A. McDonough, T. J. Krojer, W. W. Yue, S. S. Ng, L. Olsen, P. E. Brennan, U. Oppermann, S. Müller, R. J. Klose, P. J. Ratcliffe, C. J. Schofield and A. Kawamura, Chem. Sci., 2013, 4, 3110–3117 RSC
- I. K. Leung, M. Demetriades, A. P. Hardy, C. Lejeune, T. J. Smart, A. Szollossi, A. Kawamura, C. J. Schofield and T. D. Claridge, J. Med. Chem., 2013, 56, 547–555 CrossRef CAS
- C. J. Stubbs, C. Loenarz, J. Mecinovic, K. K. Yeoh, N. Hindley, B. M. Lienard, F. Sobott, C. J. Schofield and E. Flashman, J. Med. Chem., 2009, 52, 2799–2805 CrossRef CAS
- B. Raimer and T. Lindel, Chem.–Eur. J., 2013, 1521–3765 Search PubMed
Footnote |
| † Electronic supplementary information (ESI) available: Details for probe synthesis, production of PHD2, KD determination by NMR, photocrosslinking experiments, protein digestion, ESI-MS, MALDI and LC-MS/MS analysis. See DOI: 10.1039/c3sc51708j |
| This journal is © The Royal Society of Chemistry 2013 |